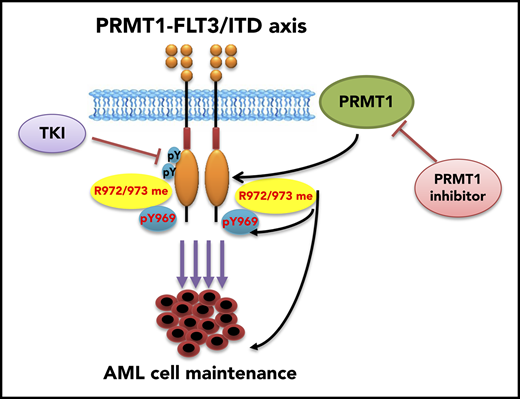
Key Points
PRMT1 promotes survival and growth of FLT3-ITD+ AML cells through methylating FLT3 protein at arginine residues 972 and 973.
PRMT1 inhibition enhances elimination of FLT3-ITD+ AML cells by FLT3 TKI treatment.

Abstract
The presence of FMS-like receptor tyrosine kinase-3 internal tandem duplication (FLT3-ITD) mutations in patients with acute myeloid leukemia (AML) is associated with poor clinical outcome. FLT3 tyrosine kinase inhibitors (TKIs), although effective in kinase ablation, do not eliminate primitive FLT3-ITD+ leukemia cells, which are potential sources of relapse. Thus, understanding the mechanisms underlying FLT3-ITD+ AML cell persistence is essential to devise future AML therapies. Here, we show that expression of protein arginine methyltransferase 1 (PRMT1), the primary type I arginine methyltransferase, is increased significantly in AML cells relative to normal hematopoietic cells. Genome-wide analysis, coimmunoprecipitation assay, and PRMT1-knockout mouse studies indicate that PRMT1 preferentially cooperates with FLT3-ITD, contributing to AML maintenance. Genetic or pharmacological inhibition of PRMT1 markedly blocked FLT3-ITD+ AML cell maintenance. Mechanistically, PRMT1 catalyzed FLT3-ITD protein methylation at arginine 972/973, and PRMT1 promoted leukemia cell growth in an FLT3 methylation–dependent manner. Moreover, the effects of FLT3-ITD methylation in AML cells were partially due to cross talk with FLT3-ITD phosphorylation at tyrosine 969. Importantly, FLT3 methylation persisted in FLT3-ITD+ AML cells following kinase inhibition, indicating that methylation occurs independently of kinase activity. Finally, in patient-derived xenograft and murine AML models, combined administration of AC220 with a type I PRMT inhibitor (MS023) enhanced elimination of FLT3-ITD+ AML cells relative to AC220 treatment alone. Our study demonstrates that PRMT1-mediated FLT3 methylation promotes AML maintenance and suggests that combining PRMT1 inhibition with FLT3 TKI treatment could be a promising approach to eliminate FLT3-ITD+ AML cells.
Introduction
To date, the overall outcome of AML has remained poor, with only 40% of younger (<60 years) and 10% of older (>60 years) patients achieving long-term survival.1 Currently available chemotherapy or targeted therapies cannot entirely eliminate leukemia clones, which underlie disease persistence and relapse. The FMS-like receptor tyrosine kinase-3 (FLT3) is highly expressed in most AML cases, suggesting its role in AML pathogenesis.2 Importantly, the activating internal tandem duplication (ITD) mutation, one of the most frequent somatic mutations in AML, is seen in mature blasts, as well as in the primitive CD34+CD38− subset.3,4 This mutation results in constant ligand-independent FLT3 kinase activation,5 making it an attractive therapeutic target.5 Currently, tyrosine kinase inhibitors (TKIs), including AC220, only partially inhibit the growth of AML cells and, when used as single agents, show only transient clinical effects.5,6 Thus, there is a pressing need to develop effective combinatory therapies, including TKI treatment, to achieve a better clinical response for FLT3-ITD+ AML patients.
Asymmetric dimethylarginine (ADMA) is formed by the addition of 2 methyl groups to a single guanidino nitrogen of an arginine (R) residue, regulating signal transduction and protein–protein interactions.7,8 Protein arginine methyltransferase 1 (PRMT1), as the predominant type I PRMT, deposits an ADMA mark onto substrates and accounts for 85% of R methylation activities in mammalian cells.9 In addition to histones, PRMT1 methylates nonhistone proteins, such as RUNX1 and EGFR, and is implicated in activities as diverse as proliferation, survival, and differentiation.10,11 In AML1-ETO9a fusion oncoprotein–transformed murine leukemia, PRMT1 methylates AML1-ETO9, promoting its transcriptional activity.11 More recently, PRMT1 was reported to be recruited by the oncogenic fusion protein MLL-GAS7 or MLL-EEN to methylate H4R3, maintaining leukemic transcriptional programs.12
Here, we first demonstrated a critical role for PRMT1 in the maintenance of FLT3-ITD+ AML through loss-of-function studies. Our proteomic and biochemical analysis revealed that PRMT1 methylates FLT3-ITD protein. Overexpression of a methylation-deficient FLT3-ITD construct in cells phenocopied PRMT1 depletion effects. Finally, we evaluated the effects of PRMT1 inhibition combined with AC220 in antagonizing AML using 2 in vivo models.
Methods
Patient samples
Samples were obtained from AML patients at the City of Hope (COH) Comprehensive Cancer Center or cord blood (CB) donors from the University of California at Los Angeles (UCLA) Center for Aids Research (CFAR) Virology Core Laboratory (supplemental Table 1, available on the Blood Web site). Sample acquisition was approved by the COH Institutional Review Board in accordance with the Declaration of Helsinki.
Mice
A PRMT1 conditional knockout (cKO) model (Mx1-Cre/PRMT1f/f) was generated by crossing PRMT1f/f mice13 with Mx1-Cre mice. Details are provided in supplemental Methods.
In vitro methylation assay
In vitro methylation was carried out using assay buffer (50 mM Tris·HCl, 5 mM MgCl2, and 4 mM dithiothreitol) at 30°C for 2 hours. For each reaction, FLT3 peptide (aa 841-993), expressed and purified from Escherichia coli, purified PRMT1 protein, and 1 µg of S-adenosyl-l-[methyl-3H] methionine were mixed simultaneously. Methylated proteins were then detected through dot blots assay using our FLT3 R972/973 dimethylation antibody (R972/973 me2a). The R972/973 me2a antibody was generated by Genemed Synthesis. Details are provided in supplemental Methods.
Statistics
Data obtained from multiple experiments were reported as the mean ± standard deviation (SD). Significance levels were determined by the Student t test, Mann-Whitney U test, or analysis of variance (ANOVA) for nonlinear distributions, and P < .05 was considered statistically significant. Other details are provided in supplemental Methods.
Results
PRMT1 inhibition blocks survival and growth of AML cells
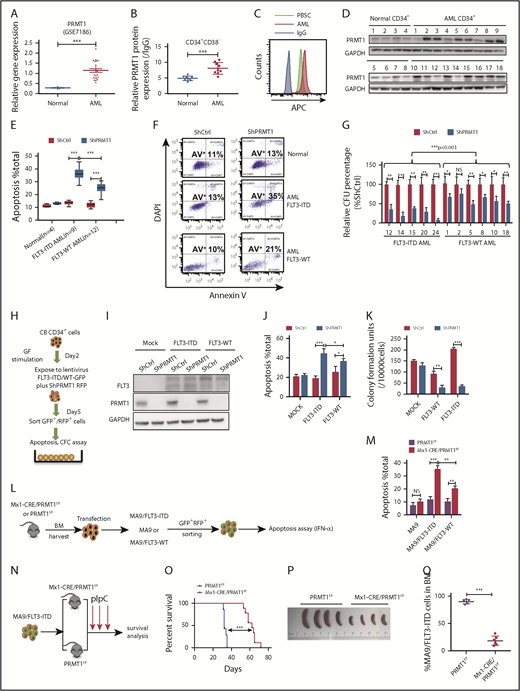
We analyzed PRMT1 expression using Gene Expression Omnibus (GEO) datasets (GSE7186, GSE13159) and found that PRMT1 levels were elevated in AML cases relative to healthy controls (Figure 1A; supplemental Figure 1A). PRMT1 messenger RNA levels were high in AML cases across all cytogenetic categories (supplemental Figure 1B). Moreover, PRMT1 levels were comparable in FLT3-ITD vs FLT3 wild-type (WT) AML (supplemental Figure 1C). Next, we assessed PRMT1 protein levels in CD34+CD38− and CD34+CD38+ subsets from AML (supplemental Table 1) and normal counterparts from mobilized peripheral blood stem cells (PBSCs) using intracellular staining (supplemental Figure 1D-F). Both AML subsets showed increased PRMT1 protein levels relative to normal counterparts (Figure 1B-C; supplemental Figure 1G). Western blot analysis confirmed elevated PRMT1 expression in AML (Figure 1D).

PRMT1 inhibition perturbs AML survival and growth. (A) Comparison of PRMT1 messenger RNA expression in mononuclear cells from BM or peripheral blood of healthy donors vs primary AML patients based on a GEO dataset (GSE7186). (B) PRMT1 protein levels in the CD34+CD38− subset from normal PBSCs (n = 8) and AML cases (n = 9), as analyzed by anti-PRMT1 intracellular staining. PRMT1 level is calculated as median fluorescence intensity of PRMT1 staining relative to immunoglobulin G (IgG) control. (C) Representative intracellular staining results are shown. (D) Western blot analysis of PRMT1 expression in primary human CD34+ cells from AML specimens (n = 18) and normal PBSC donors (n = 8). (E-F) Apoptosis of normal PBSCs (n = 4), CD34+ cells, and FLT3-ITD (n = 9) or FLT3 WT (n = 12) AML CD34+ cells transduced with ShCtrl or ShPRMT1 (targeting 3′UTR), as analyzed by Annexin V/4′,6-diamidino-2-phenylindole (DAPI) labeling. Within the FLT3-ITD+ AML and FLT3 WT AML groups, PRMT1 KD was associated with higher apoptosis levels. (E) Two-way ANOVA analyses with repeated measures revealed a statistically significant difference (P < .001) in the apoptosis increase between the 2 groups (FLT3-ITD vs FLT3 WT), indicating that PRMT1 KD induced more apoptosis in FLT3-ITD+ AML cells than in FLT3 WT AML cells. (F) Representative fluorescence-activated cell sorting plots. (G) CFC assay of FLT3-ITD (n = 5) and FLT3 WT (n = 6) AML CD34+ cells expressing ShCtrl or ShPRMT1. Colony numbers were normalized to that of ShCtrl-expressing cells. Within the FLT3-ITD+ AML and FLT3 WT AML groups, PRMT1 KD was associated with lower CFCs. Two-way ANOVA analyses with repeated measures revealed a statistically significant difference (P < .001) in the CFC decrease between the 2 groups (FLT3-ITD vs FLT3 WT). (H) CB CD34+ cells were transfected with vector control (mock), FLT3 WT, or FLT3-ITD and then further transduced with ShCtrl or ShPRMT1. Doubly transduced cells were assayed by western blotting for FLT3 and PRMT1 expression (I), for apoptosis by annexin V/DAPI labeling (J), and for CFCs (K). (L) BM cells from Mx1-Cre/PRMT1f/f or PRMT1f/f mice were transduced with a retroviral vector coexpressing MA9 plus GFP and then a lentiviral vector coexpressing FLT3-ITD or FLT3 WT plus RFP. (M) MA9, MA9/FLT3-ITD, and MA9/FLT3 WT cells, as indicated, were used to assess apoptosis in vitro after PRMT1 deletion. (N) Doubly transformed MA9/FLT3-ITD cells were transplanted into CD45.1-expressing congenic recipients to analyze leukemia progression. (O) Survival after PIPC treatment was monitored in PRMT1f/f (n = 8) and Mx1-Cre/PRMT1f/f (n = 9) groups. (P) Effects of PRMT1 deletion on splenomegaly were evaluated after the last dose of PIPC. (Q) Percentage of donor MA9/FLT3-ITD cells in BM of recipients (n = 6 per group) from the indicated group. Results represent the mean ± standard deviation. *P < .05, **P < .01, ***P < .001. NS, not statistically significant.
PRMT1 inhibition perturbs AML survival and growth. (A) Comparison of PRMT1 messenger RNA expression in mononuclear cells from BM or peripheral blood of healthy donors vs primary AML patients based on a GEO dataset (GSE7186). (B) PRMT1 protein levels in the CD34+CD38− subset from normal PBSCs (n = 8) and AML cases (n = 9), as analyzed by anti-PRMT1 intracellular staining. PRMT1 level is calculated as median fluorescence intensity of PRMT1 staining relative to immunoglobulin G (IgG) control. (C) Representative intracellular staining results are shown. (D) Western blot analysis of PRMT1 expression in primary human CD34+ cells from AML specimens (n = 18) and normal PBSC donors (n = 8). (E-F) Apoptosis of normal PBSCs (n = 4), CD34+ cells, and FLT3-ITD (n = 9) or FLT3 WT (n = 12) AML CD34+ cells transduced with ShCtrl or ShPRMT1 (targeting 3′UTR), as analyzed by Annexin V/4′,6-diamidino-2-phenylindole (DAPI) labeling. Within the FLT3-ITD+ AML and FLT3 WT AML groups, PRMT1 KD was associated with higher apoptosis levels. (E) Two-way ANOVA analyses with repeated measures revealed a statistically significant difference (P < .001) in the apoptosis increase between the 2 groups (FLT3-ITD vs FLT3 WT), indicating that PRMT1 KD induced more apoptosis in FLT3-ITD+ AML cells than in FLT3 WT AML cells. (F) Representative fluorescence-activated cell sorting plots. (G) CFC assay of FLT3-ITD (n = 5) and FLT3 WT (n = 6) AML CD34+ cells expressing ShCtrl or ShPRMT1. Colony numbers were normalized to that of ShCtrl-expressing cells. Within the FLT3-ITD+ AML and FLT3 WT AML groups, PRMT1 KD was associated with lower CFCs. Two-way ANOVA analyses with repeated measures revealed a statistically significant difference (P < .001) in the CFC decrease between the 2 groups (FLT3-ITD vs FLT3 WT). (H) CB CD34+ cells were transfected with vector control (mock), FLT3 WT, or FLT3-ITD and then further transduced with ShCtrl or ShPRMT1. Doubly transduced cells were assayed by western blotting for FLT3 and PRMT1 expression (I), for apoptosis by annexin V/DAPI labeling (J), and for CFCs (K). (L) BM cells from Mx1-Cre/PRMT1f/f or PRMT1f/f mice were transduced with a retroviral vector coexpressing MA9 plus GFP and then a lentiviral vector coexpressing FLT3-ITD or FLT3 WT plus RFP. (M) MA9, MA9/FLT3-ITD, and MA9/FLT3 WT cells, as indicated, were used to assess apoptosis in vitro after PRMT1 deletion. (N) Doubly transformed MA9/FLT3-ITD cells were transplanted into CD45.1-expressing congenic recipients to analyze leukemia progression. (O) Survival after PIPC treatment was monitored in PRMT1f/f (n = 8) and Mx1-Cre/PRMT1f/f (n = 9) groups. (P) Effects of PRMT1 deletion on splenomegaly were evaluated after the last dose of PIPC. (Q) Percentage of donor MA9/FLT3-ITD cells in BM of recipients (n = 6 per group) from the indicated group. Results represent the mean ± standard deviation. *P < .05, **P < .01, ***P < .001. NS, not statistically significant.
Next, to examine PRMT1 function, we inhibited PRMT1 expression using lentiviral vectors expressing short hairpin RNAs (ShRNAs). PRMT1 knockdown (KD) inhibited cell growth and induced apoptosis (supplemental Figure 1H-J). We also confirmed specificity of 1 ShRNA against PRMT1 (ShPRMT1) sequence targeting the 3′ (UTR) by overexpressing PRMT1 complementary DNA lacking the 3′UTR (supplemental Figure 1K-L). We then assessed the effects of PRMT1 KD in primary AML and normal PBSCs. As anticipated, PRMT1 KD reduced AML cell colony growth and potently increased apoptosis but had little effect on normal counterparts (Figure 1E-F; supplemental Figure 1M). Specifically, PRMT1 KD had more potent inhibitory effects in FLT3-ITD+ AML cells than in FLT3 WT AML cells (Figure 1E-G). Two-way ANOVA analyses with repeated measures revealed a statistically significant (P < .001) difference in apoptosis and colony formation between the FLT3-ITD and FLT3 WT groups. To further assess whether PRMT1 KD preferentially inhibited viability of FLT3-ITD–expressing cells, we cotransduced CB CD34+ cells with 2 types of lentiviral vectors: 1 type coexpressing FLT3-ITD, FLT3 WT, or vector control (mock) with GFP and the other coexpressing ShPRMT1 or control ShRNA (ShCtrl) plus RFP (Figure 1H-I). CB CD34+ cells ectopically expressing FLT3-ITD or FLT3 WT showed greater sensitivity to ShPRMT1-mediated apoptosis or colony-forming cell (CFC) inhibition than did mock cells (Figure 1J-K). Importantly, more robust inhibitory effects were seen in FLT3-ITD–transduced cells relative to FLT3 WT–transduced cells upon PRMT1 KD (Figure 1J-K).
We then assessed the effects of PRMT1 deletion in an MLL-AF9 plus FLT3 double-transformation murine AML model using bone marrow (BM) cells from PRMT1 cKO (Mx1-Cre/PRMT1f/f) mouse. Consistent with previous results,12 we observed that ex vivo deletion of PRMT1 only modestly affected apoptosis of murine BM cells transduced with MLL-AF9 (MA9) alone (Figure 1L-M). We did not exclude the possibility that in vivo deletion of PRMT1 may affect leukemogenesis induced by MA9 alone. We next used MA9 plus FLT3-ITD or FLT3 WT doubly transformed AML cells for ex vitro experiments. Briefly, we first transduced c-Kit+ BM cells from PRMT1 cKO (supplemental Figure 1N) or WT control (PRMT1f/f) mice with a retroviral vector coexpressing GFP plus MA9 (Figure 1L). We then sorted cells according to GFP expression and infected them with a lentiviral vector coexpressing RFP plus FLT3-ITD or FLT3 WT with comparable transduction efficiency (supplemental Figure 1O). We induced PRMT1 deletion in doubly transformed cells by ex vivo interferon-α administration and observed more robust apoptosis in FLT3-ITD cells than in FLT3 WT cells (Figure 1M). Moreover, we validated the functional link between FLT3-ITD and PRMT1 in FLT3-ITD–transduced NHD13+ cells.14 In this NHD13+/PRMT1f/f/Mx1Cre model, PRMT1 deletion also induced more robust apoptosis in FLT3-ITD–overexpressing NHD13+ cells than in FLT3 WT–overexpressing NHD13+ cells (supplemental Figure 1P).
To further determine whether PRMT1 deletion blocks FLT3-ITD+ AML cell growth in vivo, we transplanted MA9/FLT3-ITD doubly transduced PRMT1 cKO cells or their corresponding control cells into CD45.1-expressing congenic recipients for leukemia assessment (Figure 1N). PRMT1-deficient MA9/FLT3-ITD leukemic mice showed significantly extended survival (Figure 1O), reduced splenomegaly (Figure 1P), and leukemic chimerism (Figure 1Q) relative to controls. Furthermore, mice in the PRMT1-deficient group died of leukemia eventually, and PRMT1 expression was still detectable in leukemia cells, suggesting that leukemia reoccurrence might be due to expansion of PRMT1-expressing cells that remained after polyinosinic-polycytidylic acid (PIPC) treatment (supplemental Figure 1Q).
PRMT1 cooperates with FLT3-ITD in AML
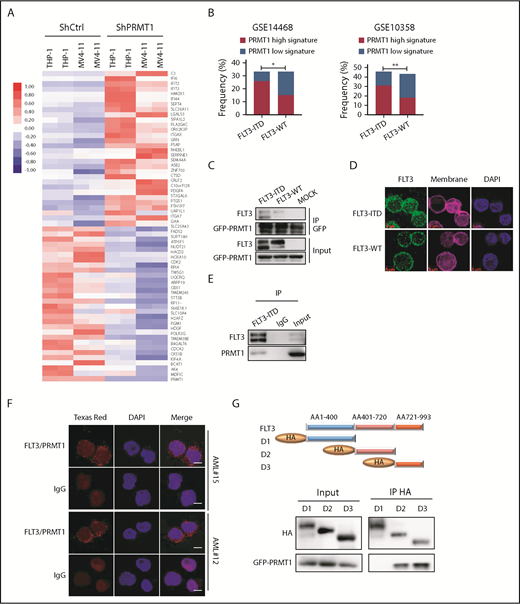
We then asked whether PRMT1 function is more relevant to the pathogenesis of FLT3-ITD+ AML than that of FLT3 WT AML. Thus, we transduced FLT3-ITD–transformed CB CD34+ cells plus AML cell lines (MV4-11 and THP-1) along with ShCtrl or ShPRMT1 and performed RNA sequencing (RNA-Seq). Efficient PRMT1 KD induced a marked change in transcriptional profile: of differentially expressed genes common to the 3 lines, 97 were significantly downregulated and 104 were significantly upregulated (fold change > 1.5, P < .05) (Figure 2A; supplemental Figure 2A-C). We defined these 201 genes as a PRMT1 signature and then used the identified PRMT1 signature (supplemental Table 2) to query 2 gene-expression datasets containing large cohorts of AML cases (GSE14468, GSE10358), each with available somatic mutation information, including FLT3-ITD status, by single sample gene set enrichment analysis, as described previously.15 In each AML case, we evaluated a PRMT1-high (normalized score ≥ 0.75) or PRMT1-low (normalized score ≤ −0.75) signature score based on single sample gene set enrichment analysis. This evaluation revealed that patients with the PRMT1-high signature were predominantly present in the AML subgroup with FLT3-ITD (Figure 2B), whereas there was no significant positive correlation between the PRMT1-high signature and other somatic mutations commonly seen in AML (supplemental Figure 2D-G). We then performed RNA-Seq on CB CD34+ cells transduced with FLT3-ITD or vector control (mock) and identified 2043 genes that were differentially expressed in FLT3-ITD vs mock cells (fold change > 1.5, P < .05) as potential FLT3-ITD targets (supplemental Figure 2H). We also identified 954 genes that were differentially expressed (fold change > 1.5, P < .05) between FLT3-ITD–expressing CB CD34+ cells transduced with ShPRMT1 and the cells transduced with ShCtrl. Among those, 282 genes (supplemental Table 3) overlapped with FLT3-ITD targets (supplemental Figure 2I). We consider those genes to be candidates regulated by the PRMT1/FLT3-ITD axis.

PRMT1 cooperates with FLT3-ITD in regulation of AML. (A) Heat map showing the top 30 upregulated and top 30 downregulated genes in THP-1 and MV4-11 lines expressing ShPRMT1 or ShCtrl, based on a fold-change > 1.5 and P < .05. (B) Bar graphs representing the percentage frequency of primary AML specimens with FLT3-ITD vs FLT3 WT and exhibiting PRMT1 high (normalized score ≥ 0.75) or PRMT1 low (normalized score ≤ −0.75) signatures, based on 2 GEO cohorts (GSE14468, GSE10358). *P < .05, **P < .01, Fisher’s exact test. (C) Co-IP of GFP from GFP-tagged PRMT1-transduced 32D cells ectopically expressing FLT3-ITD, FLT3 WT, or mock and then analyzed for FLT3 and GFP by western blotting. (D) Immunostaining for FLT3 (green), membrane (pink), and 4′,6-diamidino-2-phenylindole (DAPI; blue) in CD34+CD38− cells from primary AML specimens with or without FLT3-ITD. Scale bar, 5 μm. (E) Co-IP of endogenous FLT3 from MV4-11 cells after IP with FLT3 antibody followed by FLT3 and PRMT1. (F) Representative images of Duolink in situ proximity ligation assay in primary AML CD34+CD38- cells from 2 primary FLT3-ITD+ AML specimens. Red spots indicate PRMT1/FLT3 protein interaction (left), DAPI-stained nuclei are blue (center), and merged image is at right. Scale bar, 5 μm. (G) Hemagglutinin (HA)-tagged FLT3 (ITD 18 bp in-frame insertion at E596 within the juxtamembrane domain) fragments (D1-D3) and GFP-tagged PRMT1 were coexpressed in 293T cells, followed by pull-down with an anti-HA antibody and western blot for HA and GFP-PRMT1.
PRMT1 cooperates with FLT3-ITD in regulation of AML. (A) Heat map showing the top 30 upregulated and top 30 downregulated genes in THP-1 and MV4-11 lines expressing ShPRMT1 or ShCtrl, based on a fold-change > 1.5 and P < .05. (B) Bar graphs representing the percentage frequency of primary AML specimens with FLT3-ITD vs FLT3 WT and exhibiting PRMT1 high (normalized score ≥ 0.75) or PRMT1 low (normalized score ≤ −0.75) signatures, based on 2 GEO cohorts (GSE14468, GSE10358). *P < .05, **P < .01, Fisher’s exact test. (C) Co-IP of GFP from GFP-tagged PRMT1-transduced 32D cells ectopically expressing FLT3-ITD, FLT3 WT, or mock and then analyzed for FLT3 and GFP by western blotting. (D) Immunostaining for FLT3 (green), membrane (pink), and 4′,6-diamidino-2-phenylindole (DAPI; blue) in CD34+CD38− cells from primary AML specimens with or without FLT3-ITD. Scale bar, 5 μm. (E) Co-IP of endogenous FLT3 from MV4-11 cells after IP with FLT3 antibody followed by FLT3 and PRMT1. (F) Representative images of Duolink in situ proximity ligation assay in primary AML CD34+CD38- cells from 2 primary FLT3-ITD+ AML specimens. Red spots indicate PRMT1/FLT3 protein interaction (left), DAPI-stained nuclei are blue (center), and merged image is at right. Scale bar, 5 μm. (G) Hemagglutinin (HA)-tagged FLT3 (ITD 18 bp in-frame insertion at E596 within the juxtamembrane domain) fragments (D1-D3) and GFP-tagged PRMT1 were coexpressed in 293T cells, followed by pull-down with an anti-HA antibody and western blot for HA and GFP-PRMT1.
We then used lentivirally expressed FLT3 ShRNA to inhibit FLT3 expression in primary AML cells from a specimen harboring a homozygous FLT3-ITD mutation and observed robust apoptosis upon FLT3-KD (supplemental Figure 2J-L). By contrast, blocking FLT3 kinase activity by AC220 treatment, although effective in depleting tyrosine autophosphorylation, only modestly induced apoptosis, suggesting that FLT3 could promote leukemia cell growth/survival through kinase activity–independent mechanisms.
Next, we evaluated whether PRMT1 regulates FLT3 function through direct physical binding. To do so, we first transduced murine 32D cells overexpressing PRMT1 fused to GFP with FLT3 WT or FLT3-ITD constructs and then performed coimmunoprecipitation (co-IP) using an anti-GFP antibody. That analysis indicated that PRMT1 interacts with FLT3 protein (Figure 2C) and that the interaction was more robust with FLT3-ITD relative to FLT3 WT (Figure 2C). Also, as reported by other investigators,16,17 we observed that FLT3-ITD protein is primarily cytoplasmic, whereas WT FLT3 protein is mainly localized to the membrane (Figure 2D). We further confirmed interaction between endogenous FLT3-ITD and PRMT1 proteins by co-IP in MV4-11 cells (Figure 2E). Moreover, a proximity ligation assay confirmed interaction in situ, as evidenced by punctate red fluorescence (Figure 2F) in primary AML cells harboring the FLT3-ITD mutation. Finally, domain-mapping analysis indicated that PRMT1 preferentially interacts with the FLT3-ITD domain 3, which encodes the kinase C terminus (Figure 2G).
PRMT1 catalyzes methylation of FLT3-ITD protein at R residues 972/973
We next asked whether R residues on FLT3-ITD protein are methylated. Our mass spectrometry analysis identified 6 dimethylated R residues on endogenous FLT3 protein (Figure 3A; supplemental Figure 3A) from MV4-11 cells. Given that the FLT3 C terminus preferentially interacts with PRMT1, we further assessed only R residues at the C terminus for potential contribution to FLT3 ADMA. Accordingly, we established a series of FLT3-ITD methylation-deficient mutants (R to lysine [K]; RK). Notably, mutating R972 or R973 residues (R972K or R973K), but not R655 and R845 residues, significantly reduced FLT3 ADMA levels in 293T cells ectopically expressing FLT3-ITD (Figure 3B). Moreover, residues R972 and R973 were conserved across various species (supplemental Figure 3B). To assess endogenous FLT3-ITD methylation, we generated the R972/973 me2a antibody. Through dot blot analysis, this antibody was confirmed to specifically recognize R972/973 asymmetrically dimethylated peptide but not a corresponding unmodified or symmetrically dimethylated peptide (Figure 3C). In 293T cells ectopically expressing WT FLT3-ITD or various FLT3-ITD mutant constructs including FLT3-ITD R972/973K (R972/973K), ADMA levels recognized by the antibody were lower in the R972K or R973K single-mutation group than WT FLT3-ITD and were completely abolished in the R972/973K double-mutant group (Figure 3D).
![Figure 3. PRMT1 catalyzes FLT3-ITD protein methylation at R972/973. (A) Schematic model showing dimethylated (Di-Me) arginines identified following immunoprecipitation of endogenous FLT3 protein from MV4-11 cells. The precipitates were subjected to proteomic analysis. (B) FLT3 protein was immunoprecipitated from 293T cells ectopically expressing FLT3-ITD or FLT3-ITD methylation-deficient mutants (R655K, R845K, R972K, and R973K) and then analyzed for ADMA levels by western blotting. (C) Dot blot showing that the FLT3-R972/973 me2a antibody specifically binds asymmetric dimethylated R972/973 peptide. Amino acid sequence of peptides corresponding to the FLT3 966-980 region, in which R972 and R973 are unmodified, monomethylated, or dimethylated. Different amounts of peptides were spotted on polyvinylidene difluoride membranes and detected by control anti-FLT3 or anti-FLT3 asymmetric methylated Arg 972/973 (R972/973 me2a) antibodies. (D) 293T cells transfected with mock, WT FLT3-ITD, or FLT3-ITD methylation-deficient mutants (R655K, R845K, R972K and R973K) were analyzed by western blot using the FLT3 R972/973 me2a antibody. (E) In vitro methylation assay of GST-tagged FLT3 peptides (WT, R972K, R973K, and R972/973K) in the presence of PRMT1 protein and S-adenosyl-l-[methyl-3H] methionine (SAM). Peptide methylation was analyzed by western blotting using the methylation antibody. (F) 293T cells were transduced with lentiviral vector expressing FLT3-ITD and then transfected with mock or the indicated GFP-fused type I PRMTs. FLT3-ITD protein was immunoprecipitated from cells and analyzed for GFP by western blotting. MV4-11 cells transduced with ShCtrl, ShPRMT1 (G), or ShPRMT6 (H) were analyzed by western blotting for PRMT1 or PRMT6, FLT3 R972/973 me2a, total FLT3, and GAPDH. CT, C terminus; ECD, extracellular domain; JM juxtamembrane domain; KI, kinase insert; TK1, tyrosine kinase 1; TK2, tyrosine kinase 2; TM, transmembrane.](https://ash.silverchair-cdn.com/ash/content_public/journal/blood/134/6/10.1182_blood.2019001282/3/m_bloodbld2019001282f3.png?Expires=1767755470&Signature=x4h4MNna6MPZU51tVJYp426SfzgrYdzmuv7yKILlWdqYtgtlKErXUf5hMZ60bxSNi-aeFhhXxfsqddNFpOyKz6J4Axg~JbnuEqAYkM5Lckrq-IY8S7LNJbzIAnNH8-eJ9I1a7ZMOesEIZCNiJpWnGG9y0xmjZv-5KVKq-s~CYxNXrI-pwZbyvztGmPaTo8n9O2nDIHv3KDOcMd-o2J~TOw6-GAR9H91pwxJesYKTmdeget669m~J1hSSoM6zUIazfNxzLSXqFbFqpr1pVJK23cBl0gOTUYa7xuFMgSFzkTk8QkjfeiktPFYeU3Crf2mhn6L4IKulWCXqx66xpDHaTg__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
PRMT1 catalyzes FLT3-ITD protein methylation at R972/973. (A) Schematic model showing dimethylated (Di-Me) arginines identified following immunoprecipitation of endogenous FLT3 protein from MV4-11 cells. The precipitates were subjected to proteomic analysis. (B) FLT3 protein was immunoprecipitated from 293T cells ectopically expressing FLT3-ITD or FLT3-ITD methylation-deficient mutants (R655K, R845K, R972K, and R973K) and then analyzed for ADMA levels by western blotting. (C) Dot blot showing that the FLT3-R972/973 me2a antibody specifically binds asymmetric dimethylated R972/973 peptide. Amino acid sequence of peptides corresponding to the FLT3 966-980 region, in which R972 and R973 are unmodified, monomethylated, or dimethylated. Different amounts of peptides were spotted on polyvinylidene difluoride membranes and detected by control anti-FLT3 or anti-FLT3 asymmetric methylated Arg 972/973 (R972/973 me2a) antibodies. (D) 293T cells transfected with mock, WT FLT3-ITD, or FLT3-ITD methylation-deficient mutants (R655K, R845K, R972K and R973K) were analyzed by western blot using the FLT3 R972/973 me2a antibody. (E) In vitro methylation assay of GST-tagged FLT3 peptides (WT, R972K, R973K, and R972/973K) in the presence of PRMT1 protein and S-adenosyl-l-[methyl-3H] methionine (SAM). Peptide methylation was analyzed by western blotting using the methylation antibody. (F) 293T cells were transduced with lentiviral vector expressing FLT3-ITD and then transfected with mock or the indicated GFP-fused type I PRMTs. FLT3-ITD protein was immunoprecipitated from cells and analyzed for GFP by western blotting. MV4-11 cells transduced with ShCtrl, ShPRMT1 (G), or ShPRMT6 (H) were analyzed by western blotting for PRMT1 or PRMT6, FLT3 R972/973 me2a, total FLT3, and GAPDH. CT, C terminus; ECD, extracellular domain; JM juxtamembrane domain; KI, kinase insert; TK1, tyrosine kinase 1; TK2, tyrosine kinase 2; TM, transmembrane.
PRMT1 catalyzes FLT3-ITD protein methylation at R972/973. (A) Schematic model showing dimethylated (Di-Me) arginines identified following immunoprecipitation of endogenous FLT3 protein from MV4-11 cells. The precipitates were subjected to proteomic analysis. (B) FLT3 protein was immunoprecipitated from 293T cells ectopically expressing FLT3-ITD or FLT3-ITD methylation-deficient mutants (R655K, R845K, R972K, and R973K) and then analyzed for ADMA levels by western blotting. (C) Dot blot showing that the FLT3-R972/973 me2a antibody specifically binds asymmetric dimethylated R972/973 peptide. Amino acid sequence of peptides corresponding to the FLT3 966-980 region, in which R972 and R973 are unmodified, monomethylated, or dimethylated. Different amounts of peptides were spotted on polyvinylidene difluoride membranes and detected by control anti-FLT3 or anti-FLT3 asymmetric methylated Arg 972/973 (R972/973 me2a) antibodies. (D) 293T cells transfected with mock, WT FLT3-ITD, or FLT3-ITD methylation-deficient mutants (R655K, R845K, R972K and R973K) were analyzed by western blot using the FLT3 R972/973 me2a antibody. (E) In vitro methylation assay of GST-tagged FLT3 peptides (WT, R972K, R973K, and R972/973K) in the presence of PRMT1 protein and S-adenosyl-l-[methyl-3H] methionine (SAM). Peptide methylation was analyzed by western blotting using the methylation antibody. (F) 293T cells were transduced with lentiviral vector expressing FLT3-ITD and then transfected with mock or the indicated GFP-fused type I PRMTs. FLT3-ITD protein was immunoprecipitated from cells and analyzed for GFP by western blotting. MV4-11 cells transduced with ShCtrl, ShPRMT1 (G), or ShPRMT6 (H) were analyzed by western blotting for PRMT1 or PRMT6, FLT3 R972/973 me2a, total FLT3, and GAPDH. CT, C terminus; ECD, extracellular domain; JM juxtamembrane domain; KI, kinase insert; TK1, tyrosine kinase 1; TK2, tyrosine kinase 2; TM, transmembrane.
To assess whether PRMT1 directly catalyzes R972/973 methylation, we performed an in vitro methylation assay and detected methylation through our R972/973 me2a antibody. Notably, we observed methylation of an unmodified FLT3 peptide (aa 841-993) at R972/973 but not of an R972/973K double-mutant peptide (Figure 3E). Given that other PRMTs are also present in AML cells, we included other type I PRMTs, in addition to PRMT1, in a co-IP analysis with FLT3-ITD. Specifically, 293T cells were cotransfected with candidate GFP-tagged PRMTs plus FLT3-ITD, followed by FLT3 immunoprecipitation. Immunoblotting with GFP revealed that only PRMT1, PRMT6, and PRMT8 bound FLT3-ITD (Figure 3F). Because PRMT8 expression is restricted to brain,8 we did not analyze it further. Additionally, we did not find any interaction between endogenous PRMT6 and FLT3-ITD proteins by co-IP in MV4-11 cells (supplemental Figure 3C). More importantly, KD of endogenous PRMT1, but not PRMT6, decreased R972/973 methylation (Figure 3G-H), strongly suggesting that PRMT1 specifically methylates endogenous FLT3 at R972/973. Moreover, KD of endogenous PRMT6 did not alter FLT3 total ADMA levels (supplemental Figure 3D).
PRMT1 regulates AML cells through catalyzing FLT3-ITD R972/973 methylation
Methylation of the EGFR at R1175 reportedly modulates EGFR trans-autophosphorylation at nearby tyrosine residue 1173.18 Thus, we first assessed the impact of methylation loss on phosphorylation of nearby tyrosine residues. Because endogenous FLT3 protein is not detectable in murine BM cells transformed by MA9 alone, we further transduced MA9+ BM cells with the FLT3-ITD R972/973K mutant or corresponding control construct (WT FLT3-ITD). We observed that tyrosine 969 (Y969) phosphorylation was totally abolished in R972/973K-expressing cells, in contrast to phosphorylated (phospho)-Y589/591 and phospho-Y842 (Figure 4A). To exclude the possible effects of protein misfolding caused by mutagenesis, we assessed FLT3 Y969 phosphorylation levels using PRMT1-cKO mouse BM cells. To do so, we cotransduced MA9 and FLT3-ITD into BM cells from PRMT1-cKO mice or their corresponding control mice. Consistently, following induction of PRMT1 deletion, PRMT1-cKO cells showed decreased FLT3 Y969 phosphorylation relative to controls (Figure 4B). We also measured downstream signals in cells expressing R972/973K vs WT FLT3-ITD and included a phosphorylation-deficient (Y969F [phenylalanine]) FLT3-ITD mutant for comparison. AKT phosphorylation levels decreased to the same extent in cells expressing R972/973K or Y969F compared with WT FLT3-ITD (Figure 4A). Notably, phospho-STAT5 only decreased in cells expressing R972/973K (Figure 4A). Consistently, PRMT1 deletion also significantly reduced phospho-STAT5 levels in MA9/FLT3-ITD doubly transformed cells (Figure 4B).
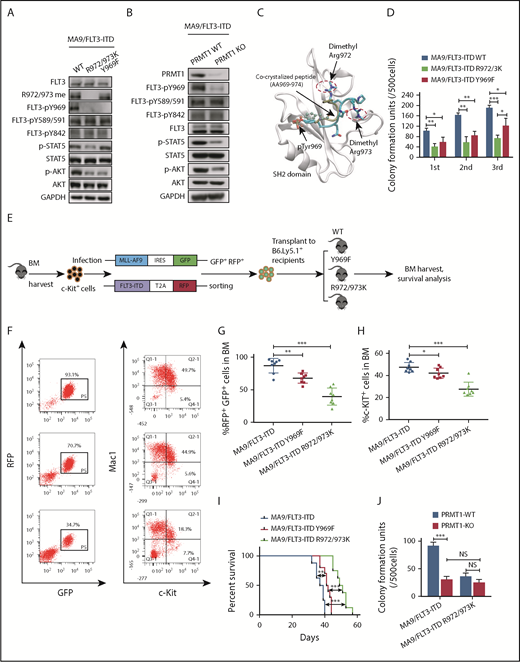
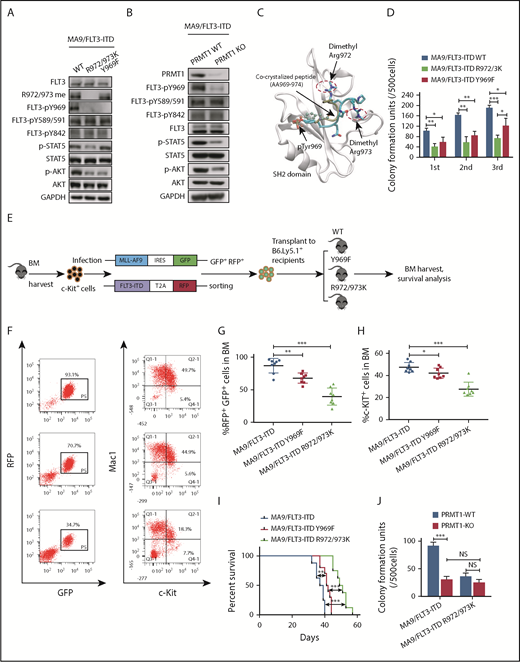
PRMT1 regulates AML cell maintenance by methylating FLT3-ITD R972/973. (A) Western blot analysis of indicated proteins in MA9-expressing murine BM cells that were further transduced with WT FLT3-ITD, FLT3-ITD Y969F, or FLT3-ITD R972/973K. (B) Following ex vivo PRMT1 deletion, Mx1-Cre/PRMT1f/f BM cells or control BM cells, both cotransduced with MA9 plus FLT3-ITD, were immunoblotted for the indicated total and phosphorylated proteins plus GAPDH. (C) The binding pose of the dimethylated peptide from molecular dynamics simulation is shown on the GRB2 SH2 domain protein (aa 56-153). Protein backbone is shown in white. Peptide backbone is shown in cyan with amino acid side chains shown in licorice mode (aa 969-974). Hydrogen atoms are omitted for clarity. Atom color: cyan, carbon; blue, nitrogen; red, oxygen. The position of the cocrystallized peptide (PDB 1BMB) is shown in tan with phospho-tyrosine shown in CPK mode. (D) MA9-expressing BM cells further transduced with FLT3-ITD, FLT3-ITD Y969F, or FLT3-ITD R972/973K were seeded for serial replating assays. (E) c-Kit+ BM cells were transduced with retroviral vectors coexpressing MLL-AF9 (MA9) plus GFP and then with lentiviral vectors coexpressing FLT3-ITD WT, FLT3-ITD Y969F, or FLT3-ITD R972/973K plus RFP. Double-positive cells were sorted and transplanted into CD45.1-expressing congenic recipients, and donor cell engraftment was analyzed 4 weeks later. (F) Representative fluorescence-activated cell sorting profile for total donor cells (GFP/RFP double-positive) and c-Kit + cells in BM of the indicated recipients. (G-H) Cumulative results are shown. (I) Survival of mice transplanted with the indicated doubly transduced murine BM cells. (J) CFC analysis of PRMT1-cKO vs PRMT1 WT doubly transformed cells. MA9-expressing BM cells from PRMT1f/f and Mx1-CRE/PRMT1f/f mice were further transduced with FLT3-ITD or FLT3-ITD R972/973K, PRMT1 deletion was induced ex vivo, and cells were seeded for CFC assays. Results represent the mean ± SD. *P < .05, **P < .01, ***P < .001.
PRMT1 regulates AML cell maintenance by methylating FLT3-ITD R972/973. (A) Western blot analysis of indicated proteins in MA9-expressing murine BM cells that were further transduced with WT FLT3-ITD, FLT3-ITD Y969F, or FLT3-ITD R972/973K. (B) Following ex vivo PRMT1 deletion, Mx1-Cre/PRMT1f/f BM cells or control BM cells, both cotransduced with MA9 plus FLT3-ITD, were immunoblotted for the indicated total and phosphorylated proteins plus GAPDH. (C) The binding pose of the dimethylated peptide from molecular dynamics simulation is shown on the GRB2 SH2 domain protein (aa 56-153). Protein backbone is shown in white. Peptide backbone is shown in cyan with amino acid side chains shown in licorice mode (aa 969-974). Hydrogen atoms are omitted for clarity. Atom color: cyan, carbon; blue, nitrogen; red, oxygen. The position of the cocrystallized peptide (PDB 1BMB) is shown in tan with phospho-tyrosine shown in CPK mode. (D) MA9-expressing BM cells further transduced with FLT3-ITD, FLT3-ITD Y969F, or FLT3-ITD R972/973K were seeded for serial replating assays. (E) c-Kit+ BM cells were transduced with retroviral vectors coexpressing MLL-AF9 (MA9) plus GFP and then with lentiviral vectors coexpressing FLT3-ITD WT, FLT3-ITD Y969F, or FLT3-ITD R972/973K plus RFP. Double-positive cells were sorted and transplanted into CD45.1-expressing congenic recipients, and donor cell engraftment was analyzed 4 weeks later. (F) Representative fluorescence-activated cell sorting profile for total donor cells (GFP/RFP double-positive) and c-Kit + cells in BM of the indicated recipients. (G-H) Cumulative results are shown. (I) Survival of mice transplanted with the indicated doubly transduced murine BM cells. (J) CFC analysis of PRMT1-cKO vs PRMT1 WT doubly transformed cells. MA9-expressing BM cells from PRMT1f/f and Mx1-CRE/PRMT1f/f mice were further transduced with FLT3-ITD or FLT3-ITD R972/973K, PRMT1 deletion was induced ex vivo, and cells were seeded for CFC assays. Results represent the mean ± SD. *P < .05, **P < .01, ***P < .001.
FLT3 phospho-Y969 reportedly acts as a docking site for the SH2 domains of cytosolic signaling molecules.19 Thus, to evaluate whether methylated R972/973 functions to recruit other factors containing an SH2 domain to phospho-Y969, we performed molecular dynamics simulations of a phospho-FLT3 C-terminal (aa 969-974) peptide pYQNRRP in complex with the SH2 domain. Our simulated binding pose of the FLT3 peptide was in excellent agreement with the binding pose of the peptide in the crystal structure (Figure 4C). The van der Waals interaction was more favorable (3.10 kcal/mol lower) in dimethylated peptide compared with nonmethylated peptide. The electrostatic interaction was also more favorable (6.26 kcal/mol lower) in dimethylated peptide (supplemental Table 4). To confirm this, we performed peptide pull-down assays and observed that greater amounts of GST-GRB2 protein were pulled down by phospho-Y969-R972/973 me2a (Y phosphorylation plus R methylation) double-modified peptide relative to phospho-Y969 (Y phosphorylation alone) peptide (supplemental Figure 4A). Taken together, these results suggest that R972/973 methylation enhances binding of the phospho-FLT3 C terminus to cytosolic signaling molecules containing an SH2 domain.
Next, we undertook phenotypic studies in MA9 cells expressing WT FLT3-ITD or R972/973K or Y969F mutants. Interestingly, murine MA9 cells harboring R972/973K showed significantly decreased CFCs compared with cells expressing Y969F in the third replating; both displayed compromised CFCs relative to cells expressing WT FLT3-ITD (Figure 4D). We further performed an in vivo transplantation assay (Figure 4E). Although expression of Y969F decreased leukemia cell engraftment relative to that of WT FLT3-ITD (Figure 4F-G), its effects on the reduction of engraftment of c-Kit+ cells in BM and extension of mouse survival were modest (Figure 4F,H-I). Notably, expression of Y969F did not alter FLT3-ITD R972/973 methylation level (Figure 4A). In contrast, expression of R972/973K significantly decreased BM engraftment of bulk, as well as c-Kit+, leukemia cells, without altering leukemia cell myeloid differentiation (supplemental Figure 4B-C). Importantly, expression of R972/973K remarkably extended animal survival (Figure 4H-I), indicating that the effects of R972/973 methylation on leukemic cell growth are not solely dependent on Y969 phosphorylation.
To further assess whether FLT3 methylation is required for PRMT1 effects, BM cells from PRMT1-cKO or control mice were cotransduced with FLT3-ITD plus MA9. Following PRMT1 deletion, we found that cells expressing the FLT3-ITD R972/973K mutant demonstrated reduced CFCs compared with those expressing WT FLT3-ITD. Interestingly, PRMT1 deletion did not reduce CFCs of R972/973K-expressing cells further (Figure 4J), suggesting that PRMT1 functions in FLT3-ITD+ AML cells primarily via R972/973 methylation.
Pharmacological inhibition of PRMT1 reduces FLT3-ITD+ AML growth and survival
We next used a type I PRMT inhibitor, MS023, that is reportedly active against PRMT1.20 We treated MV4-11 cells ex vivo with gradually increasing doses of MS023 and found that the treatment inhibited R972/973 methylation with a 50% inhibitory concentration ∼0.75 µM without altering FLT3 total tyrosine phosphorylation levels (Figure 5A; supplemental Figure 5A). We also observed significant decreases in MV4-11 cell viability at MS023 doses ranging from 0.5 to 5 μM (Figure 5B). Thus, the MS023 dose needed to inhibit FLT3 R methylation is comparable to that needed to elicit a biological response. MS023 treatment also decreased methylation levels of other known PRMT1 targets, such as histone H4R3 dimethyl asymmetric, in FLT3-ITD+ AML cells (supplemental Figure 5B). We next treated cells expressing MA9/FLT3-ITD WT or MA9/FLT3-ITD R972/973K with or without MS023 and then plated cells for CFC analysis. Although MS023 treatment greatly decreased CFCs of FLT3-ITD WT cells, it reduced CFCs of R972/973K-expressing cells only moderately (supplemental Figure 5C). Taken together, our findings suggest that, in the context of FLT3-ITD+ AML, MS023’s inhibitory effects are primarily dependent on FLT3-ITD R972/973 methylation, although additional PRMT1 targets may exist in human AML cells.
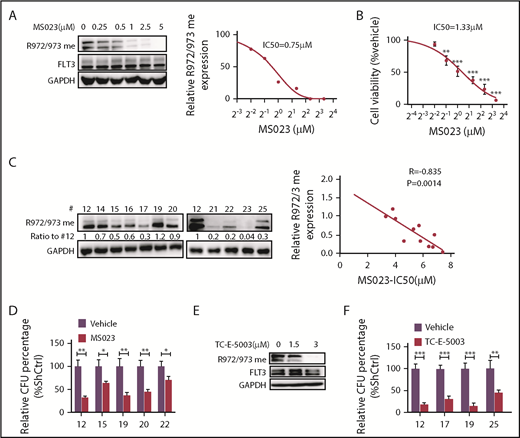
Pharmacological inhibition of PRMT1 reduces FLT3-ITD+ AML growth and survival. (A) Western blotting analysis of FLT3 R972/973 me2a in MV4-11 cells treated with the indicated doses of MS023 (left panel). Quantitative analysis of R972/973 me2a levels based on ImageJ software (right panel). (B) Viability of MV4-11 cells treated with the indicated doses of MS023, based on a CellTiter Glo assay. **P < .01, ***P < .001 vs vehicle. (C) CD34+ cells from FLT3-ITD+ AML (n = 11) were treated for 5 days with MS023 at various doses. Basal FLT3 R972/973 me2a levels are shown (left panels). Linear regression analysis of those R972/973 me2a levels and MS023 50% inhibitory concentration (right panel). Correlation was calculated as an R value, and significance was determined by Pearson correlation. (D) CFC assay of primary FLT3-ITD+ AML (n = 5) CD34+ cells treated with 5 µM MS023 or vehicle. *P < .05, **P < .01. (E) Western blotting analysis of FLT3 R972/973 me2a in MV4-11 cells treated with the indicated doses of TC-E-5003. (F) CFC assay of primary FLT3-ITD+ AML (n = 4) CD34+ cells treated with 3 µM TC-E-5003 or vehicle. **P < .01, ***P < .001. Results represent mean ± SD.
Pharmacological inhibition of PRMT1 reduces FLT3-ITD+ AML growth and survival. (A) Western blotting analysis of FLT3 R972/973 me2a in MV4-11 cells treated with the indicated doses of MS023 (left panel). Quantitative analysis of R972/973 me2a levels based on ImageJ software (right panel). (B) Viability of MV4-11 cells treated with the indicated doses of MS023, based on a CellTiter Glo assay. **P < .01, ***P < .001 vs vehicle. (C) CD34+ cells from FLT3-ITD+ AML (n = 11) were treated for 5 days with MS023 at various doses. Basal FLT3 R972/973 me2a levels are shown (left panels). Linear regression analysis of those R972/973 me2a levels and MS023 50% inhibitory concentration (right panel). Correlation was calculated as an R value, and significance was determined by Pearson correlation. (D) CFC assay of primary FLT3-ITD+ AML (n = 5) CD34+ cells treated with 5 µM MS023 or vehicle. *P < .05, **P < .01. (E) Western blotting analysis of FLT3 R972/973 me2a in MV4-11 cells treated with the indicated doses of TC-E-5003. (F) CFC assay of primary FLT3-ITD+ AML (n = 4) CD34+ cells treated with 3 µM TC-E-5003 or vehicle. **P < .01, ***P < .001. Results represent mean ± SD.
Next, we further tested MS023’s effects in a cohort of FLT3-ITD+ AML specimens and observed that growth inhibition was correlated with baseline R972/973 methylation levels (Figure 5C). MS023 treatment also significantly reduced CFCs of primary AML cells (Figure 5D). Additionally, MS023 is reported to inhibit PRMT6 activity,20 but PRMT6 KD did not alter survival or proliferation of MV4-11 cells (supplemental Figure 5D-E), THP-1 cells (supplemental Figure 5F-G), or Molm13 cells (data not shown) relative to controls, suggesting that PRMT6 function may not be essential for AML cells. We also found that PRMT6 expression level was relatively low in AML cases from The Cancer Genome Atlas database (supplemental Figure 5H). Importantly, we used another PRMT1 inhibitor, TC-E-5003,21 to treat MV4-11 cells ex vivo and found that the treatment (3 µM) effectively inhibited the R972/973 methylation signal (Figure 5E), leading to reduced CFCs of primary AML cells (Figure 5F).
PRMT1 inhibition combined with a TKI enhances elimination of FLT3-ITD+ AML cells
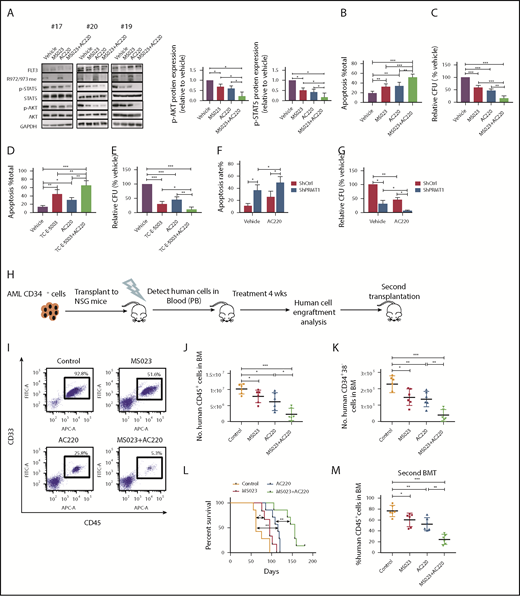
We first observed no alteration in R972/973 methylation levels (Figure 6A) in FLT3-ITD+ AML cells following AC220 treatment. Additionally, our co-IP analysis showed that AC220 treatment did not affect PRMT1/FLT3-ITD interaction (supplemental Figure 6A). These results indicate that FLT3 methylation occurs independent of its kinase activity. Treatment of FLT3-ITD+ AML cells with MS023 plus AC220 also remarkably decreased phospho-AKT and phospho-STAT5 compared with a single drug alone (Figure 6A; supplemental Figure 6B). Obviously, the ex vivo combination treatment resulted in enhanced apoptosis induction and reduced CFCs of primary AML cells (Figure 6B-C). Moreover, treatment of TC-E-5003, another PRMT1 specific inhibitor, on primary AML cells could also enhance AC220-mediated apoptosis induction and CFC inhibition (Figure 6D-E). Notably, PRMT1 KD combined with AC220 significantly inhibited AML cell survival and CFCs compared with AC220 alone (Figure 6F-G).

PRMT1 inhibition combined with AC220 treatment enhances elimination of FLT3-ITD+ AML cells. (A) Western blot analysis of the indicated total and phosphorylated proteins plus GAPDH in cells from 3 FLT3-ITD+ AML patients exposed to 5 µM MS023, 20 nM AC220, or a combination (left panels). Quantitative analysis of phospho-AKT (middle panel) and phospho-STAT5 (right panel) levels based on ImageJ software. (B-C) FLT3-ITD+ AML (n = 6) CD34+ cells were exposed to 5 µM MS023, 20 nM AC220, or a combination. (B) Apoptosis in the indicated groups, as analyzed by Annexin V/4′,6-diamidino-2-phenylindole (DAPI) labeling. (C) CFC analysis of the indicated cells. (D-E) FLT3-ITD+ AML (n = 5) CD34+ cells were exposed to 3 µM TC-E-5003, 20 nM AC220, or a combination. (D) Apoptosis in the indicated groups, as analyzed by Annexin V/DAPI labeling. (E) CFC analysis of the indicated cells. (F-G) FLT3-ITD+ AML (n = 3) CD34+ cells transduced with ShCtrl and ShPRMT1 vectors were cultured or not with AC220 (20 nM). (F) Apoptosis in the indicated groups, as analyzed by Annexin V/DAPI labeling. (G) CFC analysis of the indicated cells. (H) Selected CD34+ cells from primary human FLT3-ITD+ AML cells were injected into irradiated (250 cGy) NSGS mice (1 × 106 cells per mouse). Following confirmation of >1% engraftment, mice were treated for 4 weeks with AC220 (10 mg/kg per day, gavage), MS023 (80 mg/kg per day, intraperitoneally, twice a day), a combination of MS023 and AC220, or vehicle (control) (n = 6 mice per group). Human cell engraftment was analyzed by flow cytometry. Secondary transplantation was also performed. (I) Representative fluorescence-activated cell sorting profile for CD45 and CD33 expression in BM of treated mice. Number of human AML CD45+ cells (J) and CD34+38− cells (K) in BM of treated mice. (L) Mouse survival after treatment discontinuation. (M) Percentage of human AML cells in BM of secondary recipients at 16 weeks.*P < .05, **P < .01, ***P < .001. Results represent mean ± SD.
PRMT1 inhibition combined with AC220 treatment enhances elimination of FLT3-ITD+ AML cells. (A) Western blot analysis of the indicated total and phosphorylated proteins plus GAPDH in cells from 3 FLT3-ITD+ AML patients exposed to 5 µM MS023, 20 nM AC220, or a combination (left panels). Quantitative analysis of phospho-AKT (middle panel) and phospho-STAT5 (right panel) levels based on ImageJ software. (B-C) FLT3-ITD+ AML (n = 6) CD34+ cells were exposed to 5 µM MS023, 20 nM AC220, or a combination. (B) Apoptosis in the indicated groups, as analyzed by Annexin V/4′,6-diamidino-2-phenylindole (DAPI) labeling. (C) CFC analysis of the indicated cells. (D-E) FLT3-ITD+ AML (n = 5) CD34+ cells were exposed to 3 µM TC-E-5003, 20 nM AC220, or a combination. (D) Apoptosis in the indicated groups, as analyzed by Annexin V/DAPI labeling. (E) CFC analysis of the indicated cells. (F-G) FLT3-ITD+ AML (n = 3) CD34+ cells transduced with ShCtrl and ShPRMT1 vectors were cultured or not with AC220 (20 nM). (F) Apoptosis in the indicated groups, as analyzed by Annexin V/DAPI labeling. (G) CFC analysis of the indicated cells. (H) Selected CD34+ cells from primary human FLT3-ITD+ AML cells were injected into irradiated (250 cGy) NSGS mice (1 × 106 cells per mouse). Following confirmation of >1% engraftment, mice were treated for 4 weeks with AC220 (10 mg/kg per day, gavage), MS023 (80 mg/kg per day, intraperitoneally, twice a day), a combination of MS023 and AC220, or vehicle (control) (n = 6 mice per group). Human cell engraftment was analyzed by flow cytometry. Secondary transplantation was also performed. (I) Representative fluorescence-activated cell sorting profile for CD45 and CD33 expression in BM of treated mice. Number of human AML CD45+ cells (J) and CD34+38− cells (K) in BM of treated mice. (L) Mouse survival after treatment discontinuation. (M) Percentage of human AML cells in BM of secondary recipients at 16 weeks.*P < .05, **P < .01, ***P < .001. Results represent mean ± SD.
We then tested the effects of combination treatment on primary human CD34+ cells from a FLT3-ITD+ AML specimen engrafted in NSGS mice. CD34+ cells selected from the specimen were transplanted into NSGS mice, as described.22 Although leukemia engraftment in peripheral blood (PB) reached >1%, we treated mice with vehicle, MS023, AC220, or a combination (Figure 6H) for 4 weeks. At the end of treatment, we observed that AML burden was significantly lower in BM, spleen, or PB of MS023-treated mice compared with vehicle-treated mice, with a further reduction seen in mice that received the combination (Figure 6I-J; supplemental Figure 6C-E). Additionally, we tracked FLT3-ITD lesion to confirm the human AML origin of cells before and after xenografting (supplemental Figure 6F). Moreover, the FLT3-ITD allele signal was significantly reduced in human cells isolated from mice treated with the combination, confirming ablation of engraftment of human FLT3-ITD+ AML clones (supplemental Figure 6F). More importantly, human AML CD34+CD38− cells in BM were significantly reduced in mice treated with MS023 or AC220 alone, with a further reduction seen in mice treated with the combination (Figure 6K). Notably, mice treated with MS023 demonstrated improved survival after discontinuation of treatment compared with control mice, and the combination of MS023 and AC220 increased survival further compared with AC220 alone (Figure 6L). We then assessed combination effects on leukemia-initiating activity by secondary transplantation. Although we observed a significant AML burden in vehicle control–treated, MS023-treated, or AC220-treated transplants at 16 weeks, only minimal residual human CD45+ AML cells were found in combination-treated secondary transplants (Figure 6M). Next, we evaluated the efficacy of the combination in vivo in MA9/FLT3-ITD murine AML (supplemental Figure 6G). Combination treatment significantly reduced donor-derived AML cell engraftment (supplemental Figure 6H-K). Importantly, although AC220-treated transplants succumbed to aggressive AML with a median survival of 50 days, combination treatment significantly extended the survival of leukemia transplants (supplemental Figure 6L).
We also assessed the impact of MS023 treatment on normal hematopoietic stem and progenitor cells (HSPCs) from healthy CB engrafted in NSGS mice. To do so, we transplanted CB CD34+ cells via tail vein injection into sublethally irradiated (250 cGy) NSGS mice, as described22 (supplemental Figure 6M). The percentage and the absolute number of human CD45+ cells or certain subsets were not altered after 4 weeks of MS023 treatment, and percentages of all lineages remained the same, suggesting that MS023 treatment has minimal adverse effects on the in vivo repopulating capacity of normal HSPCs (supplemental Figure 6M-O).
Discussion
FLT3-ITD, which is one of the most frequent somatic mutations in AML, is associated with poor prognosis and occurs in 25% of AML patients.5 However, in clinical settings, treatment of AML patients with a TKI has had only modest effects, and persistence of FLT3-ITD+ AML clones is associated with relapse.5,23 Thus, new treatments to eliminate FLT3-ITD+ AML cells are needed. Here, we demonstrate that PRMT1 preferentially binds oncogenic FLT3 and catalyzes its protein methylation, eventually promoting survival and proliferation of FLT3-ITD+ AML cells. Notably, FLT3 methylation levels persist in AML cells following TKI treatment, and blocking this activity with a pharmacological inhibitor enhanced FLT3-ITD+ AML cell elimination by a TKI, suggesting that PRMT1 inhibition could serve as a therapy for AML patients with FLT3-ITD.
PRMT1 is universally overexpressed in various cancers.24-28 Nonetheless, its role in these contexts likely depends on the function of its substrate(s).24-28 Interestingly, a recent study revealed that loss of PRMT1 function blocks MLL-GAS7– or MLL-EEN–driven leukemogenesis.12 Specifically, in both contexts, PRMT1 is recruited to catalyze H4R3, and methylated H4R3 is essential for oncogenic transcriptional programs. Herein, our loss-of-function analysis in a cohort of AML specimens and a PRMT1-cKO mouse reveals that FLT3-ITD+ AML cell survival relies on PRMT1-mediated FLT3 protein R methylation. Mounting evidence15,16 suggests that the FLT3-ITD protein markedly locates intracellularly, unlike the WT receptor.16,17 PRMT1, as a predominant nuclear and cytoplasmic protein,29 preferentially interacts with FLT3-ITD relative to FLT3 WT protein, as evidenced by our co-IP analysis. Therefore, relative to FLT3 WT, FLT3-ITD protein is much more accessible to PRMT1. FLT3-ITD methylation may also be more susceptible to intervention, consistent with our results that PRMT1-KD preferentially reduced survival/growth of FLT3-ITD+ AML cells.
Protein R methylation occurs in numerous biological processes.10,27 Notably, a recent study showed signaling cross talk between EGFR R1175 methylation and Y1173 phosphorylation.18 Similarly, our results relevant to FLT3 mutant receptor show that R972/R973 methylation maintains phosphorylation of the nearby Y969 residue, a docking site for the SH2 domain containing proteins like Grb2 and GADS, which activates downstream pathways, including STAT5 and AKT.19,30 Interestingly, our results indicate that R972/973 methylation enhances Y969 phosphorylation–initiated binding of the SH2 domain with FLT3 peptide. Thus, the effects of R972/973 methylation in promoting phospho-STAT5 and phospho-AKT levels are partially due to Y969 phosphorylation. Nevertheless, our functional studies using methylation- or phosphorylation-deficient constructs suggest that loss of Y969 phosphorylation only partially phenocopies R972/973 methylation loss, suggesting that other, as yet unknown, mechanisms are responsible for FLT3 methylation biological effects and merit further analysis.
Considering the crucial role of PRMTs in cancer, small molecules, like EPZ015666 targeting PRMT5, have been extensively analyzed.31-33 Recently, MS023 was developed as a type I PRMT inhibitor mainly targeting PRMT1 and PRMT6.20 In the context of FLT3-ITD+ AML cells, we propose that MS023’s effects are primarily PRMT1 dependent, based on the following observations: PRMT6 function is not essential for AML cell survival; PRMT1, but not PRMT6, is responsible for FLT3 ADMA in AML cells; and MS023 inhibition of AML cell proliferation is positively correlated with basal FLT3 R methylation levels in those cells. Notably, MS023 treatment showed potent therapeutic efficacy in our AML mouse models while sparing normal HSPC activity (Figure 6), making it a promising compound for further preclinical testing.
Overall, our study supports the idea that FLT3-ITD+ AML cells are functionally dependent on PRMT1-mediated FLT3-ITD protein methylation. Our work demonstrates that blocking such methylation through targeting PRMT1 enzymatic activity, in combination with TKI treatment, could serve as a promising treatment of the FLT3-ITD+ AML population.
The RNA-Seq data reported in this article have been deposited in the GEO database (accession numbers GSE122435 and GSE129754).
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Acknowledgments
The authors thank the COH Comprehensive Cancer Center, as well as patients, donors, and their physicians for providing primary specimens for this study; Yanzhong Yang for providing GFP-PRMT vectors; Stéphane Richard for supplying PRMT1f/f mice; and the University of California at Los Angeles (UCLA) Center for Aids Research (CFAR) Virology core (supported by the National Institutes of Health, National Institute of Allergy and Infectious Diseases under grant 5P30 AI028697, and the UCLA AIDS Institute) for CB samples.
This work was supported in part by National Institutes of Health, National Heart, Lung, and Blood Institute grant R01 HL141336, a Research Scholar grant (RSG-19-036-01-LIB) from the American Cancer Society, the Margaret E. Early Medical Research Trust Award, a Stop Cancer Research Career Development Award, a V Scholar award, and the Gehr Family Center for Leukemia Research (L.L.). The authors acknowledge the support of the Animal Resources Center, Analytical Cytometry, Bioinformatics, Light Microscopy, Pathology (Liquid Tumor), Translational Biomarker Discovery Core, Integrative Genomics and DNA/RNA Cores at the City of Hope Comprehensive Cancer Center, supported by the National Institutes of Health, National Cancer Institute under award number P30CA33572.
The content is solely the responsibility of the authors and does not necessarily represent official views of the National Institutes of Health.
Authorship
Contribution: X.H. designed and performed the study, analyzed data, and wrote the manuscript; Y.Z. performed research, analyzed data, and reviewed the manuscript; Y.-C.L., M.L., and J.D. analyzed data; H.D., J.S., L. Zhu, Lei Zhang, Z.W., Z.D., Lianjun Zhang, D.Z., H. Wang, H. Wu, H.Z., and Y.S. performed research; Y.X., J.J., J.P., B.Z., X.Z., S.L., S.-L.X., B.S., C.-W.C., J.C., S.K., Y.-H.K., Y.L., and G.M. interpreted data and reviewed the manuscript; and L.L. initiated the topic and designed the study, analyzed and interpreted data, and wrote the manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Ling Li, Department of Hematological Malignancies Translational Science, Gehr Family Center for Leukemia Research, Hematologic Malignancies and Stem Cell Transplantation Institute, Beckman Research Institute, City of Hope Medical Center, 1500 E Duarte Rd, Duarte CA 91010; e-mail: lingli@coh.org.

![Figure 3. PRMT1 catalyzes FLT3-ITD protein methylation at R972/973. (A) Schematic model showing dimethylated (Di-Me) arginines identified following immunoprecipitation of endogenous FLT3 protein from MV4-11 cells. The precipitates were subjected to proteomic analysis. (B) FLT3 protein was immunoprecipitated from 293T cells ectopically expressing FLT3-ITD or FLT3-ITD methylation-deficient mutants (R655K, R845K, R972K, and R973K) and then analyzed for ADMA levels by western blotting. (C) Dot blot showing that the FLT3-R972/973 me2a antibody specifically binds asymmetric dimethylated R972/973 peptide. Amino acid sequence of peptides corresponding to the FLT3 966-980 region, in which R972 and R973 are unmodified, monomethylated, or dimethylated. Different amounts of peptides were spotted on polyvinylidene difluoride membranes and detected by control anti-FLT3 or anti-FLT3 asymmetric methylated Arg 972/973 (R972/973 me2a) antibodies. (D) 293T cells transfected with mock, WT FLT3-ITD, or FLT3-ITD methylation-deficient mutants (R655K, R845K, R972K and R973K) were analyzed by western blot using the FLT3 R972/973 me2a antibody. (E) In vitro methylation assay of GST-tagged FLT3 peptides (WT, R972K, R973K, and R972/973K) in the presence of PRMT1 protein and S-adenosyl-l-[methyl-3H] methionine (SAM). Peptide methylation was analyzed by western blotting using the methylation antibody. (F) 293T cells were transduced with lentiviral vector expressing FLT3-ITD and then transfected with mock or the indicated GFP-fused type I PRMTs. FLT3-ITD protein was immunoprecipitated from cells and analyzed for GFP by western blotting. MV4-11 cells transduced with ShCtrl, ShPRMT1 (G), or ShPRMT6 (H) were analyzed by western blotting for PRMT1 or PRMT6, FLT3 R972/973 me2a, total FLT3, and GAPDH. CT, C terminus; ECD, extracellular domain; JM juxtamembrane domain; KI, kinase insert; TK1, tyrosine kinase 1; TK2, tyrosine kinase 2; TM, transmembrane.](https://ash.silverchair-cdn.com/ash/content_public/journal/blood/134/6/10.1182_blood.2019001282/3/m_bloodbld2019001282f3.png?Expires=1767755471&Signature=DtVqao1CUgiozD8FDoY9cti-6IAPYzHUhgFP8skKW-V4BWshxaCTjIREHhKw6CpiB20GgE84sLjmOs1pk4powgvwoXJJu7LcaaC3UvzflUh1aQ8k9PGoa5IjMACjeuRuTW5amCKYvRBUvAxPVgsBjJ98-v5N0PFj-F1Im87e-KbNY-QRzBAF6soUff-6cZHtCDvZl-MdFOpydjkdSWlFXPT8RI4NoygV~j2wfHBN~YO0ozf7hkVchOTl3CkZ30QPjZ-jb3rJEAioHDngxqzP~UadecSr5YiPkTGthli7xWSDofLgdU92yVxXtZzIfxQ7UBZ7gVfNqMv9srgcghjT8w__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)