

Key Points
RUNX-1 haploinsufficiency results in a deficiency of megakaryocyte-biased hematopoietic progenitor cells (HPCs).
RUNX-1 haploinsufficiency elevates druggable proinflammatory and TGFβR-1–related pathways in HPCs.

Abstract
Patients with familial platelet disorder with a predisposition to myeloid malignancy (FPDMM) harbor germline monoallelic mutations in a key hematopoietic transcription factor, RUNX-1. Previous studies of FPDMM have focused on megakaryocyte (Mk) differentiation and platelet production and signaling. However, the effects of RUNX-1 haploinsufficiency on hematopoietic progenitor cells (HPCs) and subsequent megakaryopoiesis remains incomplete. We studied induced pluripotent stem cell (iPSC)–derived HPCs (iHPCs) and Mks (iMks) from both patient-derived lines and a wild-type (WT) line modified to be RUNX-1 haploinsufficient (RUNX-1+/−), each compared with their isogenic WT control. All RUNX-1+/− lines showed decreased iMk yield and depletion of an Mk-biased iHPC subpopulation. To investigate global and local gene expression changes underlying this iHPC shift, single-cell RNA sequencing was performed on sorted FPDMM and control iHPCs. We defined several cell subpopulations in the Mk-biased iHPCs. Analyses of gene sets upregulated in FPDMM iHPCs indicated enrichment for response to stress, regulation of signal transduction, and immune signaling-related gene sets. Immunoblot analyses in FPDMM iMks were consistent with these findings, but also identified augmented baseline c-Jun N-terminal kinase (JNK) phosphorylation, known to be activated by transforming growth factor-β1 (TGF-β1) and cellular stressors. These findings were confirmed in adult human CD34+-derived stem and progenitor cells (HSPCs) transduced with lentiviral RUNX1 short hairpin RNA to mimic RUNX-1+/−. In both iHPCs and CD34+-derived HSPCs, targeted inhibitors of JNK and TGF-β1 pathways corrected the megakaryopoietic defect. We propose that such intervention may correct the thrombocytopenia in patients with FPDMM.
Introduction
The runt-related transcription factor 1 (RUNX-1) is critical in definitive hematopoiesis, lymphopoiesis, and myelopoiesis.1-4 Patients with familial platelet disorder with a predisposition to myeloid malignancy (FPDMM) harbor germline monoallelic mutations in RUNX1, often presenting with thrombocytopenia and bleeding.5 These individuals also have an increased risk of acquiring myeloid dysplasias and malignancies.6 Investigations of patients with FPDMM have focused on the role of RUNX-1 haploinsufficiency (RUNX-1+/−) in megakaryopoiesis and number and function of platelets.7-18 The effect of RUNX-1+/− on hematopoietic stem and progenitor cell (HSPC) differentiation has not been characterized, although such cells may contribute to the megakaryocyte (Mk) and platelet defects and oncogenic evolution.
HSPCs sit atop the hematopoietic hierarchy,19-21 but are not composed of homogenous populations transitioning through sequential progenitor stages into mature blood lineages. Single-cell transcriptomics, single-cell functional assays, and lineage-tracing studies have shown that phenotypic HSPCs contain subpopulations with variable long-term self-renewal potential that are transcriptionally and functionally unipotent or biased toward production of certain blood lineages and undergo early lineage committment in hematopoiesis.22-26 In particular, von Willebrand factor–expressing (VWF+) hematopoietic stem cells in mice have long-term multilineage reconstitution potential skewed toward the Mk lineage.22,25 In human hematopoiesis, there are Mk-restricted progenitors in the immunophenotypic HSPCs and common myeloid progenitor compartments that display in vivo self-renewal and robust platelet-producing activity after xenotransplantation.27,28 Mk-biased HSPCs are responsive to stress and inflammation.25,26,29 The degree and direction of HSPC lineage bias is linked to levels of transcription factor, such as GATA2 and NFE2,24,28 which, together with RUNX-1, form key regulatory complexes in HSPCs.30,31
Heterozygous mutant Runx1 mice do not recapitulate FPDMM.4,32,33 Unlike FPDMM, conditional Runx1 mutant mice display myeloid differentiation defects and paradoxical expansion of HSPCs.3,32,34 These species differences have focused etiologic studies of FPDMM to RUNX-1+/− human induced-pluripotent stem cells (iPSCs) that seem to replicate the megakaryocytic defects seen in patients with FPDMM.35-37 RUNX-1+/− results in an ∼50% level of final Mks per initial iPSC-derived hematopoietic progenitor cell (iHPC), compared with isogenic controls.36 A defect in NOTCH signaling has been proposed to be linked to the observed decrease in Mk yield.37
We have similarly studied iHPCs and the resultant Mks (iMks) from affected individuals to understand the relationship between the decrease in Mk yield and a defect in HPCs. Our data confirmed previously observed defects in FPDMM megakaryopoiesis,11,35,36 but also identified a marked deficiency of an Mk-biased iHPC population. By single-cell RNA sequencing (scRNA-SEQ), this deficiency was connected with an increase in gene sets associated with response to stress, regulation of signal transduction, and response to cytokines. We also observed an increase in the stress pathway by upregulation of c-Jun N-terminal kinase (JNK)-2 phosphorylation and increased sensitivity to transforming growth factor-β1 (TGF-β1). Drugs that block these pathways correct megakaryopoiesis. Because iPSCs are embryonic and may generate an immature blood developmental program, we used a short hairpin RNA (shRNA) approach to mimic RUNX-1 haploinsufficiency in adult CD34+-HSPCs, referred to herein as RUNX-1 insufficiency (RUNX-1in), and observed Mk deficiency associated with a decrease in an Mk-biased HSPC population. Similar stress pathways were upregulated in the RUNXin Mk-biased HSPCs and were corrected by blocking JNK and TGFβ receptor 1 (TGFβR-1) signaling. The mechanistic and therapeutic implications of these findings in patients with FPDMM are discussed.
Materials and methods
iPSC lines and mobilized, peripheral-blood, adult CD34+ cells
All iPSC constructs studied are noted in supplemental Figure 1 (available on the Blood Web site). Line L1 was established with Institutional Review Board approval in accordance with the principles of the Declaration of Helsinki. Granulocyte colony-stimulating factor–mobilized CD34+ peripheral blood HSPCs were provided by the Fred Hutchinson Cancer Research Center.38,39
Flow cytometry and cell sorting
Flow cytometric analyses were performed on a CytoFLEX LX flow cytometer (Beckman-Coulter) and analyzed with FlowJo software, v10.6 (Becton-Dickinson). iHPCs were CD235a+CD41+, whereas adult CD34+ HSPCs were CD34+CD38−CD45RA−CD41+. The absolute number of iPSC- or CD34+-derived Mks was determined by using CD41 and CD42 cell surface markers. A full list of the labeled markers, antibodies, and their sources is in supplemental Table 1. A FACS Jazz (Becton-Dickinson) with a 100-µm nozzle was used to sort the cells. Gating schemes for iHPCs and CD34+ HSPCs are provided in supplemental Figures 2 and 3.
Statistical analysis
Statistical analysis was performed with 1- and 2-way ANOVA, and data are reported as ±1 standard error of the mean (SEM), with GraphPad Prism, version 8 (GraphPad Software). Differential expression analysis of the input genes and P values used to generate volcano plots were computed in the Loupe Cell Browser by the negative binomial exact test, with P values adjusted for multiple tests using the Benjamini-Hochberg procedure to decrease false discovery.40 Differences were considered significant at P < .05.
Additional methodology
See the supplemental Methods for additional details of RUNX-1+/− CD34+ HSPC creation, analysis of hematopoietic differentiation, scRNA-SEQ library preparation, data preprocessing and analysis, western blot analysis, and immunofluorescence experiments.
Results
Effect of RUNX-1+/− on Mk-biased iHPCs
Patients with FPDMM display defects in Mks and platelets.11-13,16,17,36 Whether this phenotype stems from defective early HSPC populations is unclear. HSPCs displaying commitment to Mk-biased lineage have been reported in mice and humans.22,25,27,28 We examined whether these early progenitors are affected in RUNX-1+/− by using 3 distinct RUNX-1+/− iPSC lines and their isogenic control (supplemental Figure 1). L1 was from a patient with FPDMM harboring a previously characterized monoallelic splice-site acceptor mutation in RUNX1 intron 341 (supplemental Figure 1A) and its isogenic mutation–corrected line, L1-C. A second set included a previously established wild-type (WT) line altered to introduce this splice mutation (WT-L1). The premature stop in RUNX1 in these lines has been found to activate nonsense-mediated decay of messenger RNA (mRNA).42,43 A third set included RUNX-1+/− L2 with a C→A transversion in RUNX1 exon 7 that introduces a stop codon in the RUNX-1 transactivation domain35 (supplemental Figure 1A) and its isogenic control L2-C.35 These 6 lines (supplemental Figure1B) were exposed to a hematopoietic differentiation program to generate iHPCs, which arose as free-floating CD235a+CD41+ cells on day 7 of culture (Figure 1A)44,45 , to enrich for multipotent progenitor cells.45,46 They were then differentiated into Mks (Figure 1A).47 All 3 RUNX-1+/− lines showed decreased iMk yield per iHPC (supplemental Figure 2A). Reduction in RUNX-1 protein levels in terminal iMks was validated for lines L1 and L2 (supplemental Figure 2B), consistent with patients with FPDMM11 and prior studies of iMks.35-37
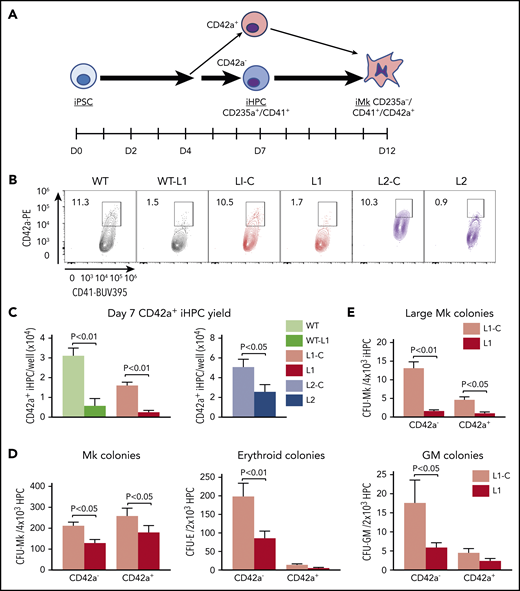
Deficiency in an Mk-biased CD42a+ subpopulation in RUNX-1+/− iHPCs. (A) Megakaryopoiesis in a human iPSC system highlighting generation of iHPCs and iMks. On day 7 after exposure of adherent iPSCs to a directed differentiation protocol, free-floating, multipotent CD235a+CD41+ iHPCs emerged that included CD42a− and CD42a+ iHPC subpopulations. (B) Flow cytometric analysis of day 7 iHPCs showing the CD42a+ iHPC subpopulation that is present at similar frequencies in all control lines, but is severely reduced in all RUNX-1+/− lines. (C) Quantitation of panel B, showing the mean number of CD42+ iHPCs detected and normalized per well for iPSCs seeded ±1 SEM (n = 5-15 experiments per study arm). (D) Colony-forming assays of L1-C vs L1 iHPCs, showing quantitation of the mean number of Mk, erythroid, and granulocyte/macrophage (GM) colonies, normalized to the number of day 7 iHPCs seeded ±1 SEM (n = 4 experiments per study arm). (E) Same as panel D, but for large Mk colonies (>20 Mks/colony). All P values were determined by Student t test.
Deficiency in an Mk-biased CD42a+ subpopulation in RUNX-1+/− iHPCs. (A) Megakaryopoiesis in a human iPSC system highlighting generation of iHPCs and iMks. On day 7 after exposure of adherent iPSCs to a directed differentiation protocol, free-floating, multipotent CD235a+CD41+ iHPCs emerged that included CD42a− and CD42a+ iHPC subpopulations. (B) Flow cytometric analysis of day 7 iHPCs showing the CD42a+ iHPC subpopulation that is present at similar frequencies in all control lines, but is severely reduced in all RUNX-1+/− lines. (C) Quantitation of panel B, showing the mean number of CD42+ iHPCs detected and normalized per well for iPSCs seeded ±1 SEM (n = 5-15 experiments per study arm). (D) Colony-forming assays of L1-C vs L1 iHPCs, showing quantitation of the mean number of Mk, erythroid, and granulocyte/macrophage (GM) colonies, normalized to the number of day 7 iHPCs seeded ±1 SEM (n = 4 experiments per study arm). (E) Same as panel D, but for large Mk colonies (>20 Mks/colony). All P values were determined by Student t test.
We tested whether we could detect an Mk-biased iHPC population, analogous to the adult Mk-biased HSPC population,27,28,48 and whether this population is deficient in RUNX-1+/−. We evaluated iHPCs for intracellular stores of vWF, a known Mk- and endothelium-associated marker, known to be enriched in murine Mk-biased HSPCs.22 Using intracellular flow cytometry and immunofluorescence microscopy, we detected VWF expression in all normal and RUNX-1+/− iHPCs, with no significant difference (supplemental Figure 3A-B). Similarly, there was no significant subpopulations of iHPCs based on the Mk-associated α-granule protein TREM-like transcript-1 (TLT-1) nor was there a difference between the controls and RUNX-1+/− iHPCs (supplemental Figure 3C and data not shown).
We did identify a CD235a+CD41+CD42a+ iHPC subpopulation (Figure 1A-C; supplemental Figure 4A-B). CD42a is the Mk-specific protein, glycoprotein (GP) IX, a component of the GPIb/IX receptor.49,50 We sorted CD42a− and CD42a+ iHPC subpopulations and seeded them in equal numbers in Mk, erythroid, and myeloid colony-forming assays. Both control and RUNX-1+/− CD42a− iHPCs, displayed trilineage potential, whereas CD42a+ iHPCs produced near-exclusive Mk colonies (Figure 1D). For all RUNX-1+/− iPSC lines, there was a 50% to 80% decrease in CD42a+ iHPCs (Figure 1B-C; supplemental Table 2). RUNX-1+/− iHPCs had a near-absence of large Mk colony formation (Figure 1E), suggestive of a proliferative defect. Indeed, RUNX-1+/− iHPCs displayed reduced cell cycling (supplemental Figure 5A-B). Whether this defect delayed CD42a+ iHPC emergence from RUNX-1+/− iPSCs was tested and the results showed defective CD42+ iHPCs continuing beyond day 7 (supplemental Figure 6). These results suggest that RUNX-1+/− severely affects the emergence of Mk-biased progenitors.
scRNA-SEQ identifies novel deregulated pathways in RUNX-1+/− iHPCs
To gain insight into the global and local transcriptomic changes underlying defective production of CD42a+ iHPCs in RUNX-1+/−, we performed scRNA-SEQ on sorted CD42a− and CD42a+ iHPCs from L1 and L1-C. Analysis of distinct subpopulations in the control and RUNX-1+/− iHPCs by dimension reduction and cell clustering using uniform manifold approximation and projection (UMAP).51 Based on differentially expressed (DE) genes, 8 transcriptomic cell clusters were defined in CD42a+ iHPCs in both control and RUNX-1+/− iHPCs (Figure 2A; supplemental Table 3). To identify changes related to RUNX-1+/−, we compared the number of DE genes between clusters. L1-C and L1 CD42a+ iHPCs, clusters 1, 3, 4, and 7, showed the highest number of DE genes (supplemental Figure 7). Functional enrichment profiling of genes upregulated in L1-C iHPC clusters52 was consistent with previous scRNA-SEQ studies that showed a large contribution of cell cycle genes to transcriptional heterogeneity in differentiating hematopoietic cells,53 particularly stem and progenitor cells.54,55 L1-C CD42a+ iHPCs, clusters 1 and 3, were enriched in cell cycle–related biological processes, whereas cluster 7 showed enrichment in myeloid- and, especially, Mk/platelet-associated genes (Figure 2B).
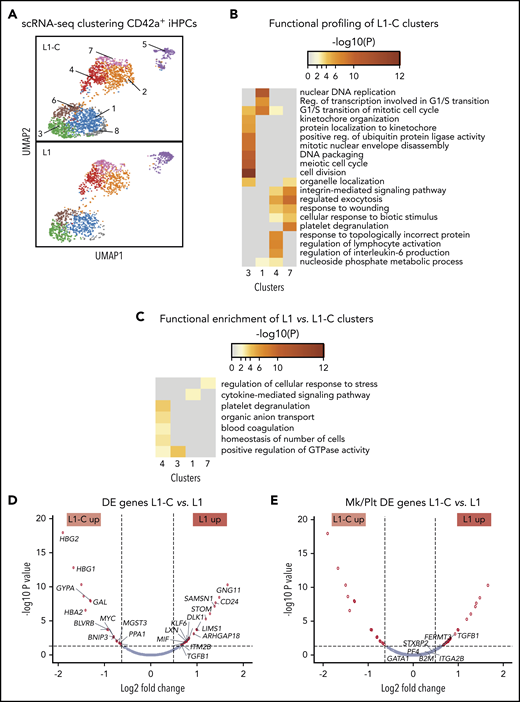
scRNA-SEQ analysis of Mk-biased CD42+ iHPCs. (A) Identification and visualization of transcriptional heterogeneity within sorted L1-C and L1 CD42+ iHPCs by dimensionality reduction with UMAP. (B-C) Eight populations were identified in both L1-C (n = 2335) and L1 (n = 2302) cells by grouping of cells based on DE genes and gene ontology analyses, using lists of genes from select clusters with the highest detected number of DE genes to show functional enrichment in control CD42a+ iHPC clusters only (B) or processes (C) that are upregulated in L1 vs L1-C CD42+ iHPCs. (D-E) Volcano plots showing significantly changed genes among all DE genes (D) or focusing on Mk/platelet (Plt)-associated genes (E), when L1-C to L1 CD42a+ iHPCs were compared across all clusters. To improve clarity, only select genes are shown and labeled. (See supplemental Tables 5 and 6 for a full list.) Filled red circles denote significantly changed genes; unfilled red circles indicate DE genes that are not Mk/platelet associated.
scRNA-SEQ analysis of Mk-biased CD42+ iHPCs. (A) Identification and visualization of transcriptional heterogeneity within sorted L1-C and L1 CD42+ iHPCs by dimensionality reduction with UMAP. (B-C) Eight populations were identified in both L1-C (n = 2335) and L1 (n = 2302) cells by grouping of cells based on DE genes and gene ontology analyses, using lists of genes from select clusters with the highest detected number of DE genes to show functional enrichment in control CD42a+ iHPC clusters only (B) or processes (C) that are upregulated in L1 vs L1-C CD42+ iHPCs. (D-E) Volcano plots showing significantly changed genes among all DE genes (D) or focusing on Mk/platelet (Plt)-associated genes (E), when L1-C to L1 CD42a+ iHPCs were compared across all clusters. To improve clarity, only select genes are shown and labeled. (See supplemental Tables 5 and 6 for a full list.) Filled red circles denote significantly changed genes; unfilled red circles indicate DE genes that are not Mk/platelet associated.
Most genes from a curated list of 133 known Mk-associated genes (supplemental Table 4) did not show significant differences in DE analysis between CD42a− and CD42a+ iHPCs subpopulations (Figures 7C, 8, and 9; supplemental Table 5). We believe that clusters enriched in cell cycle genes were proliferating iHPCs, whereas those deficient were likely to be further committed iHPCs (supplemental Figures 10 and 11; supplemental Table 6). In support, gene set enrichment analysis indicated that, in both lines, CD42a+ iHPCs showed enrichment of Mk progenitor- and myeloid-associated genes and depletion of erythroid-associated genes, consistent with an early Mk bias (supplemental Figure 12). An analysis of gene sets upregulated in L1 CD42a+ iHPC clusters revealed enrichment of several processes, including platelet degranulation, cytokine-mediated signaling, and regulation of cellular stress response (Figure 2C; supplemental Table 7). Many genes found in these enriched processes were also DE in global analyses of all cell clusters from L1-C and L1 CD42+ iHPCs (Figure 2D-E; supplemental Table 8).
For CD42a− iHPCs, scRNA-SEQ UMAP clustering defined 6 transcriptomically distinct cell clusters (supplemental Figure 13A). Functional profiling of control L1-C CD42a− iHPC clusters showed a similar pattern of distinct cluster-specific enrichment of cell cycle and erythroid, myeloid, and Mk/Plt gene sets (supplemental Figure 13B). DE genes in RUNX-1+/− CD42a− iHPC clusters showed enrichment for stress-associated processes (supplemental Figure 13C), although L1 CD42+ iHPCs showed a greater number of DE and upregulated genes than did the L1 CD42a− iHPCs (supplemental Figures 3D, 7, and 13E). Functional profiling revealed a similar enrichment of stress-associated and proinflammatory processes (supplemental Figure 9B).
Our analysis of Mk/platelet-associated genes in RUNX-1+/− CD42+ iHPCs indicated that TGFB1 was among the highest DE genes (Figure 2E) and that thrombospondin 1 (THBS1), a negative regulator of megakaryopoiesis involved in regulating TGF-β1 activity,56,57 was also among the highest DE genes in cluster-specific analyses (supplemental Figure 14). Gene set enrichment analysis of TGFβ receptor signaling pathway genes upregulated in L1 CD42+ iHPCs supported these findings (supplemental Figure 15). Together, local and global analyses of genes in RUNX-1+/− L1 iHPCs revealed upregulated TGF-β1–related processes.
Augmented TGF-β1 and JNK signaling in RUNX-1+/−
Many of the augmented cytokine and cellular stress–response genes in the RUNX-1+/− CD42a+ iHPCs involve TGFβR-1–associated pathways.58-63 Therefore, their deficiency could be caused by elevated TGF-β1 and/or TGFβR-1–associated levels or signaling. As TGF-β1 is released in the marrow predominantly by Mks,64,65 we tested whether RUNX-1+/− iMks release excess TGF-β1; however, instead of elevated levels, we found decreased TGF-β1 levels in RUNX-1+/− iMks (Figure 3A). With correction for iMk yield, TGF-β1 yield per iMk was similar in L1 and L1-C iMk cultures (Figure 3B). We next tested whether RUNX-1+/− CD42+ iHPC iMk yield was related in part to enhanced TGF-β1 sensitivity; isogenic and RUNX-1+/− iHPCs were exposed to recombinant human TGF-β1 (rhTGF-β1) protein, known to inhibit Mk proliferation.66 RUNX-1+/− cultures showed greater sensitivity to recombinant human GF-β1 (rhTGF-β1; Figure 3C), which was rescued by anti-TGF-β1–blocking antibodies67 (Figure 3D).

Augmented TGF-β1 and JNK signaling in RUNX-1+/− iHPCs and iMks. (A-B) ELISA of TGF-β1 levels in iMk culture conditioned medium. Cell culture supernatants were collected from L1-C and L1 day 13 iMk liquid cultures and used to measure levels of active TGF-β1. Mean ±1 SEM of TGF-β1 without normalization for differences in the number of iMks (A) and with normalization for iMks (B) (n = 5 experiments per study arm). (C) The effect of TGF-β1 exposure on Mk yield from L1-C and L1 iHPCs. Cultures were exposed to rhTGF-β1 for 5 days at the indicated doses, and Mk yield was quantified per input iHPC, as in Figure 1C. Results are shown as a percentage of untreated iMks. (D) The effect of 20 µg/mL anti-α-TGF-β–blocking antibodies (Abs) on suppression of Mk yield resulting from TGF-β1 exposure. Mean ±1 SEM (n = 6 experiments per study arm; P values calculated by 2-way analysis of variance. (E) Day 13 iMk proteins with antibodies directed against TGFβR-1, p21, p57, and pSMAD2/3 with GAPDH as a loading control. (F) Day 7 iHPC immunostained with antibodies against phosphorylated JNK (pJNK). Mean ±1 SEM (n = 4 experiments per study arm). (G) Elevated JNK phosphorylation in day 13 L1 iMks. The iMks were solubilized and immunoblotted with antibodies against pJNK, total JNK, and vinculin (as a loading control). (H) Experiments in panel G were scanned and optical density was quantified. Mean ±1 SEM (n = 6 experiments per study arm). All P values by Student t test, unless stated otherwise.
Augmented TGF-β1 and JNK signaling in RUNX-1+/− iHPCs and iMks. (A-B) ELISA of TGF-β1 levels in iMk culture conditioned medium. Cell culture supernatants were collected from L1-C and L1 day 13 iMk liquid cultures and used to measure levels of active TGF-β1. Mean ±1 SEM of TGF-β1 without normalization for differences in the number of iMks (A) and with normalization for iMks (B) (n = 5 experiments per study arm). (C) The effect of TGF-β1 exposure on Mk yield from L1-C and L1 iHPCs. Cultures were exposed to rhTGF-β1 for 5 days at the indicated doses, and Mk yield was quantified per input iHPC, as in Figure 1C. Results are shown as a percentage of untreated iMks. (D) The effect of 20 µg/mL anti-α-TGF-β–blocking antibodies (Abs) on suppression of Mk yield resulting from TGF-β1 exposure. Mean ±1 SEM (n = 6 experiments per study arm; P values calculated by 2-way analysis of variance. (E) Day 13 iMk proteins with antibodies directed against TGFβR-1, p21, p57, and pSMAD2/3 with GAPDH as a loading control. (F) Day 7 iHPC immunostained with antibodies against phosphorylated JNK (pJNK). Mean ±1 SEM (n = 4 experiments per study arm). (G) Elevated JNK phosphorylation in day 13 L1 iMks. The iMks were solubilized and immunoblotted with antibodies against pJNK, total JNK, and vinculin (as a loading control). (H) Experiments in panel G were scanned and optical density was quantified. Mean ±1 SEM (n = 6 experiments per study arm). All P values by Student t test, unless stated otherwise.
We examined which components of the TGF-β1 signaling pathway were responsible for enhanced TGF-β1 sensitivity in RUNX-1+/− cells by immunoblot studies, focusing on canonical TGF-β1 signaling pathways (Figure 3E). We did not detect differences in levels of TGFβR-1, p21 and p57. SMAD-2/3 protein or phosphorylation levels were not detected in iMks from both L1-C and L1 by immunoblot analysis and intracellular flow cytometry of iHPCs (Figure 3E; supplemental Figure 16). We next evaluated phosphorylation of JNK, a stress-responsive MAPK that is known to be activated by TGF-β1.68 We identified a significant elevation of JNK phosphorylation in RUNX-1+/− CD42+ iHPCs (Figure 3F) and iMks (Figure 3G-H) that was not related to JNK protein levels (Figure 3G).
Drug intervention to correct iMK deficiency from RUNX-1+/− iHPCs
To determine whether decreasing JNK signaling in differentiating RUNX-1+/− iHPCs would correct defective megakaryopoiesis, we evaluated the effect of JNK inhibition on iMk, using the JNK inhibitors, J-IN869 and J-IX.70 Treatment of L1 iHPCs with J-IN8 improved iMk yield, although J-IX did not (Figure 4A-B), which may be related to its reduced potency and increased reversibility.70
![Drug intervention to correct iMK deficiency from RUNX-1+/− iHPCs. (A) Representative flow cytometric analyses of L1-C and L1 day 13 iMk cultures treated with DMSO or J-IN8 and then stained with antibodies against the Mk surface markers CD42a and CD41. (B) Mean ±1 SEM quantitation of the effect of 0.6 μM of JNK inhibitors (J-IN8 or J-IX) on iMk yield as a percentage of DMSO-treated L1-C control cells (n = 4 experiments per study arm; P values by 1-way analysis of variance [ANOVA]). (C) Quantitation of the effect of 0.1 µM RS on iMk yield, as in panel B (n = 4-6 experiments per study arm; P values by 1-way ANOVA). (D-F) Colony-forming assays performed with sorted L1-C and L1 CD42a− and CD42a+ iHPCs treated with DMSO or RS (0.1 μM). (D-F) Sorted cells were seeded in MegaCult medium for Mk colonies (D) or MethoCult medium for erythroid colonies (E) and GM colonies (F). Mean ±1 SEM (n = 4 experiments per study arm; P values by 2-way ANOVA). (G) Quantitation of response of agonist-induced PAC-1 binding in drug-treated L1-C and L1 iMks. After 5 days of culture in the presence of DMSO, RS, or J-IN8, day 13 iMks were collected, stained with Mk surface markers and PAC-1, and stimulated with 0.1 U/mL thrombin. Mean ±1 SEM (n = 3 experiments per study arm; P values by 2-way ANOVA).](https://ash.silverchair-cdn.com/ash/content_public/journal/blood/137/19/10.1182_blood.2020006389/2/m_bloodbld2020006389f4.png?Expires=1769110190&Signature=uWd~cHjKzdnFSJlYMTrFR-gNxGjtinos~xSpN~H9km2nlgf5j7~6T3bCjgbHGFfF5je4IVPqqlHaFHtR5smA7x0aA-DKgQU0Gbd0LZm28OW0Q-UF1F1QfflsqFJPsM2XV8I95J9B3uqeaNIOsB2A-g5rpGneVP0pRbmsVV-jQ4HB37lRnVstfoTDH1FlKneayWTh8LloTqQ9Vow~sqZEXKbrLHQXgeK1PhoqGztJEZ0qb904Y~afQH928tg2aktnGO~htfwfECumL9htEkwlkGJKhgsISbjtEamMH1gzESWK3Mjl3pU~U~W7-PU4u2akPVDjIJem1c0wL8UQa2Vs1g__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
Drug intervention to correct iMK deficiency from RUNX-1+/− iHPCs. (A) Representative flow cytometric analyses of L1-C and L1 day 13 iMk cultures treated with DMSO or J-IN8 and then stained with antibodies against the Mk surface markers CD42a and CD41. (B) Mean ±1 SEM quantitation of the effect of 0.6 μM of JNK inhibitors (J-IN8 or J-IX) on iMk yield as a percentage of DMSO-treated L1-C control cells (n = 4 experiments per study arm; P values by 1-way analysis of variance [ANOVA]). (C) Quantitation of the effect of 0.1 µM RS on iMk yield, as in panel B (n = 4-6 experiments per study arm; P values by 1-way ANOVA). (D-F) Colony-forming assays performed with sorted L1-C and L1 CD42a− and CD42a+ iHPCs treated with DMSO or RS (0.1 μM). (D-F) Sorted cells were seeded in MegaCult medium for Mk colonies (D) or MethoCult medium for erythroid colonies (E) and GM colonies (F). Mean ±1 SEM (n = 4 experiments per study arm; P values by 2-way ANOVA). (G) Quantitation of response of agonist-induced PAC-1 binding in drug-treated L1-C and L1 iMks. After 5 days of culture in the presence of DMSO, RS, or J-IN8, day 13 iMks were collected, stained with Mk surface markers and PAC-1, and stimulated with 0.1 U/mL thrombin. Mean ±1 SEM (n = 3 experiments per study arm; P values by 2-way ANOVA).
Drug intervention to correct iMK deficiency from RUNX-1+/− iHPCs. (A) Representative flow cytometric analyses of L1-C and L1 day 13 iMk cultures treated with DMSO or J-IN8 and then stained with antibodies against the Mk surface markers CD42a and CD41. (B) Mean ±1 SEM quantitation of the effect of 0.6 μM of JNK inhibitors (J-IN8 or J-IX) on iMk yield as a percentage of DMSO-treated L1-C control cells (n = 4 experiments per study arm; P values by 1-way analysis of variance [ANOVA]). (C) Quantitation of the effect of 0.1 µM RS on iMk yield, as in panel B (n = 4-6 experiments per study arm; P values by 1-way ANOVA). (D-F) Colony-forming assays performed with sorted L1-C and L1 CD42a− and CD42a+ iHPCs treated with DMSO or RS (0.1 μM). (D-F) Sorted cells were seeded in MegaCult medium for Mk colonies (D) or MethoCult medium for erythroid colonies (E) and GM colonies (F). Mean ±1 SEM (n = 4 experiments per study arm; P values by 2-way ANOVA). (G) Quantitation of response of agonist-induced PAC-1 binding in drug-treated L1-C and L1 iMks. After 5 days of culture in the presence of DMSO, RS, or J-IN8, day 13 iMks were collected, stained with Mk surface markers and PAC-1, and stimulated with 0.1 U/mL thrombin. Mean ±1 SEM (n = 3 experiments per study arm; P values by 2-way ANOVA).
We next tested whether targeting TGF-β1 signaling would improve iMk yield from RUNX-1+/− iHPCs. RepSox (RS), a small compound TGFβR-1 inhibitor, promotes human megakaryopoiesis and increases RUNX-1 expression in vitro and in mice.71 RS treatment improved iMk yield from L1− iHPCs to near L1-C levels (Figure 4C). Further, in colony-forming assays on sorted CD42− and CD42+ iHPCs, RS increased the number of iMk colonies, but not erythroid and myeloid colony yield (Figure 4D-F). We also tested the effect of these compounds on agonist-induced integrin αIIbβ3 activation, as determined by monoclonal PAC-1 binding. Thrombin-stimulated day 13 iMks cultured with J-IN8 or RS showed no suppression of PAC-1 binding compared with dimethyl sulfoxide (DMSO) vehicle-only controls (Figure 4G), suggesting that these drugs improve differentiation, but do not interfere with agonist-induced integrin signaling.
Studies of RUNX-1in adult CD34+ HSPCs
FPDMM platelet defect and oncogenic risk involve adult hematopoiesis.72 To determine whether our findings in iHPCs apply to adult hematopoiesis, we transduced mobilized, peripheral-blood CD34+ HSPCs with lentiviruses expressing RUNX1-targeting shRNAs (supplemental Figures 17 and 18). Both RUNX1 lentiviral shRNAs 386 and 813 reduced by ∼50% both RUNX1 message (Figure 5A) and Mk yield in liquid culture conditions (Figure 5B), compared with nontargeting (NT) shRNA controls.

Studies of RUNX-1in adult CD34+ HSPCs. NT- or RUNX1-targeting shRNA lentiviral-transduced (supplemental Figure 2) CD34+ HSCs. (A) Quantitation of RUNX1 mRNA levels in day 14 Mk cultures by quantitative PCR with RUNX1 levels in NT control shRNA-treated cells set as 1 (n = 3-5 experiments per study arm). (B) Day 14 Mk yield ±1 SEM from RUNX1 shRNA-transduced CD34+ HSPC cultures as a percentage of the NT control shRNA (n = 3-5 experiments per study arm). (C) Colony-forming assays performed in sorted NT- and RUNX1 shRNA-expressing CD41− and CD41+ HPSCs. Sorted cells were seeded in MegaCult medium for Mk colonies or MethoCult medium for erythroid and GM colonies (n = 4 experiments per study arm). (D) Quantitation of the number (mean ± 1 SEM) of terminal day 14 Mks from sorted CD41− and CD41+ HPSCs as a percentage of the NT control (n = 4 experiments per study arm). (E) Quantitation of the percentage of annexin V+ cells from sorted CD41− and CD41+ HSPCs in NT and RUNX1 shRNA lentiviral-transduced cells. Mean ±1 SEM (n = 3 experiments per study arm). All data are expressed as the mean ±1 SEM, with P values by Student t test.
Studies of RUNX-1in adult CD34+ HSPCs. NT- or RUNX1-targeting shRNA lentiviral-transduced (supplemental Figure 2) CD34+ HSCs. (A) Quantitation of RUNX1 mRNA levels in day 14 Mk cultures by quantitative PCR with RUNX1 levels in NT control shRNA-treated cells set as 1 (n = 3-5 experiments per study arm). (B) Day 14 Mk yield ±1 SEM from RUNX1 shRNA-transduced CD34+ HSPC cultures as a percentage of the NT control shRNA (n = 3-5 experiments per study arm). (C) Colony-forming assays performed in sorted NT- and RUNX1 shRNA-expressing CD41− and CD41+ HPSCs. Sorted cells were seeded in MegaCult medium for Mk colonies or MethoCult medium for erythroid and GM colonies (n = 4 experiments per study arm). (D) Quantitation of the number (mean ± 1 SEM) of terminal day 14 Mks from sorted CD41− and CD41+ HPSCs as a percentage of the NT control (n = 4 experiments per study arm). (E) Quantitation of the percentage of annexin V+ cells from sorted CD41− and CD41+ HSPCs in NT and RUNX1 shRNA lentiviral-transduced cells. Mean ±1 SEM (n = 3 experiments per study arm). All data are expressed as the mean ±1 SEM, with P values by Student t test.
To identify an immunophenotypic Mk-biased HSPC subpopu-lation from the overall CD34+ HSPC population, we sorted for CD34+CD38−CD45RA−CD41+ HSPCs (CD41+ HSPCs) and CD34+CD38−CD45RA−CD41− HSPCs (CD41− HSPCs). We demonstrated that CD41+ HSPCs almost exclusively form Mk colonies relative to the multilineage potential of CD41− HSPCs (Figure 5C; supplemental Figure 19). RUNX-1in, introduced by shRNA-transduction, resulted in reduced Mk yield both in both CD41− and CD41+ HSPCs (Figure 5D), and defects in proliferation were noted in RUNX-1in CD34+ HSPC Mk cultures (supplemental Figure 20) Thus, both Mk-biased and unbiased HSPCs contribute to the observed decrease in megakaryopoiesis. Although RUNX1-386 shRNA significantly reduced the number of Mk colonies, erythroid and myeloid yields were minimally affected (Figure 5C). This finding differs from the panreduction in terminal hematopoietic lineages seen using RUNX-1+/− iHPCs (Figure 2D), but similar to what is observed in patients with FPDMM.73 Sorted RUNX-1in HSPCs on day 6 of culture showed a marked selective enhancement of annexin V+, presumably apoptotic cells (Figure 5E; supplemental Figure 21).
To gain insights into the global and local transcriptomic changes underlying defects in adult RUNX-1in HSPCs, we performed scRNA-SEQ on sorted CD41a− and CD41a+ HSPCs from cultures expressing NT or RUNX1-targeting shRNA. Single-cell transcriptomic analysis defined 7 populations in both control NT and RUNX-1in CD41+ HSPCs (Figure 6A). To understand the physiological relevance of these cell populations, we compared each cell cluster gene expression profile to a previously reported dataset of healthy human multilineage hematopoietic progenitors, committed progenitors, and mature hematopoietic cells.74 Cell type prediction analyses indicated that clusters 1, 2, 4, and 6 primarily matched the expression profile of CD34+ Mk progenitors (MkPs). In contrast, clusters 3 and 5 primarily matched CD34+ megakaryocyte–erythroid progenitors and cluster 7 to CD34+ multilineage progenitors and HSCs (supplemental Figure 22). In support of these findings, functional profiling showed broad enrichment of myeloid-related genes in cluster 7 and Mk/Plt gene sets in cluster 4 (Figure 6B). To identify changes related to RUNX-1in, we compared the number of DE genes between clusters. NT and RUNX1 CD41+ HSPCs, clusters 1, 3, and 4 showed the highest number of DE genes (supplemental Figure 23; supplemental Table 10). Most genes from a curated list of 133 known Mk-associated genes (supplemental Table 4) did not show significant differences in global DE analysis between CD41− and CD41+ HSPCs subpopulations (supplemental Figure 23C; supplemental Table 11). Analysis of gene sets upregulated in RUNX-1in CD41+ HSPC clusters revealed enrichment of proinflammatory signaling, SMAD signaling and immune and myeloid cell-related processes (supplemental Figures 24A and 25A). Many genes found in these enriched processes were also DE in global analyses of all cell clusters from NT and RUNX-1in CD41+ HSPCs (Figure 6C-D; supplemental Table 12).
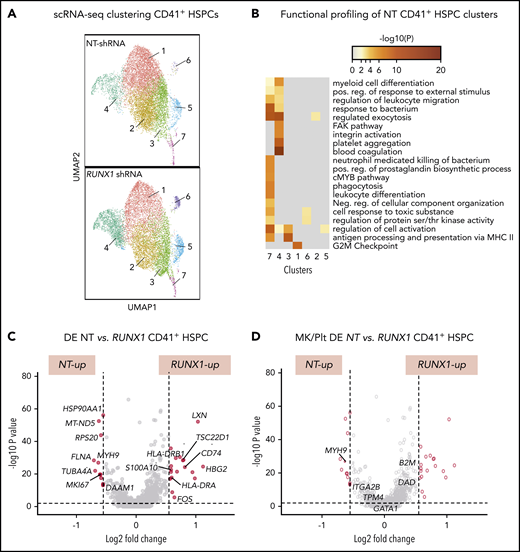
scRNA-SEQ and local and global DE genes analysis in adult CD34-derived RUNX-1in CD41+ HSPCs. (A) Identification and visualization of transcriptional heterogeneity within sorted NT and L1 RUNX-1in CD41+ HSPCs by dimensionality reduction with UMAP. Seven clusters were identified in both NT (n = 7855 cells) and RUNX-1in CD41+ HSPCs (n = 6429 cells) by grouping of cells based on DE. (B) Gene ontology analyses using lists of genes from select clusters with the highest detected number of DE genes, to show functional enrichment in NT CD41+ HSPC clusters. (C-D) Volcano plots show significantly changed genes (C) among all DE genes or focus only on DE of 133 Mk/platelet-associated genes (D), when comparing RUNX-1in to NT HSPCs across all clusters. Filled red circles denote significantly changed genes; unfilled red circles indicate DE genes that are not Mk/platelet associated.
scRNA-SEQ and local and global DE genes analysis in adult CD34-derived RUNX-1in CD41+ HSPCs. (A) Identification and visualization of transcriptional heterogeneity within sorted NT and L1 RUNX-1in CD41+ HSPCs by dimensionality reduction with UMAP. Seven clusters were identified in both NT (n = 7855 cells) and RUNX-1in CD41+ HSPCs (n = 6429 cells) by grouping of cells based on DE. (B) Gene ontology analyses using lists of genes from select clusters with the highest detected number of DE genes, to show functional enrichment in NT CD41+ HSPC clusters. (C-D) Volcano plots show significantly changed genes (C) among all DE genes or focus only on DE of 133 Mk/platelet-associated genes (D), when comparing RUNX-1in to NT HSPCs across all clusters. Filled red circles denote significantly changed genes; unfilled red circles indicate DE genes that are not Mk/platelet associated.
For CD41− HSPCs, UMAP clustering defined 8 transcriptomic clusters in both NT and RUNX-1in cells (supplemental Figure 26A). CD41− HSPC clusters 5 and 7 were enriched for gene sets involved in mRNA metabolism and ribosome assembly (supplemental Figures 24B and 26B). Cell type prediction analyses indicated that their gene expression profile matched CD34+ HSCs and multilineage progenitors (supplemental Figure 27). Similar to embryonic RUNX-1+/− iHPCs, adult RUNX-1in CD41+ HSPCs showed an increased number of DE and upregulated genes, compared with the RUNX-1in CD41a− HSPCs (supplemental Figures 7 and 23). Functional profiling revealed a similar enrichment for stress-associated and proinflammatory processes (supplemental Figure 28B). In support of dysregulated immune cell and TGF-β1–associated pathway analysis, RUNX-1+/− iHPCs and RUNX-1in HSPCs display conserved upregulation of genes involved in TGF-β1–associated and proinflammatory signaling (supplemental Figure 29).
We next determined whether small-molecule inhibitors of JNK and TGFβR-1 signaling would promote Mk yield from RUNX-1in HSPCs introduced by transduction with lentiviral shRNA. NT or RUNX1-targeted shRNA CD34+ HSPCs showed that both J-IN8 and RS corrected Mk yield (Figure 7A; supplemental Figure 30). RS corrected the yield of Mks from both RUNX-1in CD41− and CD41+ HPCs (Figure 7B), but not erythroid or myeloid yield (Figure 7C-D). Another TGFβR-1 inhibitor, galunisertib, in clinical development,75 also corrected Mk yield (Figure 7A).
![Effect of drugs on megakaryopoiesis in RUNX-1in adult CD34+ HSPCs. Experiments are as described in Figure 6, but with added J-IN8, RS, GS, and DAPT. Mean ±1 SEM (n = 4 experiments per study arm; P values by 2-way analysis of variance [ANOVA]). (A) Drug effects correcting the Mk yield per HSPC of NT vs RUNX1 shRNA lentiviral-transduced CD34+ HSPCs after exposure to J-IN8 (0.1 μM), RS (0.1 μM), GS (0.1 μM), or DAPT (10 μM), added on days 5 to 14. (B-D) Effect of RS on colony-forming assays performed using sorted NT- and RUNX1 shRNA-expressing CD41− and CD41+ HPSCs. Sorted cells were seeded in MegaCult medium for Mk colonies (B) or MethoCult medium for erythroid (C) and GM colonies (D).](https://ash.silverchair-cdn.com/ash/content_public/journal/blood/137/19/10.1182_blood.2020006389/2/m_bloodbld2020006389f7.png?Expires=1769110190&Signature=oH6nfP1m6tBVGqUp5XdNelUNP-Qty3E34bc7vtKSNcGk1OZxwZnn73C5Gy7PJdzQclxiSlDoHZLwOhOnOiBE5lDsxWdZ697zvP~NZgNdfZs8SA99a4m1yWDLkcKrG-RuTdVGm4k9u2o2pXUiaRUXH1LRah51ZrtYqCHqRJk50fqfy9WYmAf6neIHxYJLX0m0~HlafHB1xIehkGrDJrO~pNgzxwgccqnvGymNyA8TgcEvxvWzQx6quTsnRnfzvfOKL7Sbu84wh4mY~OvDN30FLREA9MLiF1HU1u3BjH1GI6X7S2~byZTv0oplsbMeqyN0tDGg3SiYILNOqCXVTS7~-A__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
Effect of drugs on megakaryopoiesis in RUNX-1in adult CD34+ HSPCs. Experiments are as described in Figure 6, but with added J-IN8, RS, GS, and DAPT. Mean ±1 SEM (n = 4 experiments per study arm; P values by 2-way analysis of variance [ANOVA]). (A) Drug effects correcting the Mk yield per HSPC of NT vs RUNX1 shRNA lentiviral-transduced CD34+ HSPCs after exposure to J-IN8 (0.1 μM), RS (0.1 μM), GS (0.1 μM), or DAPT (10 μM), added on days 5 to 14. (B-D) Effect of RS on colony-forming assays performed using sorted NT- and RUNX1 shRNA-expressing CD41− and CD41+ HPSCs. Sorted cells were seeded in MegaCult medium for Mk colonies (B) or MethoCult medium for erythroid (C) and GM colonies (D).
Effect of drugs on megakaryopoiesis in RUNX-1in adult CD34+ HSPCs. Experiments are as described in Figure 6, but with added J-IN8, RS, GS, and DAPT. Mean ±1 SEM (n = 4 experiments per study arm; P values by 2-way analysis of variance [ANOVA]). (A) Drug effects correcting the Mk yield per HSPC of NT vs RUNX1 shRNA lentiviral-transduced CD34+ HSPCs after exposure to J-IN8 (0.1 μM), RS (0.1 μM), GS (0.1 μM), or DAPT (10 μM), added on days 5 to 14. (B-D) Effect of RS on colony-forming assays performed using sorted NT- and RUNX1 shRNA-expressing CD41− and CD41+ HPSCs. Sorted cells were seeded in MegaCult medium for Mk colonies (B) or MethoCult medium for erythroid (C) and GM colonies (D).
Previously defined defects in RUNX-1+/− Mks
Most groups studying RUNX-1+/− have focused on the observed final Mk yield from iHPCs36,76 and from CD34+ HSPCs.11,18 Our group and others have defined defects in late cytoskeletal proteins36 and granule-related proteins,15,17 platelet-activation signaling proteins,10 or Mk-lineage–enriched transcription factors.13 A recent study of L2 iPSCs showed that NOTCH-4 downregulation in iMks was defective in RUNX-1+/−, and targeting NOTCH signaling with the γ-secretase inhibitor DAPT was corrective.37 We confirmed that RUNX-1 regulates PF4 levels using lentiviral shRNA transduction in adult CD34+ HSPCs to introduce RUNX-1in (supplemental Figure 31A). However, we did not detect elevation of NOTCH-4 in RUNX-1+/− iMks in line L1 (supplemental Figure 31B), and DAPT did not improve the Mk yield from RUNX1-targeted shRNA CD34+ HSPCs (Figure 7A). This difference in the role of NOTCH-4 in RUNX-1+/− iMk yield is unclear.
Discussion
Patients with FPDMM are at risk of high oncogenic progression, but predominantly manifest platelet defects initially. How the platelet defects translate into other aspects of the disease is unclear. Recent reports have shown that the Mk lineage emerges, in part, early at the HSPC level.21 We reasoned that RUNX-1 activity in HSPCs promotes Mk-biased differentiation and that targeting affected pathways elevated in these cells may correct at least the Mk and platelet defects. To this end, we explored an FPDMM iPSC–derived model of Mk-biased differentiation and shRNA-derived RUNX-1in in adult CD34+ HSPCs. Both investigations supported that RUNX-1+/− results in a deficiency of Mk-biased progenitors that contribute to the observed Mk defect and that these Mk-biased progenitor defects are associated with elevations in druggable stress and proinflammatory pathways.
Prior studies of single-cell transcriptomics, lineage tracing, and in vivo xenotransplantation assays provided evidence that the Mk lineage can be specified at the level of HSPCs19,21 and that differentiation directly toward the Mk/platelet lineage occurs under both steady-state and inflammatory conditions.25,29 In mice, VWF expression marks adult HSCs with Mk-biased differentiation.22 In humans, we detected VWF expression in both Mk-biased and -unbiased iHPCs, and both were unaffected by RUNX-1 levels. Because CD41 and CD42 surface markers had been reported to enrich for human common myeloid progenitors with Mk-bias27 and our iPSC system produced functionally multilineage primitive common myeloid progenitors,45,46 we examined whether CD42 surface expression would enrich for Mk-biased CD235+CD41+ HPCs and found that CD42+ Mk-biased iHPCs emerged concurrently with multipotent iHPCs. Our single-cell transcriptomic data revealed the heterogenous nature of CD42+ iHPCs, along with enrichment for several Mk-associated genes and depletion of cell cycle–associated genes. A comparison of our control iHPC transcriptomic data with published gene sets of terminal Mks,77,78 Plts,78 and MkPs,74,79 monocyte dendritic cell progenitors, granulocytes, and erythrocyte progenitors74 suggests that although most known Mk-associated genes are not significantly upregulated, CD42+ iHPCs are enriched for MkP- and granulocyte-associated genes, supporting that these cells are not just early-maturing Mks (supplemental Figure 12; supplemental Table 5).
We also investigated Mk bias using adult CD34+ HSPCs mobilized into the peripheral blood. We found that a subpopulation of immunophenotypic CD34+CD38− HSPCs express CD41 and are Mk-biased in colony assays. The frequency of adult CD34+CD38−CD41+ progenitor cells (∼25%) reported herein is higher than that in prior reports in freshly isolated bone marrow HSPCs,27,80 likely because of the longer time in HSPC expansion culture medium containing fms-like tyrosine kinase 3 ligand, thrombopoietin, and stem cell factor, which promote Mk differentiation. The lentiviral shRNA approach that we used achieved a 50% to 70% reduction of RUNX-1 levels in terminal adult Mks and in Mk yield.
scRNA-SEQ revealed that both CD42+RUNX-1+/− iHPCs and adult CD34+CD41+RUNX-1in HSPCs display transcriptional deregulation associated with stress, immune, and cytokine response pathways. Among DE Mk-associated genes we identified upregulation of TGFB1 and THBS1, both known for playing a role in TGFβR-1 signaling,56,57 suggesting that elevated TGF-β1 signaling contributes to the underlying defect in RUNX-1+/− iHPCs. Other upregulated genes in RUNX-1+/− iHPCs, such as KLF6, TIMP3, and HIPK2 are reportedly associated with or induced by TGF-β1 signaling.58,59,62 Importantly, comparing DE genes in CD42+RUNX-1+/− iHPCs to DE genes in adult CD34+CD41+RUNX-1in HSPCs revealed a common set of upregulated genes that are known to play roles in TGF-β1 and proinflammatory signaling pathways.62,81-83 Although we saw no elevation of TGF-β1 levels in conditioned medium, RUNX-1+/− iHPCs displayed enhanced sensitivity to TGF-β1 and enhanced baseline JNK-2 phosphorylation, known to be activated by TGF-β1.68 Further investigation is needed to determine how RUNX-1 regulates these genes, the role of these molecules in the HPC defect, and whether these pathways offer druggable targets in patients with FPDMM that would improve the platelet defect and perhaps the oncogenic proclivity.
In addition, apoptosis may have a role in defective early megakaryopoiesis in patients with FPDMM at the level of HSPCs. We observed that CD41+RUNX-1in HSPCs display a selectively enhanced apoptotic phenotype (supplemental Figure 21). We speculate that RUNX-1 is important for promoting survival and/or transition of CD41− to CD41+ HSPCs, a phenomenon reminiscent of the abortive hematopoietic differentiation phenotype reported during early embryonic hematopoiesis in Runx-1–deficient zebrafish.84 In addition, RUNX-1 deficiency has been reported to dysregulate BM niche Mk-HSC interactions dependent on TEK/angiopoietin signaling,85 a pathway linked to regulation of HSC quiescence and apoptosis.86
RUNX-1+/− HPCs were treated with TGFβR-1 and JNK inhibitors to determine whether TGF-β1–associated signaling played a role in our embryonic model of FPDMM and found a significant correction of megakaryopoiesis. In addition, treatment of RUNX-1+/− HPCs with a TGFβR-1 inhibitor in colony assays showed selective improvement of the Mk progenitor defect. The response of adult CD34+ HSPC controls to TGFβR-1 that inhibition was greater than CD34+RUNX-1in HSPCs in colony assays, suggesting that additional TGFβR-1–independent pathways may be involved. Nonetheless, these results support our hypothesis that elevated TGF-β1–associated and JNK-2 signaling pathways in RUNX-1+/− contribute to defective megakaryopoiesis.
In summary, we have demonstrated that RUNX-1+/− causes defects in early hematopoietic cells and in Mk lineage differentiation. RUNX-1 activity in these cells is important for repression of TGFβR-1/JNK-2–associated signaling, and proinflammatory pathways that inhibit Mk progenitor differentiation and expansion. The findings in this study, which extends RUNX-1 function in terminal megakaryopoiesis to promoting the earliest stages of Mk specification from HSPCs, has implications for developing better models of FPDMM and provide insights that may lead to therapeutic strategies for alleviating the platelet defects and perhaps prevent leukemic evolution in FPDMM.
Original data are available by e-mail request to the corresponding author Mortimer Poncz (poncz@chop.edu).
The data reported in this article have been deposited in the Gene Expression Omnibus database for Mk-biased iHPCs and HSPCs (iHPC accession number GSE149136; HSPC accession number GSE166042).
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Acknowledgments
This work was supported by National Institutes of Health (NIH)/National Heart, Lung, and Blood Institute grants T32HL007150 (B.E.) and R35HL150698 (M.P.), and NIH/National Institute of Diabetes and Digestive and Kidney Diseases grant U54DK106829 to the Fred Hutchinson Cancer Research Center Cooperative Centers Excellence in Hematology and the RUNX-1 Research Program in association with Alex’s Lemonade Stand Foundation (M.P., D.F., P.G., W.T., and N.S.).
Authorship
Contribution: B.E. proposed, performed, and evaluated the results of many of the experiments; and prepared the first draft and subsequent revisions; S. Borst constructed the L1, L1-C, and WT-L1 cell lines, contributed to the methodology section, and participated in useful discussions; D.J. helped in early experiments and the interpretation of results, as well as manuscript editing; V.S. performed many of the western blot analyses; M.G. and P. Gao provided assistance in design and technical considerations of scRNA-SEQ experiments, and in preparation of the scRNA-SEQ libraries; M.G., H.H., and J.G. assisted in data preparation and analyses; K.T. provided guidance in bioinformatic analyses; P.L. provided the L2 and L2-C cell lines and contributed to the editing of the manuscript; S. Bagga, N.H., and W.T. provided assistance with technical considerations of generating lentiviral shRNA, as well as construction and testing of the NT- and RUNX1-targeting lentiviral vectors; N.S. provided guidance of the study and assisted in editing the manuscript; D.L.F. and P. Gadue guided the construction of cell lines L1, L1-C, WT, and WT-L1, and helped provide overall guidance, as well as manuscript editing; and M.P. provided overall project organization and direction, data interpretation, and manuscript preparation.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Mortimer Poncz, 3615 Civic Center Blvd, Children’s Hospital of Philadelphia, ARC, Rm 317, Philadelphia, PA 19104; e-mail: poncz@chop.edu.
