

Key Points
IL7-responsive CD127+ T-ALL and T-LBL are sensitive to in vivo PIM inhibition.
Combination of the PIM inhibitor PIM447 with induction chemotherapy improves leukemic survival in a PDX model of CD127+ T-ALL.

Abstract
T-cell acute lymphoblastic leukemia (T-ALL) and T-cell acute lymphoblastic lymphoma (T-LBL) are aggressive hematological malignancies that are currently treated with high-dose chemotherapy. Over the last several years, the search toward novel and less-toxic therapeutic strategies for T-ALL/T-LBL patients has largely focused on the identification of cell-intrinsic properties of the tumor cell. However, non–cell-autonomous activation of specific oncogenic pathways might also offer opportunities that could be exploited at the therapeutic level. In line with this, we here show that endogenous interleukin 7 (IL7) can increase the expression of the oncogenic kinase proviral integration site for Moloney-murine leukemia 1 (PIM1) in CD127+ T-ALL/T-LBL, thereby rendering these tumor cells sensitive to in vivo PIM inhibition. In addition, using different CD127+ T-ALL/T-LBL xenograft models, we also reveal that residual tumor cells, which remain present after short-term in vivo chemotherapy, display consistent upregulation of PIM1 as compared with bulk nontreated tumor cells. Notably, this effect was transient as increased PIM1 levels were not observed in reestablished disease after abrogation of the initial chemotherapy. Furthermore, we uncover that this phenomenon is, at least in part, mediated by the ability of glucocorticoids to cause transcriptional upregulation of IL7RA in T-ALL/T-LBL patient-derived xenograft (PDX) cells, ultimately resulting in non–cell-autonomous PIM1 upregulation by endogenous IL7. Finally, we confirm in vivo that chemotherapy in combination with a pan-PIM inhibitor can improve leukemia survival in a PDX model of CD127+ T-ALL. Altogether, our work reveals that IL7 and glucocorticoids coordinately drive aberrant activation of PIM1 and suggests that IL7-responsive CD127+ T-ALL and T-LBL patients could benefit from PIM inhibition during induction chemotherapy.
Introduction
T-cell acute lymphoblastic leukemia (T-ALL) and T-cell acute lymphoblastic lymphoma (T-LBL) are aggressive hematological malignancies that arise from abnormal activation of oncogenes and/or inactivation of tumor-suppressor genes, followed by a differentiation arrest and uncontrolled clonal expansion of immature thymocytes.1
Proviral integration site for Moloney-murine leukemia 1 (PIM1) is a known JAK-STAT target gene that recently emerged as a therapeutic target for the treatment of T-ALL and T-LBL.2-7 PIM1 activation in primary T-ALL/T-LBLs can occur through T-cell receptor–driven translocations5,6 or activating mutations targeting IL7RA, JAK1, JAK3, or STAT5B.5,6 Based on these cell-intrinsic mechanisms, the number of patients that might eventually benefit from PIM-inhibitor therapy remains relatively low. However, recent work suggested that activation of JAK-STAT signaling in T-ALL could also be achieved through non–cell-autonomous mechanisms, such as stimulation by interleukin 7 (IL7), a cytokine abundantly present in the leukemic microenvironment.7-9 Notably, the ability of T-ALL cells to increase STAT5 phosphorylation upon IL7 stimulation solely depended on surface IL7R (CD127) expression and corresponded with in vivo responses of patient-derived xenografts (PDXs) toward the JAK1/2 inhibitor ruxolitinib, irrespective of the presence of activating IL7R/JAK/STAT pathway mutations.9 This cell-extrinsic mechanism of JAK-STAT pathway activation, mediated by endogenous IL7, suggests that the fraction of T-ALL and T-LBL patients that might benefit from in vivo PIM-inhibitor therapy could be more substantial than originally anticipated.
Current T-ALL and T-LBL treatment protocols consist of induction, consolidation, intensification, and maintenance therapy using a variety of different chemotherapeutic agents.10-13 During induction, the bulk of leukemic cells are supposed to be eradicated by a combination of glucocorticoids, asparaginase, daunorubicin, and vincristine. However, in most cases, some level of minimal residual disease (MRD) persists after induction and these MRD measurements are used to guide risk stratification and tailor consolidation treatment intensity in T-ALL/T-LBL. Currently, the molecular mechanisms by which these residual malignant cells escape the effects of chemotherapy remain poorly understood and, so far, no MRD-specific therapeutic targets have been identified in human T-ALL/T-LBL.
Notably, previous research has shown that glucocorticoids, 1 of the core components of T-ALL/T-LBL induction therapy, can directly induce Il7r expression in murine T cells through binding at an enhancer near the Il7r locus.14,15 Also in the human context, mature CD4+ and CD8+ T cells were shown to induce IL7RA expression upon glucocorticoid treatment16 through a glucocorticoid-response element located about 2000 bp upstream of the IL7RA TATA box.17 Glucocorticoids could therefore potentially drive therapy-induced and non–cell-autonomous activation of the JAK-STAT pathway, eventually leading to downstream PIM1 activation. Given that hyperactivation of JAK-STAT signaling has recently emerged as a mechanism of glucocorticoid resitance,7-9 we hypothesized that T-ALL/T-LBL cells that escape induction therapy could also be characterized by increased PIM1 levels.
Here, we investigated the therapeutic relevance of both cytokine- and chemotherapy-induced PIM1 activation in T-ALL and T-LBL.
Methods
Cell lines and patient samples
Cell lines were purchased from the German Collection of Microorganisms and Cell Cultures GmbH (DSMZ) repository (Braunschweig, Germany) and cultured in RPMI 1640 medium supplemented with 10% fetal calf serum, 100 U/mL penicillin, 100 mg/mL streptomycin, and 2 mM l-glutamine (hereafter referred to as 10% RPMI) at 37°C with 5% CO2. Primary T-ALL/T-LBL cells for establishing PDX models were acquired by informed consent through the Department of Pediatric Hematology-Oncology at Ghent University Hospital, Leuven University Hospital, and Perth Children’s Hospital. The primary T-ALL/T-LBL samples were assigned to a specific molecular genetic subclass based on real-time polymerase chain reaction (RT-PCR) of SIL-TAL1, TLX1/TLX3, or MLL fusion transcripts, or fluorescence in situ hybridization analysis of the LMO2 locus.
Phospho-STAT5 flow cytometry
PDX spleen cells were stimulated ex vivo for 30 minutes with 100 ng/mL human IL7 (PeproTech) in 10% RPMI, washed, and subsequently extracellularly stained with a fixable viability dye (eBioscience) and human CD45 (hCD45)–fluorescein isothiocyanate (Miltenyi Biotec) to gate for human leukemic blasts. The cells were then fixed and permeabilized with the FIX & PERM Cell Permeabilization kit (Invitrogen), using a methanol fixation modification to maintain the phosphorylated (phospho) sites, as described in the standard Invitrogen protocol. Samples were stained for 30 minutes at room temperature with phospho-STAT5 (pSTAT5; Tyr694) monoclonal antibody (SRBCZX), which was allophycocyanin (APC) conjugated (eBioscience). After washing away excess antibody, cells were analyzed by flow cytometry and data analysis was performed using FlowJo software.
In vivo treatment of xenografts with PIM inhibitors and chemotherapy
Nonobese diabetic/severe combined immunodeficient γ (NSG) mice were injected by tail vein at 6 weeks of age with 150 µL of phosphate-buffered saline containing 2.5 × 106 human T-ALL cells harvested from the bone marrow or human T-LBL cells isolated from pleural fluid. At regular time points, leukemia engraftment was monitored by flow cytometry, with hCD45 staining (CD45–fluorescein isothiocyanate antibody; Miltenyi Biotec) of the peripheral blood after red blood cell lysis. Upon establishment of disease, human leukemic cells were isolated from the spleen and retransplanted into secondary recipients.
For in vivo monotherapy treatment with the PIM inhibitors AZD1208 and PIM447 dihydrochloride, a secondary xenograft was established for PDX#6 (supplemental Table 1, available on the Blood Web site). Upon leukemic cell engraftment (>1% hCD45 in peripheral blood), the mice were randomly divided into 4 groups of 3 mice and treatment was started. Mice were treated with 100 mg/kg PIM447 dihydrochloride,18 or with 30 mg/kg AZD1208,19 or with vehicle via oral gavage for 2 weeks (5 days on, 2 days off). Both drugs were used at their maximum tolerated dose and according to previously reported treatment schedules.18,20 PIM447 was formulated in 50 mM sodium acetate buffer, pH 4, and AZD1208 was formulated in dimethyl sulfoxide/PEG400/0.5% methylcellulose at a 10:45:45 ratio. The percentage of hCD45+ cells in the peripheral blood was measured weekly. Immediately after the last treatment dose, animals were euthanized and the spleen weight and percentage of hCD45+ leukemic blasts in bone marrow and spleen were determined by flow cytometry.
For combination therapy with PIM447 dihydrochloride and an induction chemotherapy cocktail composed of vincristine, dexamethasone, and l-asparaginase (VXL) or dexamethasone alone, tertiary xenograft injections were performed in a cohort of 20 NSG mice. Upon detection of >1% hCD45+ leukemic blasts in the peripheral blood, mice were randomized into 4 groups and treated with vehicle, 80 mg/kg PIM447 via oral gavage for 3 weeks (5 days on, 2 days off), VXL via intraperitoneal injection for 5 days, or both PIM447 and VXL using the same concentrations and schedules as per the monotherapy groups. VXL was formulated in 100% phosphate-buffered saline, with the VXL treatment comprising 10 µg of vincristine on day 1, 100 IU of l-asparaginase on day 1, and 10 mg/kg dexamethasone on days 1 to 5. The percentage of hCD45+ was followed weekly in the peripheral blood. After treatment, survival of the mice was defined by means of humane ethical end points. Mice were euthanized when they suffered from >20% weight loss or when >90% leukemic blasts were detected in the peripheral blood for PDX#6, or when >40% blasts were detected for PDX#1.
All in vivo experiments were approved by the ethical committee on animal welfare at Ghent University Hospital.
Statistical analysis
GraphPad Prism 7.0 (La Jolla, CA) was used for statistical analyses. The Mann-Whitney U test was used to analyze differences between subgroups. P values <.05 were considered statistically significant. All error bars represent the standard error of the mean (SEM). All in vitro experiments were run at least in duplicate.
Additional materials and methods are provided as supplemental Methods.
Results
Cytokine-induced PIM1 activation in CD127+ T-ALL and T-LBL
Previous studies have shown that a subset of T-ALL patients is able to activate JAK-STAT signaling in response to exogenous IL7, regardless of the mutational status of IL7R-pathway genes.7-9,21 Although initially reported for early T-cell precursor ALL, the exact genetic features of T-ALL/T-LBL patients that show this IL7 responsiveness remain largely unknown.
Given this, we analyzed pSTAT5 (Tyr694) levels upon IL7 stimulation in a series of genetically well-characterized PDX samples obtained from 7 T-ALL and 4 T-LBL pediatric patient samples (supplemental Table 1). T-ALL/T-LBL PDX models are known to mirror the genomic landscape of primary pediatric samples, as previously shown by Richter-Pechańska et al.22 Supplemental Figure 1 depicts similarities between primary and PDX samples in our series. From these 11 PDX samples, 4 T-ALL and 2 T-LBL cases showed IL7-induced pSTAT5 induction, which was not the case for the remaining 5 T-ALL/T-LBL samples (Figure 1A). IL7-responsive T-ALL/T-LBL samples covered a broad spectrum of molecular-genetic subtypes, including immature, TLX1+, TLX3+, NKX2.1+, HOXA+ and TAL/LMO+, and were mostly characterized by higher CD127 expression as compared with NKX2.5+ or TAL/LMO+ nonresponders (Figure 1B). No samples had mutations in IL7RA or other members of the JAK/STAT pathway (cooperative somatic mutations identified in these T-ALL/T-LBL patient samples are summarized in supplemental Table 1).
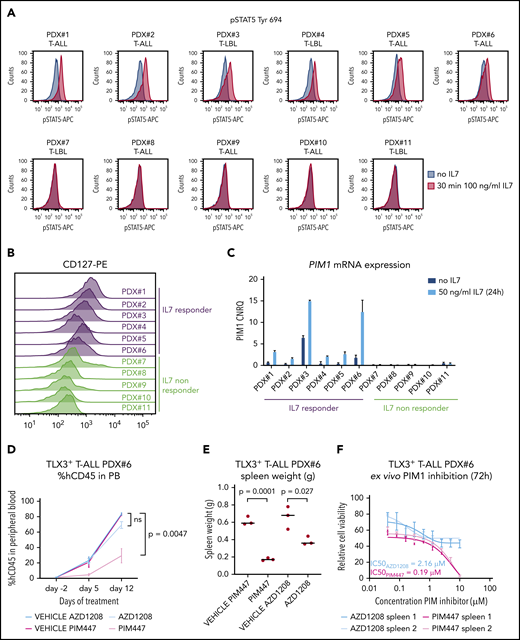
A subset of T-ALL/T-LBL patients upregulates PIM1 expression in response to IL7. (A) PDX spleen samples were stimulated with 100 ng/mL IL7 for 30 minutes, after which phosphorylation sites were fixed with methanol and pSTAT5 (Tyr 694) was measured by flow cytometry. (B) CD127 protein expression levels, analyzed by flow cytometry, for 11 PDX spleen samples. (C) PDX spleen samples were stimulated with 50 ng/mL IL7 for 24 hours and were subsequently collected for RNA isolation. PIM1 messenger RNA (mRNA) expression levels are shown, measured by quantitative RT-PCR (RT-qPCR). (D) A PDX model of PDX#6 was established, after which 3 mice per group were treated for 2 weeks (5 days on/2 days off) with either 30 mg/kg AZD1208 or 100 mg/kg PIM447 or their respective vehicles. The percentages of human CD45+ (%hCD45+) leukemic blasts in peripheral blood (PB) are shown per group for 3 time points. (E) PDX#6 mice were euthanized after 2 weeks of treatment with either AZD1208 or PIM447. Spleen weights are shown per treatment group. (F) Ex vivo treatment of PDX#6 spleen cells. Fifty thousand cells were treated per condition for 72 hours in 10% RPMI supplemented with 10 ng/mL IL7, 50 ng/mL stem cell factor (SCF), 20 ng/mL FLT3, and 100 ng/mL IL2, in duplicate. Adenosine triphosphate (ATP) content was measured by means of a CellTiter-Glo viability assay. CNRQ, calibrated normalized relative quantity; IC50, 50% inhibitory concentration; PE, phycoerythrin.
A subset of T-ALL/T-LBL patients upregulates PIM1 expression in response to IL7. (A) PDX spleen samples were stimulated with 100 ng/mL IL7 for 30 minutes, after which phosphorylation sites were fixed with methanol and pSTAT5 (Tyr 694) was measured by flow cytometry. (B) CD127 protein expression levels, analyzed by flow cytometry, for 11 PDX spleen samples. (C) PDX spleen samples were stimulated with 50 ng/mL IL7 for 24 hours and were subsequently collected for RNA isolation. PIM1 messenger RNA (mRNA) expression levels are shown, measured by quantitative RT-PCR (RT-qPCR). (D) A PDX model of PDX#6 was established, after which 3 mice per group were treated for 2 weeks (5 days on/2 days off) with either 30 mg/kg AZD1208 or 100 mg/kg PIM447 or their respective vehicles. The percentages of human CD45+ (%hCD45+) leukemic blasts in peripheral blood (PB) are shown per group for 3 time points. (E) PDX#6 mice were euthanized after 2 weeks of treatment with either AZD1208 or PIM447. Spleen weights are shown per treatment group. (F) Ex vivo treatment of PDX#6 spleen cells. Fifty thousand cells were treated per condition for 72 hours in 10% RPMI supplemented with 10 ng/mL IL7, 50 ng/mL stem cell factor (SCF), 20 ng/mL FLT3, and 100 ng/mL IL2, in duplicate. Adenosine triphosphate (ATP) content was measured by means of a CellTiter-Glo viability assay. CNRQ, calibrated normalized relative quantity; IC50, 50% inhibitory concentration; PE, phycoerythrin.
As expected, IL7 stimulation resulted in PIM1 upregulation in responsive T-ALL/T-LBL patient samples (Figure 1C), suggesting that CD127+ T-ALL/T-LBLs, lacking IL7RA/JAK/STAT mutations, might also benefit from PIM inhibition. To test this, we performed in vivo evaluation of the PIM inhibitors PIM447 and AZD1208 using T-ALL PDX#6 (TLX3+), which lacks cell-intrinsic genetic abnormalities targeting PIM1 or the IL7R pathway but has the ability to increase PIM1 expression upon IL7 stimulation (Figure 1A; supplemental Figure 1). In vivo treatment with 2 PIM inhibitors PIM447 and AZD1208 at their maximum tolerated dose (100 mg/kg and 30 mg/kg, respectively) for 2 weeks (5 days on, 2 days off) resulted in a significant reduction of the leukemic burden, as measured by the percentage of blasts in peripheral blood (Figure 1D) or spleen size (Figure 1E) and revealed that PIM447 was more effective in vivo as compared with AZD1208. Supplemental Table 2 provides an overview of different PIM inhibitors that are currently investigated in clinical trials.
The superior activity of PIM447, which shows promising results in clinical trials for hematological malignancies,23,24 was also confirmed ex vivo on PDX#6 cells obtained from the spleen, with 50% inhibitory concentration values for PIM447 and AZD1208 of 0.19 µM and 2.16 µM, respectively (Figure 1F). Moreover, a recent study by Chen et al confirmed the higher activity of PIM447 compared with AZD1208 due to stronger binding of PIM447 to PIM kinases,25 whereas Burger et al confirmed that this compound only targets the PIM kinases with slight aspecificity for GSK3β, PKN1, and PKCτ at higher concentrations.23 Next, we confirmed thatex vivo PIM447 treatment triggered a block in the G1 phase of the cell cycle with a concomitant increase in early and late apoptotic cells (supplemental Figure 2).2,3 In addition, we performed RNA sequencing on ex vivo–treated PDX#6 spleen cells, with or without PIM447. In line with other PIM inhibitors (such as AZD1208 and TP-3654), we observed changes in cell cycle and Myc targets (supplemental Figure 3A-B).6 Finally, ex vivo treatment of PDX#1 spleen cells with PIM447 in the presence or absence of IL7 showed a greater effect of PIM447 in the presence of IL7 (supplemental Figure 4).
Thus, cytokine-induced PIM1 activation in IL7-responsive CD127+ T-ALL/T-LBL patient samples can be therapeutically targeted by the PIM inhibitor PIM447.
Simultaneous PIM1 activation by combined cell-intrinsic and -extrinsic effects in human T-ALL/T-LBL
PIM1 overexpression in T-ALL/T-LBL can be caused by genomic translocations involving T-cell receptor β (TCRβ) or activating mutations targeting the IL7R/JAK/STAT signaling pathway. However, PIM1 can also become activated through non–cell-autonomous stimulation by exogenous IL7 in CD127+ T-ALL/T-LBL.
Here, we evaluated whether these different mechanisms of PIM1 activation are mutually exclusive or not. Stimulation of HPB-ALL cells, which already express considerable amounts of PIM1 due to the presence of a JAK1 mutation, showed clear pSTAT5 induction that coincided with a significant upregulation of PIM1 (Figure 2A). Similarly, IL7 stimulation of ex vivo–treated PDX cells obtained from a TCRβ-PIM1+ T-LBL case6 (PDX#3) resulted in pSTAT5 activation and simultaneous upregulation of PIM1 (Figure 2B).
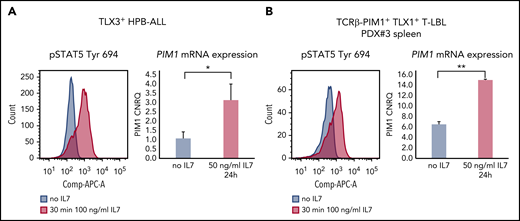
Intrinsic and extrinsic PIM1 activation can co-occur. (A) HPB-ALL cells were stimulated with 100 ng/mL IL7 for 30 minutes and stained for pSTAT5 (Tyr 694) conjugated to APC (left panel). For downstream differential transcriptional analysis via PIM1 RT-qPCR, cells were stimulated with 50 ng/mL IL7 for 24 hours (right panel). (B) PDX#3 spleen cells were stimulated with 100 ng/mL IL7 for 30 minutes and stained for pSTAT5 (Tyr 694) conjugated to APC (left panel). For downstream differential transcriptional analysis via PIM1 RT-qPCR, cells were stimulated with 50 ng/mL IL7 for 24 hours (right panel). *P < .05; **P < .01
Intrinsic and extrinsic PIM1 activation can co-occur. (A) HPB-ALL cells were stimulated with 100 ng/mL IL7 for 30 minutes and stained for pSTAT5 (Tyr 694) conjugated to APC (left panel). For downstream differential transcriptional analysis via PIM1 RT-qPCR, cells were stimulated with 50 ng/mL IL7 for 24 hours (right panel). (B) PDX#3 spleen cells were stimulated with 100 ng/mL IL7 for 30 minutes and stained for pSTAT5 (Tyr 694) conjugated to APC (left panel). For downstream differential transcriptional analysis via PIM1 RT-qPCR, cells were stimulated with 50 ng/mL IL7 for 24 hours (right panel). *P < .05; **P < .01
Therefore, the IL7R-JAK-STAT-PIM1 activation status in human T-ALL/T-LBL is defined by a combination of both cell-intrinsic and cell-extrinsic effects affecting this signaling pathway.
Glucocorticoids induce PIM1 in steroid-sensitive T-ALL/T-LBL by transcriptional activation of IL7RA
Glucocorticoids, such as prednisone and dexamethasone, are core components of T-ALL treatment.26 Their mechanism of action is based on binding to the glucocorticoid receptor (NR3C1) and subsequent translocation to the nucleus, where NR3C1 binds to a broad spectrum of specific target genes. Interestingly, previous research showed that glucocorticoids are able to bind an enhancer of the IL7RA locus, thereby upregulating IL7RA expression in thymocytes.14,15,27
Given this, we evaluated IL7RA expression in our panel of 11 T-ALL/T-LBL PDX samples upon ex vivo dexamethasone treatment (100 nM, 24 hours). Notably, this analysis confirmed that glucocorticoids can induce IL7RA expression in most T-ALL/T-LBL PDX samples (Figure 3A). As expected, the 1 T-ALL patient sample that failed to show IL7RA upregulation (PDX#5) was intrinsically resistant to glucocorticoids (supplemental Figure 5).
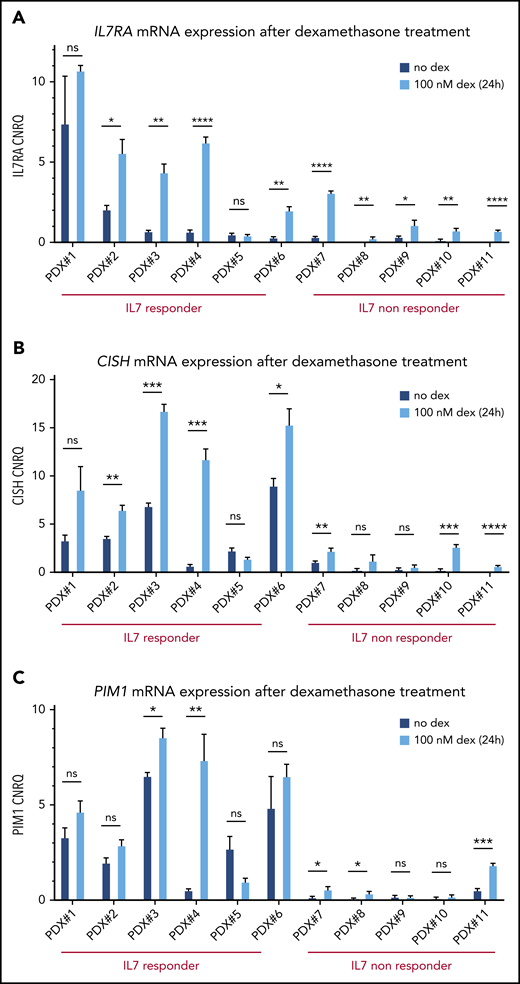
Glucocorticoid-responsive T-ALL/T-LBL PDX samples show IL7R/CISH/PIM1 upregulation upon dexamethasone treatment. (A) IL7R mRNA expression levels are shown for 11 PDX spleen samples with and without 100 nM dexamethasone (dex) treatment of 24 hours, in 10% RPMI supplemented with 50 ng/mL IL7. (B) CISH mRNA expression levels are shown for 11 PDX spleen samples with and without 100 nM dexamethasone treatment of 24 hours, in 10% RPMI supplemented with 50 ng/mL IL7. (C) PIM1 mRNA expression levels are shown for 11 PDX spleen samples with and without 100 nM dexamethasone treatment of 24 hours, in 10% RPMI supplemented with 50 ng/mL IL7. *P < .05; **P < .01; ***P < .001; ****P < .0001. ns, non significant.
Glucocorticoid-responsive T-ALL/T-LBL PDX samples show IL7R/CISH/PIM1 upregulation upon dexamethasone treatment. (A) IL7R mRNA expression levels are shown for 11 PDX spleen samples with and without 100 nM dexamethasone (dex) treatment of 24 hours, in 10% RPMI supplemented with 50 ng/mL IL7. (B) CISH mRNA expression levels are shown for 11 PDX spleen samples with and without 100 nM dexamethasone treatment of 24 hours, in 10% RPMI supplemented with 50 ng/mL IL7. (C) PIM1 mRNA expression levels are shown for 11 PDX spleen samples with and without 100 nM dexamethasone treatment of 24 hours, in 10% RPMI supplemented with 50 ng/mL IL7. *P < .05; **P < .01; ***P < .001; ****P < .0001. ns, non significant.
Finally, we determined whether glucocorticoid-induced IL7RA activation would also result in JAK-STAT pathway activation with concomitant upregulation of PIM1. Indeed, ex vivo dexamethasone treatment of PDX samples in combination with IL7 caused significant upregulation of both CISH and PIM1 expression along the JAK-STAT axis in most T-ALL/T-LBL PDX samples (Figure 3B-C). However, the eventual PIM1 expression after steroid treatment in the IL7-nonresponder group remains relatively low as compared with the levels obtained in the IL7-responsive T-ALL/T-LBL PDX samples (Figure 3B-C).
To check whether PIM1 can be directly upregulated by NR3C1, we analyzed publicly available chromatin immunoprecipitation data for NR3C1 (SRX2900586, 100 nM dexamethasone, 1 hour) and H3K27ac (SRX583005) in the acute monocytic leukemia cell line THP-1. As expected, NR3C1 was readily detected at the enhancer region of the well-established NR3C1 target gene GILZ. Similarly, NR3C1 binding was observed at the promoter region of IL7RA, but was absent from the PIM1 locus, suggesting that PIM1 is not a direct transcriptional target of the glucocorticoid receptor itself (supplemental Figure 6).
Increased levels of PIM1 in T-ALL/T-LBL xenografts upon in vivo induction therapy
To determine whether this mechanism of glucocorticoid-induced PIM1 activation would also take place when leukemic cells are exposed to induction therapy in an in vivo IL7-producing microenvironment, we performed additional T-ALL/T-LBL xenograft experiments in which we treated mice for 1 week with a chemotherapeutic cocktail of VXL or vehicle control (Figure 4A). After 1 week, both control and chemotherapy-treated mice were euthanized and hCD45+ leukemic cells were isolated by fluorescence-activated cell sorting from the bone marrow. On average, 2.5 million bone marrow cells were sorted out from control mice, whereas only 100 000 to 300 000 leukemic cells remained present in the bone marrow after the 4-day chemotherapy regimen. Notably, quantitative RT-PCR revealed significant upregulation of both PIM1 and CISH expression in residual leukemic blasts as compared with controls in 4 CD127+ PDX samples analyzed, confirming that induction chemotherapy can trigger in vivo JAK-STAT pathway activation in human T-ALL and T-LBL (Figure 4B). Interestingly, for 1 PDX sample (PDX#6), we waited for tumor cells to reappear after initial chemotherapy treatment, resulting in reestablishment of disease 8 weeks after induction therapy. Notably, CISH and PIM1 levels in these postchemotherapy relapse samples were comparable to the initial levels seen in the tumor xenograft material prior to treatment (Figure 4C). Thus, induction chemotherapy triggers PIM1 induction in residual human T-ALL/T-LBL cells in vivo, but these effects are lost upon disease recurrence after the treatment has been abrogated.
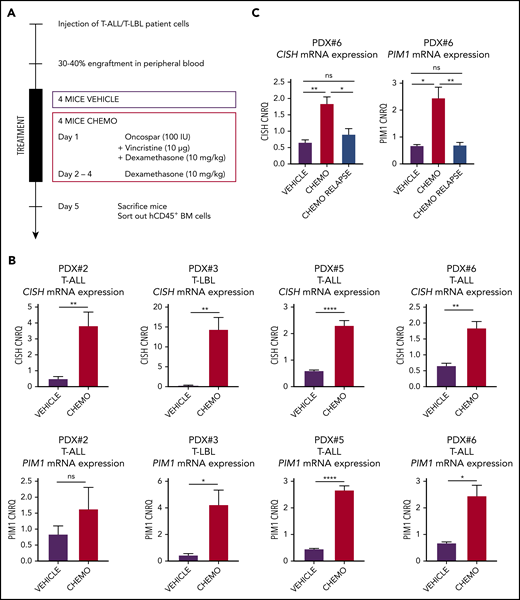
Chemotherapy-resistant blasts are characterized by high PIM1 expression. (A) In vivo chemotherapeutic (Chemo) treatment schedule. Four mice per group per PDX were treated via intraperitoneal injection and euthanized on day 5. Leukemic blasts (hCD45+ cells) were sorted from bone marrow, after which RNA was isolated. (B) RT-qPCR for CISH and PIM1 on residual blasts in the bone marrow after in vivo chemotherapy treatment. (C) RT-qPCR for CISH and PIM1 on bone marrow samples from nontreated (vehicle) mice, mice that were treated for 1 week with the chemotherapy schedule from panel A (chemo), and mice that relapsed after chemotherapy treatment (chemo relapse). *P < .05; **P < .01; ****P < .0001.
Chemotherapy-resistant blasts are characterized by high PIM1 expression. (A) In vivo chemotherapeutic (Chemo) treatment schedule. Four mice per group per PDX were treated via intraperitoneal injection and euthanized on day 5. Leukemic blasts (hCD45+ cells) were sorted from bone marrow, after which RNA was isolated. (B) RT-qPCR for CISH and PIM1 on residual blasts in the bone marrow after in vivo chemotherapy treatment. (C) RT-qPCR for CISH and PIM1 on bone marrow samples from nontreated (vehicle) mice, mice that were treated for 1 week with the chemotherapy schedule from panel A (chemo), and mice that relapsed after chemotherapy treatment (chemo relapse). *P < .05; **P < .01; ****P < .0001.
Preclinical evaluation of PIM447 in combination with chemotherapy in a CD127+ T-ALL PDX model that lacks cell-intrinsic JAK-STAT pathway activation
Previously, we have shown that in vivo combination therapy of a PIM inhibitor with glucocorticoids resulted in significantly prolonged survival using a PDX model of a TCRβ-PIM1+ T-LBL case.6 Of note, PDX cells from this T-LBL patient sample have also been included in this study (PDX#3) and these cells showed both cytokine and therapy-induced PIM1 activation. Therefore, the previously published synergistic effects of this combination therapy in PDX#3 are most probably mediated by a combination of both cell-intrinsic and cell-extrinsic effects, which ultimately resulted in robust in vivo PIM1 activation.
To further test this, we evaluated the combination of PIM447 and dexamethasone ex vivo in PDX cells from both IL7 responder and nonresponder T-ALL/T-LBL samples in the presence of exogeneous IL7. Interestingly, synergism was observed in the IL7-responder group, whereas no beneficial effect was seen in IL7 nonresponders (Figure 5A-B). Next, we evaluated this combination therapy in vivo using PDX#1 with a treatment schedule as visualized in Figure 5C. This analysis revealed a significant improvement in survival for the PIM447/dexamethasone combination as compared with both monotherapy treatments (P = .0034; Figure 5D).
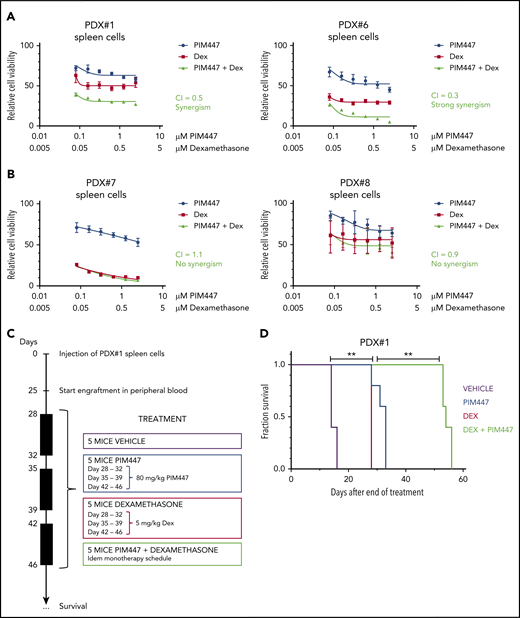
PIM447-glucocorticoid combination therapy is synergistic in IL7-responsive T-ALL/T-LBL. (A) Ex vivo combination treatment with PIM447 and dexamethasone (Dex) of IL7-responsive PDX spleen cells. Fifty thousand cells were treated per condition for 72 hours in 10% RPMI supplemented with 10 ng/mL IL7, 50 ng/mL SCF, 20 ng/mL FLT3, and 100 ng/mL IL2, in duplicate. ATP content was measured by means of a CellTiter-Glo viability assay. Combination indexes (CI) were calculated with CalcuSyn software. (B) Ex vivo combination treatment with PIM447 and dexamethasone of IL7-nonresponsive PDX spleen cells. The same conditions were used as per panel A. (C) In vivo combination treatment schedule with PIM447 and dexamethasone. Five mice per group were treated with either vehicle, or 80 mg/kg PIM447 via oral gavage, or 5 mg/kg dexamethasone via intraperitoneal injection, or the combination of PIM447 with dexamethasone. The frequency of treatment was once daily for 5 days followed by 2 days off. Mice were monitored and euthanized when weight dropped by >20%, or leukemic blasts in the peripheral blood reached 90%, or when mice developed leukemia-associated morbidity (ruffled fur, heavy breathing, slow to respond). (D) Survival curve for in vivo treatment of PDX#1 with PIM447 and dexamethasone. **P < .01.
PIM447-glucocorticoid combination therapy is synergistic in IL7-responsive T-ALL/T-LBL. (A) Ex vivo combination treatment with PIM447 and dexamethasone (Dex) of IL7-responsive PDX spleen cells. Fifty thousand cells were treated per condition for 72 hours in 10% RPMI supplemented with 10 ng/mL IL7, 50 ng/mL SCF, 20 ng/mL FLT3, and 100 ng/mL IL2, in duplicate. ATP content was measured by means of a CellTiter-Glo viability assay. Combination indexes (CI) were calculated with CalcuSyn software. (B) Ex vivo combination treatment with PIM447 and dexamethasone of IL7-nonresponsive PDX spleen cells. The same conditions were used as per panel A. (C) In vivo combination treatment schedule with PIM447 and dexamethasone. Five mice per group were treated with either vehicle, or 80 mg/kg PIM447 via oral gavage, or 5 mg/kg dexamethasone via intraperitoneal injection, or the combination of PIM447 with dexamethasone. The frequency of treatment was once daily for 5 days followed by 2 days off. Mice were monitored and euthanized when weight dropped by >20%, or leukemic blasts in the peripheral blood reached 90%, or when mice developed leukemia-associated morbidity (ruffled fur, heavy breathing, slow to respond). (D) Survival curve for in vivo treatment of PDX#1 with PIM447 and dexamethasone. **P < .01.
Finally, we assessed the combination of PIM447 with VXL induction chemotherapy in vivo using another PDX sample (PDX#6) that lacked cell-intrinsic genetic abnormalities targeting PIM1 or the IL7R pathway, but had the ability to activate PIM1 upon IL7 stimulation (Figure 3C) and induction chemotherapy (Figure 4B-C). The treatment schedule is shown in supplemental Figure 7A. Notably, this analysis also revealed a significant improvement in survival in the PIM447/VXL combination group as compared with VXL monotherapy (P = .0103; supplemental Figure 7B).
Discussion
We and others have recently shown that PIM1 might serve as a valuable therapeutic target for the treatment of human T-ALL and T-LBL.2-7
PIM1 is a proto-oncogene that encodes a constitutively active serine/threonine kinase.28,29 Its expression is regulated through the JAK-STAT signaling pathway.30,31 PIM1 mediates cellular activities through the phosphorylation of a myriad of substrates involved in cell-cycle progression (eg, p21 and p27), transcription (eg, Myc), apoptosis (eg, Bad), drug resistance (eg, Pgp), and cellular metabolism (eg, PRAS40).32-37 Moreover, PIM1 plays a role in homing and migration of hematopoietic stem cells through regulating surface expression of CXCR4 chemokine.38 In vivo studies with Pim knockout mice showed that PIM kinase depletion resulted in subtle hematological changes such as anemia and reduced peripheral T- and B-cell numbers.39,40 Therefore, it should be noted that potential hematological side effects should be carefully monitored during PIM inhibitor treatment.
PIM1 translocations,5,6 activating mutations in IL7RA, JAK1, JAK3, or STAT5B5,6 or loss-of-function alterations targeting PTPN2, can drive cell-intrinsic PIM1 activation, rendering human T-ALL/T-LBL cells susceptible to pan-PIM inhibitors such as AZD12083 or TP3654.6 However, non–cell-autonomous mechanisms of PIM1 activation might also contribute to the potential sensitivity of T-ALL/T-LBL cells toward PIM inhibition. Indeed, in this study, we show that cell-extrinsic PIM1 activation by IL7 can render CD127+ T-ALL/T-LBL cells sensitive to in vivo PIM inhibition by PIM447. Of note, a recent phase 1 trial in relapsed or refractory multiple myeloma recently revealed that PIM447 is well tolerated in humans, suggesting that this agent might have the potential to be translated toward the clinic in the future.24
Interestingly, the surface IL7R/CD127high T-ALL/T-LBL cells in this study belonged to different molecular genetic subtypes of human T-ALL/T-LBL,41 including both diagnostic and/or relapse samples from immature, TLX1+, TLX3+, HOXA+, NKX2.1+, and SIL-TAL1+ cases. In contrast, IL7R/CD127low tumors were TAL/LMO+ or NKX2.5+, suggesting that these samples, which lacked the potential for non–cell-autonomous cytokine-induced PIM1 activation, might be enriched for the TAL/LMO subtype of human T-ALL/T-LBL.
Although we show that the ability of tumor cells to activate PIM1 upon IL7 stimulation depends on the presence of surface IL7R/CD127 expression,8,9 the actual level of this oncogenic kinase in each T-ALL/T-LBL patient sample will be defined by a combination of cell-intrinsic and cell-extrinsic effects. Indeed, the baseline expression of IL7R/CD127 in normal T-cell progenitors can be modulated and enhanced by additional cell-intrinsic genetic defects in T-ALL/T-LBL cells. For example, loss-of-function alterations targeting DNM2 will cause impaired clathrin-mediated endocytosis of IL7R/CD127, ultimately resulting in enhanced receptor density on the surface of T-ALL/T-LBL cells.42 Alternatively, Weijenborg Campos et al recently described a new class of IL7RA mutations in human T-ALL that increased the sensitivity of the mutated receptor toward IL7.43 Furthermore, a recent study also showed that T-ALLs can execute autocrine production of IL7,44 a phenomenon that could further increase IL7 levels in the tumor microenvironment, subsequently causing enhanced PIM1 levels in T-ALL/T-LBL tumor cells. Also in this study, we show that exogenous IL7 can further increase PIM1 levels in a IL7R/CD127+ T-LBL case that already displayed a cell-intrinsic TCRβ-PIM1 translocation. With this in mind, we believe that targeting PIM1 might serve as a better and more comprehensive therapeutic strategy for IL7R/CD127+ human T-ALL/T-LBL as compared with JAK inhibitors, such as ruxolitinib or tofacitinib. Indeed, although both strategies will be able to target cytokine-induced activation of the IL7R-JAK-STAT-PIM1 signaling axis, only the PIM inhibitors will be functional against tumor-specific alterations targeting molecules downstream of JAK proteins, such as STAT5B or PIM1 itself. Alternatively, both therapeutic strategies could also be combined,4 a notion that is currently evaluated in a trial of PIM447 in combination with ruxolitinib for patients with myelofibrosis (NCT02370706).
Over the last several decades, overall survival rates for pediatric T-ALL and T-LBL patients have gradually improved toward almost 90% with the most recent MRD-based treatment protocols. Indeed, patients with a high MRD load after induction therapy receive an intensified treatment regimen potentially followed by hematopoietic stem cell transplantation. Although this dose-escalation strategy has led to improved overall survival rates, it has also been associated with more severe toxic side effects and an increased rate of treatment-related mortalities. Therefore, specific therapeutic interventions that could reduce MRD tumor load during induction therapy might serve as valuable therapeutic strategies to reduce the need for therapy intensification, eventually causing less toxic side effects and potentially lower the relapse rates in human T-ALL or T-LBL.
Here, we used 4 different IL7R/CD127+ T-ALL/T-LBL PDX models to show that residual tumor cells, which remain present after 4 days of short-term in vivo VXL induction chemotherapy, show consistent upregulation of PIM1 as compared with bulk nontreated tumor cells. Of note, these effects were transient as recurrent disease that reestablished after abrogation of initial short-term in vivo chemotherapy again displayed lower PIM1 expression similar to the levels observed in bulk nontreated control tumor cells. Therefore, these effects are most probably mediated by a direct transcriptional response driven by the short-term chemotherapy regimen itself. In line with this notion, we find that this phenomenon is, at least in part, mediated by the steroids present in the induction therapy treatment regimen. Indeed, in agreement with previous data showing that glucocorticoids can directly bind a glucocorticoid response element upstream of the IL7RA TATA box in human CD4+ and CD8+ T cells,17 we show that dexamethasone treatment in T-ALL/T-LBL PDX cells results in transcriptional upregulation of IL7RA. Of note, recent studies have shown that both cell-intrinsic41 as well as cell-extrinsic7-9 mechanisms of JAK-STAT pathway activation can render T-ALL cells less sensitive to steroid therapy. Therefore, one could hypothesize that this therapy-induced IL7RA activation eventually leads to non–cell-autonomous stimulation of the JAK-STAT-PIM1 signaling pathway by endogenous IL7, ultimately resulting in residual PIM1high T-ALL/T-LBL cells that survive initial debulking through their reduced sensitivity toward glucocorticoids. With this in mind, we finally show that ex vivo combination therapy of PIM447 and dexamethasone is synergistic in IL7-responsive IL7R/CD127+ T-ALL and T-LBL PDX cells. Interestingly, it was previously shown that murine IL7 is able to bind and activate human IL7R/CD127.45 Therefore, we performed an in vivo study, by using a PDX model from a IL7R/CD127+ T-ALL that lacked cell-intrinsic IL7R-JAK-STAT-PIM1 abnormalities, and confirmed that combination PIM447 with VXL induction chemotherapy improves leukemia survival.
Altogether, our study shows that cytokine- and therapy-induced PIM1 activation can be therapeutically targeted by the pan-PIM inhibitor PIM447 in human T-ALL/T-LBL. In addition, our work suggests that IL7 responsiveness in CD127+ T-ALL/T-LBL could serve as a valuable biomarker to identify patients that might benefit from PIM inhibition during induction chemotherapy.
The data reported in this article have been deposited in the Gene Expression Omnibus database (accession number GSE145165).
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Acknowledgments
This work was supported by the following funding agencies: the Fund for Scientific Research Flanders (FWO [P.V.V. and R.D.S.]), Kom op tegen Kanker (Stand up to Cancer, the Flemish cancer society [P.V.V. and S.G.]), Kinderkankerfonds (a nonprofit childhood cancer foundation under Belgian law [P.V.V., T.L., and B.D.M.]), the European Hematology Association (EHA; J.M.), the European Research Council (StG-639784 [P.V.V.] and CoG-648455 [J.T.B.]), and Projects of National Interest (PRIN) 2017 (2017PPS2X4 [C.M.]). R.S.K. was supported by a Fellowship from the National Health and Medical Research Council of Australia (NHMRC APP1142627).
Authorship
Contribution: R.D.S., L.R., J.M., V.B., J.T.B., R.L.S., S.G., and A.A. performed experiments; R.D.S., P.V.V., J.R., C.M., T.T., and W.V.L. performed analyses; L.R. and B.L. provided technical assistance; L.C.C., R.S.K., M.R.M., A.U., P.V., T.L., N.V.R., and B.D.M. collected and provided primary T-ALL patient material; P.V.V. and R.D.S. designed research and wrote the paper, with help from the other authors; and all authors have seen and reviewed the manuscript and approved the final version.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Pieter Van Vlierberghe, Department of Biomolecular Medicine, Ghent University, Medical Research Building 2, Building 38, Room 110.006, Corneel Heymanslaan 10, 9000 Ghent, Belgium; e-mail: pieter.vanvlierberghe@ugent.be.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal