

In this issue of Blood, demonstrate that glucocorticoid treatment of a subset of T-cell acute lymphoblastic leukemia/T-cell acute lymphoblastic lymphoma (T-ALL/T-LBL) cells upregulates PIM1 expression, creating a synergistic vulnerability and potent antileukemic effect with PIM1 inhibitors in a preclinical model.1
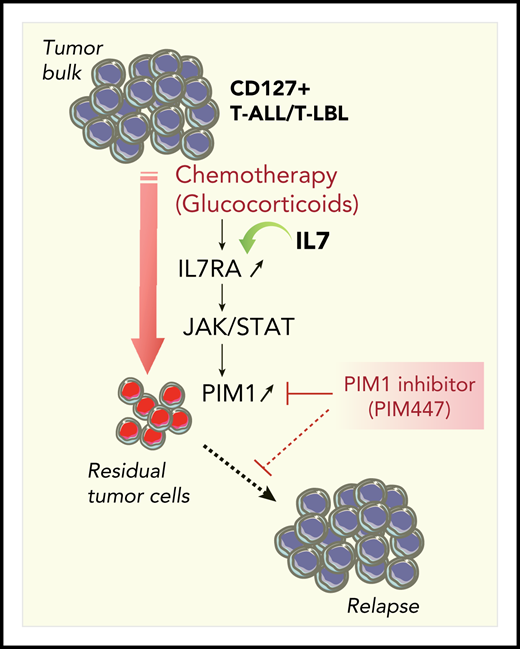
Synthetic lethality approach to killing residual tumor cells in CD127+ T-ALL/T-LBL. Glucocorticoids efficiently reduce the tumor burden and induce acute in vivo PIM1 expression in residual tumor cells through the cell-non-autonomous activation of the IL7RA/JAK-STAT signaling pathway. Combination therapy with the PIM inhibitor PIM447 allows for a synergistic antileukemic effect on residual tumor cells.
Synthetic lethality approach to killing residual tumor cells in CD127+ T-ALL/T-LBL. Glucocorticoids efficiently reduce the tumor burden and induce acute in vivo PIM1 expression in residual tumor cells through the cell-non-autonomous activation of the IL7RA/JAK-STAT signaling pathway. Combination therapy with the PIM inhibitor PIM447 allows for a synergistic antileukemic effect on residual tumor cells.
Initially described in the context of a murine T-cell lymphoma, PIM1 is an oncogenic serine/threonine kinase involved in several cellular processes, including cell-cycle progression, transcription, apoptosis, and drug resistance. PIM1 is transcriptionally activated by the canonical JAK-STAT pathway in response to extracellular ligand stimulation. Not surprisingly, activating mutations within the IL7RA-JAK-STAT pathway in human T-ALL/T-LBL were reported to cooperate in activating PIM1 in these cells. In rare cases, the PIM1 locus itself can be activated by chromosomal rearrangements. Indeed, De Smedt et al recently identified the TCRβ-PIM1 translocation in a case of pediatric T-LBL and demonstrated the preclinical benefit of using a second-generation PIM1 inhibitor combined with a glucocorticoid to treat such patients.2 De Smedt et al report an unexpected mechanism of PIM1 activation and propose that a broader subset of CD127+ T-ALL/T-LBL patients lacking known mutations of the IL7RA pathway could benefit from treatment with a PIM1 inhibitor.
By using patient-derived xenograft (PDX) models of CD127+ T-ALL/T-LBL, the authors uncovered a cell-non-autonomous mechanism that occurs in drug-resistant cells after treatment with glucocorticoids. They demonstrated that glucocorticoids upregulate the IL7RA/JAK-STAT/PIM1 axis. Two notable features emerge from this study. First, glucocorticoid-triggered upregulation of PIM1 occurs in the absence of detectable known mutations in genes of the interleukin-7 receptor (IL-7R) pathway, indicating that PIM1 activation may not be the result of the well-studied mutational burden within the tumor. Second, PIM1 upregulation was observed exclusively in PDX samples that respond to IL-7 stimulation and did not occur in nonresponders, which strongly suggests that PIM1 upregulation was the result of IL-7 signaling. In line with this hypothesis, an analysis of published chromatin immunoprecipitation sequencing (ChIP-seq) data shows that the glucocorticoid receptor NR3C1 occupies the IL7RA but not the PIM1 locus, thereby upregulating IL7RA expression.
Finally, PIM1 upregulation in residual leukemic blasts after chemotherapy is transient, further supporting the view that the upregulation is not due to constitutive mutations within the pathway in leukemic blasts. Remarkably, this cell-non-autonomous mechanism provides a window of therapeutic opportunity for killing drug-resistant cells with PIM1 inhibitors (see figure). Indeed, using an elegant in vivo preclinical approach, the authors demonstrated a synergistic antileukemic effect of combination therapy of glucocorticoids and the PIM inhibitor PIM447. A survey of clinical trials indicates that several PIM1 inhibitors are currently in clinical trials for myeloma, myelofibrosis and, more recently, for diffuse large B-cell lymphoma and solid tumors. The study by De Smedt suggests that PIM447 could be a new drug candidate for clinical treatment of CD127+ T-ALL/T-LBL patients.
In addition to its translational impact, the work by De Smedt et al brings into question whether there are additional mechanisms of drug resistance that should be considered. First, despite the clear synergistic antileukemic effect of PIM447 and glucocorticoids, as well as the substantial delays in leukemia onset in preclinical models, this 2-drug combination does not prevent long-term leukemia development in PDX models. This could be a result of the presence of a persisting minor subclone within residual leukemic cells that is insensitive to PIM447. Given the possibility of subclonal diversity3,4 and the emerging evidence of therapy-induced mutations in ALL,5 drawing the genetic and the molecular landscape of leukemic cells after glucocorticoids and/or PIM447 treatment in PDX models should bring additional understanding of the biology of short- and long-term relapse. Second, tumor cell interaction with its microenvironment could modify drug response, as illustrated by the recent report in Blood6 of mitochondria transfer from activated stromal cells to rescue ALL cells from drug-induced oxidative stress. The possibility of tumor cell-stromal cell interactions shaping drug response has led to the design of niche-based drug screening strategies in acute myeloid leukemia (AML)7 and T-ALL.8
Last but not least, pioneering work using xenotransplantation of primary AML blasts into immune-deficient mice revealed the heterogeneity of leukemic cells and the presence of leukemic stem cells (LSCs) that are able to maintain the neoplastic process. Those original xenografting experiments clearly demonstrated that administering human cytokines to murine hosts is essential for human LSC engraftment.9 In T-ALL, it was shown using PDX models that the interactions between tumor cells and their microenvironment are critical to promote leukemia progression.10 Taken in context, the work reported by De Smedt et al is another “cue” that dependency on cell-non-autonomous signals can be a tractable vulnerability in leukemias and lymphomas.
Conflict-of-interest disclosure: The authors declare no competing financial interests

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal