

Key Points
Functional genomic screens reveal SHMT2 as a drug target in BL, and SHMT2 inhibitors synergize with methotrexate to induce anti-BL effects.
SHMT2 inhibition disrupts the BL survival program by triggering autophagic degradation of TCF3 and a subsequent collapse of BCR signaling.

Abstract
Burkitt lymphoma (BL) is an aggressive lymphoma type that is currently treated by intensive chemoimmunotherapy. Despite the favorable clinical outcome for most patients with BL, chemotherapy-related toxicity and disease relapse remain major clinical challenges, emphasizing the need for innovative therapies. Using genome-scale CRISPR-Cas9 screens, we identified B-cell receptor (BCR) signaling, specific transcriptional regulators, and one-carbon metabolism as vulnerabilities in BL. We focused on serine hydroxymethyltransferase 2 (SHMT2), a key enzyme in one-carbon metabolism. Inhibition of SHMT2 by either knockdown or pharmacological compounds induced anti-BL effects in vitro and in vivo. Mechanistically, SHMT2 inhibition led to a significant reduction of intracellular glycine and formate levels, which inhibited the mTOR pathway and thereby triggered autophagic degradation of the oncogenic transcription factor TCF3. Consequently, this led to a collapse of tonic BCR signaling, which is controlled by TCF3 and is essential for BL cell survival. In terms of clinical translation, we also identified drugs such as methotrexate that synergized with SHMT inhibitors. Overall, our study has uncovered the dependency landscape in BL, identified and validated SHMT2 as a drug target, and revealed a mechanistic link between SHMT2 and the transcriptional master regulator TCF3, opening up new perspectives for innovative therapies.
Introduction
Burkitt lymphoma (BL) is an aggressive disease1 that often responds well to intensive immunochemotherapy.2-4 However, relapsed or refractory BL and therapy-related toxicity are remaining clinical challenges, emphasizing the need for innovative therapies that would be expected to emerge on the basis of an improved mechanistic understanding of this disease.3,5,6
MYC deregulation was identified as a key driver in BL, causing high proliferation rates and leading to genomic instability.7-10 The most frequently recurring somatic mutations cooperating with MYC were more recently found in the transcription factor TCF3 (also known as E2A) and its negative regulator ID3. These mutations induce a complex rewiring of the TCF3 transcriptional network toward promotion of BL cell survival.10-12 Mechanistically, BL cell dependence on aberrant TCF3 function has been attributed to the molecular link between TCF3 and tonic B-cell receptor (BCR) signaling,11 a key survival pathway in BL. Aberrant TCF3 function has been shown to promote pro-survival BCR signaling, particularly by activating the PI3-kinase pathway.12 Despite these recent advances in our understanding of BL pathophysiology, treatment of BL has for decades relied on conventional cytostatic drugs, among which methotrexate (MTX) has been found to be particularly effective.3,13,14 MTX is a competitive inhibitor of the dihydrofolate reductase; it acts as a functional folate antagonist, thereby interfering with one-carbon metabolism. From this perspective, it seems warranted to study one-carbon metabolism further with regard to its therapeutic implications.
In this study, we have systematically identified vulnerabilities in BL that may be therapeutically exploitable. We found that serine hydroxymethyltransferase 2 (SHMT2), a mitochondrial enzyme involved in one-carbon metabolism,15-17 is a potential drug target in BL. SHMT2 inhibition caused apoptosis in BL cells, mainly by autophagic degradation of TCF3, which in turn led to a collapse of cell survival promoting BCR signaling. Hence, SHMT2 function is essential to maintain a TCF3-driven transcriptional survival program, making it an attractive therapeutic target in BL.
Methods
Cell culture
Origin of cell lines and cell culture conditions are described in supplemental Methods, available on the Blood Web site.
Ethical approval
Ethical approval for using the human tissue and cryoconserved patient samples was obtained from the Ethics Committee of the University Medical Center Göttingen (#19-2-16) and from the Ethics Committee of the Goethe University of Frankfurt (SHN-9-2017). Xenograft experiments using BL60 cells were approved by the National Cancer Institute Animal Care and Use Committee (NCI-ACUC). Transplantation experiments using M2121 cells were approved by the local animal care committee and the relevant authorities (Landesamt für Natur, Umwelt und Verbraucherschutz Nordrhein-Westfalen, 84-02.04.2017.A131).
Method description
For genome wide screening, we used the human Brunello CRISPR knockout pooled library (gift from David Root and John Doench [Addgene #73178]). CRISPR-Cas9 screens were performed as previously described18 and detailed in the supplemental Methods. Mass spectrometry was performed as previously described.19 All experiments, statistics, and codes are described in the supplemental Methods.
Results
The dependency landscape of BL cells
To identify vulnerabilities and tumor suppressors in BL, we performed genome-wide CRISPR-Cas9 loss-of-function screens in 3 BL cell lines (BL60, Ramos, Raji) using the Brunello small guide RNA (sgRNA) library (supplemental Table 1). The data were analyzed in a comparative manner together with CRISPR-Cas9 screening data previously published for activated B-cell–like diffuse large B-cell lymphoma (ABC-DLBCL) cells (HBL1, TMD8, HLY1, and U2932 cells),18 another aggressive lymphoma type dependent on BCR signaling (Figure 1). To this end, we computed for each gene a CRISPR screen score (CSS; supplemental Methods).18 We performed 2 replicates per screen, and these measurements were highly reproducible (R2 = 0.73-0.88; supplemental Figure 1A). Similar to our previous screens in ABC-DLBCL,18 nontargeting control sgRNAs were not toxic, whereas sgRNAs targeting pan-essential genes were depleted in all BL cell lines (supplemental Figure 1B). BL and ABC-DLBCL cells exhibited lymphoma-type specific dependencies on a set of B-cell transcription factors (Figure 1). Whereas BL cells were dependent on TCF3, MEF2B, and SPI1 (Figure 1; supplemental Figure 1C), a dependency pattern that has been previously described for germinal-center–like (GCB) DLBCL,18 ABCs were dependent on SPIB and BATF. This differential dependency profile reflects the cell of origin of both lymphoma types, with BL originating from GC centroblasts and ABC-DLBCL likely originating from more differentiated plasmablasts.1 Other B-cell transcription factors such as IRF4 and PAX5 were shared dependencies between BL and ABC-DLBCL cells. From a signaling perspective, both lymphoma types relied on the expression of the BCR subunits CD79A and CD79B; however, their dependencies on downstream BCR signaling pathways were only partly overlapping. While both BL and ABC-DLBCL cells relied on the PI3K/mTOR pathway, BL cells were particularly dependent on the PI3K activators LYN and CD19, in contrast to ABC-DLBCL cells, which showed a strong dependency on SYK and PIK3AP1. In line with previous findings,11,12 BL cells did not depend on NF-κB and JAK1/STAT3 signaling (the main survival pathways in ABC-DLBCL), highlighting the fact that BL cells mainly rely on tonic BCR-driven PI3K survival signals. Our screens also revealed dependencies beyond BCR signaling, especially on metabolic pathways such as one-carbon metabolism. Several mitochondrial (SHMT2, MTHFD2, and MTHFD1L) and cytoplasmic (MTHFD1) effectors of this pathway were discovered as vulnerabilities in BL cells, nominating them as potential drug targets (Figure 1; supplemental Figure 1D,F). Interestingly, some of these vulnerabilities were also found in ABC-DLBCL but, according to public resource data sets, not in most screened cancer cell lines (supplemental Figure 1E,G). Given the known clinical efficacy of MTX, a folate antagonist, in BL treatment, we further focused on this pathway to investigate its therapeutic implications. Among the identified essential effectors of one-carbon metabolism, the mitochondrial enzyme SHMT2 qualified as a potential drug target because it is not a pan-essential gene according to several genome-scale CRISPR-Cas9 and RNAi-based loss-of-function screens (supplemental Figure 1G); moreover, compounds that inhibit its enzymatic function have already been developed.15,20,21
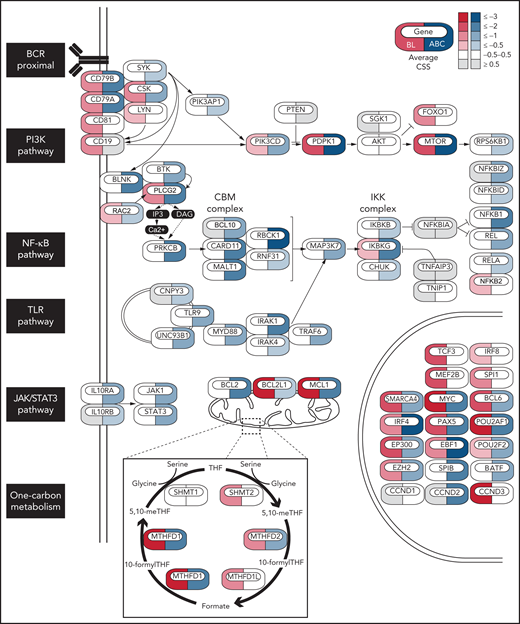
Essential genes in BL and ABC-DLBCL. Icons indicate essential genes from CRISPR screens colored by the average CSS in BL (red) or ABC-DLBCL (blue) cell lines. BL cell lines: BL60, RAJI, Ramos, ABC-DLBCL cell lines: HBL-1, TMD8, U2932, HLY1.
Essential genes in BL and ABC-DLBCL. Icons indicate essential genes from CRISPR screens colored by the average CSS in BL (red) or ABC-DLBCL (blue) cell lines. BL cell lines: BL60, RAJI, Ramos, ABC-DLBCL cell lines: HBL-1, TMD8, U2932, HLY1.
Validation of SHMT2 as a drug target in BL
Next, we validated SHMT2 as a potential therapeutic target. SHMT2 protein expression was found in 100% of the BL cases analyzed, 84% of the DLBCL cases, and 100% of the gray zone lymphoma cases (Figure 2A-B). We then validated our CRISPR-Cas9 screen results by performing a constitutive shRNA-based SHMT2 knockdown in BL cell lines. In competitive growth assays, SHMT2 knockdown BL cells, similar to MTHFD1 knockdown BL cells, were outcompeted by control cells, indicating a toxic effect of SHMT2 downregulation in all tested BL cell lines (Figure 2C; supplemental Figure 2A). These results were confirmed by a doxycycline-inducible SHMT2 (iSHMT2) knockdown (supplemental Figure 2B),22 although it reduced SHMT2 expression less efficiently, as well as by a targeted CRISPR-Cas9-based SHMT2 knockout in BL cell lines (supplemental Figure 2C-D). BrdU cell cycle analyses 5 days after SHMT2 knockdown revealed a significant G0/G1 arrest upon SHMT2 depletion in BL cell lines (P < .001 in Student t test; Figure 2D; supplemental Figure 2E). Moreover, a significant increase in poly (ADP-ribose) polymerase (PARP) and caspase 3 cleavage was observed (P < .05 in Student t test; Figure 2E, supplemental Figure 2F); hence, impaired cell growth of SHMT2 knockdown cells was caused by a combination of cell cycle arrest and increased apoptotic rate. To confirm the on-target activity of the shRNA used, we transduced the BL60 iSHMT2 knockdown cells that expressed green fluorescent protein (GFP) with a lentiviral vector encoding the fluorescent reporter blue fluorescent protein (BFP) and shRNA-resistant SHMT2 variants23 or BFP only as control (empty vector). By confocal imaging, we ensured regular localization of the overexpressed SHMT2 variants (supplemental Figure 2G). In these experiments, the shRNA-resistant SHMT2 construct significantly rescued the cell toxic effect of SHMT2 shRNAs, confirming their on-target activity (P < .001 Student t test; Figure 2F). In contrast, a catalytically inactive SHMT2 mutant (SHMT2 K280A)24 failed to rescue iSHMT2 knockdown cells, indicating that the enzymatic function of SHMT2 is required to maintain the fitness of BL cells (Figure 2F). To finally test whether SHMT2 also critically regulates BL tumor growth in vivo, we subcutaneously transplanted BL60 iSHMT2 knockdown cells into NSG (non-obese diabetic/severe combined immunodeficient/Il2rg−/−) mice and induced the knockdown of SHMT2 in established tumors with a tumor volume >140 mm3. In agreement with our in vitro experiments, the inducible SHMT2 knockdown reduced tumor growth by 70.1% compared with transplanted control cells (P = .0017 in two-way analysis of variance [ANOVA] test; Figure 2G; supplemental Figure 2H).
![SHMT2 enzymatic activity is essential for BL cell growth and viability. (A) Immunohistochemical staining of SHMT2 in lymphoma biopsies and lymph nodes from healthy individuals. Magnification is indicated with 20× and 63×, respectively. Scale bar for 100 µM is indicated. (B) Quantification of IHC with classification into negative, moderate, or strong expression. GC, germinal center; LN, lymph node; MZ, marginal zone. (C) Competitive growth assay of BL cell lines expressing a constitutive shRNA vector targeting SHMT2 (shSHMT2#1) or a nontargeting control shRNA (shCtrl) together with RFP in coculture with wild-type cells. Percentages of RFP-positive cells were measured at indicated times, and obtained values were normalized to the values of control shRNA expressing cells (mean ± standard error of the mean [SEM]; n = 3; P < .001 compared with nontargeting control shRNA transduced cells [two-way ANOVA]). (D) Cell cycle analyses of the BL cell lines BL60 and Ramos constitutively expressing an shRNA against SHMT2 (shSHMT2#1) or a nontargeting control shRNA (shCtrl) (mean ± SEM is shown; n = 4 to 5; ***P < .001 in Student t test). (E) Western blot analyses of cleaved PARP (cPARP), total PARP, and cleaved and total Caspase-3 in BL60 and Ramos cells constitutively expressing an shRNA against SHMT2 (shSHMT2#2) or a nontargeting control shRNA (shCtr). GAPDH or β-actin served as loading controls (n = 3-4). For quantification see supplemental Figure 2F. (F) Rescue experiment in which BL60 cells carrying a doxycycline-inducible shRNA against SHMT2 (shSHMT2#2) or a nontargeting control shRNA (shCtrl) with GFP as fluorescent reporter were transduced with a lentiviral vector coding for the fluorescent reporter BFP only (empty vector) or BFP and an shRNA-resistant version of SHMT2 (SHMT2res) or a catalytically inactive mutant thereof (SHMT2res-K280A). Percentages of GFP/BFP positive cells were determined over time by flow cytometry and normalized to the values of day 2 after doxycycline induction of shRNA expression. The diagram displays the values obtained at day 6 after induction of knockdown (mean ± SEM; n = 3). ***P < .001 in Student t test. Western blot validation for successful SHMT2 reconstitution is shown below. GAPDH served as loading control (n = 3; for empty vector/shCtrl vs shSHMT2 P < .001 in paired Student t test; for SHMT2res and SHMT2res-K280A, P = ns in paired Student t test; P = .51 and P = .37, respectively). (G) Tumor volume in NSG mice that were subcutaneously transplanted with BL60 carrying a doxycycline-inducible shRNA against SHMT2 (shSHMT2#2) or a nontargeting control shRNA (shCtrl). Tumor volumes were measured upon intraperitoneal doxycycline induction (once per day) of shRNAs (mean ± SEM; n = 5; P < .01 in two-way ANOVA; **P < .01 in Bonferroni posttest; ***P < .001 in Bonferroni posttest).](https://ash.silverchair-cdn.com/ash/content_public/journal/blood/139/4/10.1182_blood.2021012081/3/m_bloodbld2021012081f2.png?Expires=1769084841&Signature=bH~qHT6Zk-BRfAv5QkA2Faymg3XKjIHUYIhc6HFG3c97GvkwBuoUvvwXn-vUCOr7SxWhnPPkGfKXnIo-A7cn1buT~Sqp5ycj8R4PEZ3iek-JuwtBfXmitzntIY1UP43Fj5nUNb8WBoM1~yM5t0nCTX5bnoo9pDF-3-xqmIM8QREVTV5upqS1qXipgad0IPnQnFk4yfmU67fOsHo4lnix1zk14mB22PKimf3ythbCZySjQ0NVIn1i4Fk5hpezx9yn4iWY2Vl80vUP0wNvbNmCY~gPBItQzeBcLo7rUz5mYTkoHhC83nmdANqovWHPjUxLmBGpTmN58P4R2VH4mvdUPA__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
SHMT2 enzymatic activity is essential for BL cell growth and viability. (A) Immunohistochemical staining of SHMT2 in lymphoma biopsies and lymph nodes from healthy individuals. Magnification is indicated with 20× and 63×, respectively. Scale bar for 100 µM is indicated. (B) Quantification of IHC with classification into negative, moderate, or strong expression. GC, germinal center; LN, lymph node; MZ, marginal zone. (C) Competitive growth assay of BL cell lines expressing a constitutive shRNA vector targeting SHMT2 (shSHMT2#1) or a nontargeting control shRNA (shCtrl) together with RFP in coculture with wild-type cells. Percentages of RFP-positive cells were measured at indicated times, and obtained values were normalized to the values of control shRNA expressing cells (mean ± standard error of the mean [SEM]; n = 3; P < .001 compared with nontargeting control shRNA transduced cells [two-way ANOVA]). (D) Cell cycle analyses of the BL cell lines BL60 and Ramos constitutively expressing an shRNA against SHMT2 (shSHMT2#1) or a nontargeting control shRNA (shCtrl) (mean ± SEM is shown; n = 4 to 5; ***P < .001 in Student t test). (E) Western blot analyses of cleaved PARP (cPARP), total PARP, and cleaved and total Caspase-3 in BL60 and Ramos cells constitutively expressing an shRNA against SHMT2 (shSHMT2#2) or a nontargeting control shRNA (shCtr). GAPDH or β-actin served as loading controls (n = 3-4). For quantification see supplemental Figure 2F. (F) Rescue experiment in which BL60 cells carrying a doxycycline-inducible shRNA against SHMT2 (shSHMT2#2) or a nontargeting control shRNA (shCtrl) with GFP as fluorescent reporter were transduced with a lentiviral vector coding for the fluorescent reporter BFP only (empty vector) or BFP and an shRNA-resistant version of SHMT2 (SHMT2res) or a catalytically inactive mutant thereof (SHMT2res-K280A). Percentages of GFP/BFP positive cells were determined over time by flow cytometry and normalized to the values of day 2 after doxycycline induction of shRNA expression. The diagram displays the values obtained at day 6 after induction of knockdown (mean ± SEM; n = 3). ***P < .001 in Student t test. Western blot validation for successful SHMT2 reconstitution is shown below. GAPDH served as loading control (n = 3; for empty vector/shCtrl vs shSHMT2 P < .001 in paired Student t test; for SHMT2res and SHMT2res-K280A, P = ns in paired Student t test; P = .51 and P = .37, respectively). (G) Tumor volume in NSG mice that were subcutaneously transplanted with BL60 carrying a doxycycline-inducible shRNA against SHMT2 (shSHMT2#2) or a nontargeting control shRNA (shCtrl). Tumor volumes were measured upon intraperitoneal doxycycline induction (once per day) of shRNAs (mean ± SEM; n = 5; P < .01 in two-way ANOVA; **P < .01 in Bonferroni posttest; ***P < .001 in Bonferroni posttest).
SHMT2 enzymatic activity is essential for BL cell growth and viability. (A) Immunohistochemical staining of SHMT2 in lymphoma biopsies and lymph nodes from healthy individuals. Magnification is indicated with 20× and 63×, respectively. Scale bar for 100 µM is indicated. (B) Quantification of IHC with classification into negative, moderate, or strong expression. GC, germinal center; LN, lymph node; MZ, marginal zone. (C) Competitive growth assay of BL cell lines expressing a constitutive shRNA vector targeting SHMT2 (shSHMT2#1) or a nontargeting control shRNA (shCtrl) together with RFP in coculture with wild-type cells. Percentages of RFP-positive cells were measured at indicated times, and obtained values were normalized to the values of control shRNA expressing cells (mean ± standard error of the mean [SEM]; n = 3; P < .001 compared with nontargeting control shRNA transduced cells [two-way ANOVA]). (D) Cell cycle analyses of the BL cell lines BL60 and Ramos constitutively expressing an shRNA against SHMT2 (shSHMT2#1) or a nontargeting control shRNA (shCtrl) (mean ± SEM is shown; n = 4 to 5; ***P < .001 in Student t test). (E) Western blot analyses of cleaved PARP (cPARP), total PARP, and cleaved and total Caspase-3 in BL60 and Ramos cells constitutively expressing an shRNA against SHMT2 (shSHMT2#2) or a nontargeting control shRNA (shCtr). GAPDH or β-actin served as loading controls (n = 3-4). For quantification see supplemental Figure 2F. (F) Rescue experiment in which BL60 cells carrying a doxycycline-inducible shRNA against SHMT2 (shSHMT2#2) or a nontargeting control shRNA (shCtrl) with GFP as fluorescent reporter were transduced with a lentiviral vector coding for the fluorescent reporter BFP only (empty vector) or BFP and an shRNA-resistant version of SHMT2 (SHMT2res) or a catalytically inactive mutant thereof (SHMT2res-K280A). Percentages of GFP/BFP positive cells were determined over time by flow cytometry and normalized to the values of day 2 after doxycycline induction of shRNA expression. The diagram displays the values obtained at day 6 after induction of knockdown (mean ± SEM; n = 3). ***P < .001 in Student t test. Western blot validation for successful SHMT2 reconstitution is shown below. GAPDH served as loading control (n = 3; for empty vector/shCtrl vs shSHMT2 P < .001 in paired Student t test; for SHMT2res and SHMT2res-K280A, P = ns in paired Student t test; P = .51 and P = .37, respectively). (G) Tumor volume in NSG mice that were subcutaneously transplanted with BL60 carrying a doxycycline-inducible shRNA against SHMT2 (shSHMT2#2) or a nontargeting control shRNA (shCtrl). Tumor volumes were measured upon intraperitoneal doxycycline induction (once per day) of shRNAs (mean ± SEM; n = 5; P < .01 in two-way ANOVA; **P < .01 in Bonferroni posttest; ***P < .001 in Bonferroni posttest).
SHMT2 inhibition reduces glycine and formate levels in BL cells
Having validated SHMT2 as a potential therapeutic target in BL cells, we investigated how SHMT2 controls BL cell survival. SHMT2 has been described as a key effector in one-carbon metabolism because it catalyzes the transfer of a carbon unit from tetrahydrofolate to 5,10-methylenetetrahydrofolate and converts serine to glycine.15-17 Hence, SHMT2 activity is central within one-carbon metabolism and contributes to the generation of important products such as inosine monophosphate (IMP), glycine, glutathione (GSH), NADPH, and methionine. To characterize the metabolic changes induced by SHMT2 inhibition, we performed a mass spectrometry–based metabolome profiling in iSHMT2 BL60 cells. A total of 98 metabolites was reproducibly quantified upon SHMT2 knockdown, of which 15 showed significantly altered abundance (6 up and 9 downregulated; Figure 3A-B; supplemental Figure 3A-C). In line with previous studies, SHMT2 downregulation led to reduced levels of glycine (the direct product of the reaction catalyzed by SHMT2 (Figure 1; supplemental Figure 1D), IMP, as well as GSH (Figure 3A-B). Moreover, we found a significant downregulation of formate in an enzyme-based colorimetric assay upon pharmacological SHMT2 inhibition by the SHMT inhibitor SHIN1 (supplemental Figure 3D). To identify the metabolic products that are critical for the fitness of BL cells, we next performed supplementation analyses. The impairment in cellular growth of BL cells that were treated with SHIN1 could be reversed by supplementation with glycine and formate while serine, nucleosides, or leucovorin had no effect (Figure 3C; supplemental Figure 3E).
![The metabolic effect of SHMT2 inhibition. (A) Metabolites significantly regulated upon inducible SHMT2 knockdown (shSHMT2#2, d3 upon doxycyline induction) from LC/MS analysis (n = 6 each). The heatmap is color coded by row-wise z-scores of metabolite abundances. AICAR, 5-aminoimidazole-4-carboxamide-1-β-d-ribofuranoside; GSH, glutathione; IMP, inosine monophosphate. (B) Metabolite abundance for selected significantly regulated metabolites from LC/MS analysis (up to n = 6 each; median is shown). (C) Cell viability assay (MTT assay) for BL60 and SU-DHL4 after 48 hours of SHIN1 treatment and supplementation with indicated metabolites. (0.1 mM glycine and 0.3 mM serine correspond to regular medium concentration.) Nucleoside components are described in the supplemental Methods. Data were normalized to medium control (n = 3-4; mean ± SEM is shown). Glycine supplementation: [BL60] P = .0052, [SU-DHL4] ns (two-way ANOVA), Glycine/formate supplementation: [BL60] P = .0032, [SU-DHL4] ns (two-way ANOVA). Bonferroni posttest for glycine supplementation at 3.3 mM: [BL60] 1 µM SHIN1 P < .01; 5 and 10 µM SHIN1 P < .001; [SU-DHL4] 10 µM SHIN1 P < .001; for glycine/formate supplementation at 3.3/2 mM: [BL60] 1 µM SHIN1 P < .01; 5 to 10 µM SHIN1 P < .001; [SU-DHL4] 5 µM SHIN1 P < .05, 10 µM SHIN1 P < .001.](https://ash.silverchair-cdn.com/ash/content_public/journal/blood/139/4/10.1182_blood.2021012081/3/m_bloodbld2021012081f3.png?Expires=1769084841&Signature=ir1qhgbB3A39Sb3XByg57rF5OKfgk0xLT70mwxslg~Mt4XcWTT7QGsJoKDJDYPJBBSw2NzAgeTxO-erla3zEY4g7ReAdOuY5lyfNbxdiHKUBu1uwHhOSIiQMkIz0xEmT1p7oBQ9xeE-tyVqOI5vZ~Ht7zXkybB6x2XYn9pAgwkN-vndKUdZ5LujLt7wX-PDirMA7CejpdOS0s8E1rokPLPlz8rYQ5ekJeD07zwGpv2x4gDIROaBJogd5EryO6LXFhrsq0cRY9xdHcHPwMMtWxpUvZxlysGz06OUCC7KNQlzBIn0B0EzYtBVK4R7Lr7KAvUt6EFtvDuA6-8Jgp1uzaQ__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
The metabolic effect of SHMT2 inhibition. (A) Metabolites significantly regulated upon inducible SHMT2 knockdown (shSHMT2#2, d3 upon doxycyline induction) from LC/MS analysis (n = 6 each). The heatmap is color coded by row-wise z-scores of metabolite abundances. AICAR, 5-aminoimidazole-4-carboxamide-1-β-d-ribofuranoside; GSH, glutathione; IMP, inosine monophosphate. (B) Metabolite abundance for selected significantly regulated metabolites from LC/MS analysis (up to n = 6 each; median is shown). (C) Cell viability assay (MTT assay) for BL60 and SU-DHL4 after 48 hours of SHIN1 treatment and supplementation with indicated metabolites. (0.1 mM glycine and 0.3 mM serine correspond to regular medium concentration.) Nucleoside components are described in the supplemental Methods. Data were normalized to medium control (n = 3-4; mean ± SEM is shown). Glycine supplementation: [BL60] P = .0052, [SU-DHL4] ns (two-way ANOVA), Glycine/formate supplementation: [BL60] P = .0032, [SU-DHL4] ns (two-way ANOVA). Bonferroni posttest for glycine supplementation at 3.3 mM: [BL60] 1 µM SHIN1 P < .01; 5 and 10 µM SHIN1 P < .001; [SU-DHL4] 10 µM SHIN1 P < .001; for glycine/formate supplementation at 3.3/2 mM: [BL60] 1 µM SHIN1 P < .01; 5 to 10 µM SHIN1 P < .001; [SU-DHL4] 5 µM SHIN1 P < .05, 10 µM SHIN1 P < .001.
The metabolic effect of SHMT2 inhibition. (A) Metabolites significantly regulated upon inducible SHMT2 knockdown (shSHMT2#2, d3 upon doxycyline induction) from LC/MS analysis (n = 6 each). The heatmap is color coded by row-wise z-scores of metabolite abundances. AICAR, 5-aminoimidazole-4-carboxamide-1-β-d-ribofuranoside; GSH, glutathione; IMP, inosine monophosphate. (B) Metabolite abundance for selected significantly regulated metabolites from LC/MS analysis (up to n = 6 each; median is shown). (C) Cell viability assay (MTT assay) for BL60 and SU-DHL4 after 48 hours of SHIN1 treatment and supplementation with indicated metabolites. (0.1 mM glycine and 0.3 mM serine correspond to regular medium concentration.) Nucleoside components are described in the supplemental Methods. Data were normalized to medium control (n = 3-4; mean ± SEM is shown). Glycine supplementation: [BL60] P = .0052, [SU-DHL4] ns (two-way ANOVA), Glycine/formate supplementation: [BL60] P = .0032, [SU-DHL4] ns (two-way ANOVA). Bonferroni posttest for glycine supplementation at 3.3 mM: [BL60] 1 µM SHIN1 P < .01; 5 and 10 µM SHIN1 P < .001; [SU-DHL4] 10 µM SHIN1 P < .001; for glycine/formate supplementation at 3.3/2 mM: [BL60] 1 µM SHIN1 P < .01; 5 to 10 µM SHIN1 P < .001; [SU-DHL4] 5 µM SHIN1 P < .05, 10 µM SHIN1 P < .001.
SHMT2 inhibition disrupts a TCF3-controlled survival program in BL
To resolve how SHMT2 regulates BL cell survival and what the molecular consequences of the detected metabolic changes are, we quantified the changes in protein expression that occur upon inducible SHMT2 downregulation by SILAC (stable isotope labeling by amino acids in cell culture)-based mass spectrometry. To identify SHMT2-dependent proteins that are essential in BL cells, we compared the expression of each protein quantified by mass spectrometry with its respective CSS score (Figure 4A). This resulted in 42 essential genes whose protein expression was downregulated upon SHMT2 knockdown. A pathway enrichment analysis revealed that most of these proteins were involved in cell cycle regulation; among them, we found proteins involved in DNA replication (GINS1 complex members) and proteins regulating the mitotic checkpoint, such as SPC24 and SPC25, both being members of the NDC80 complex (supplemental Figure 4A). Interestingly, we found that TCF3 was significantly downregulated after SHMT2 knockdown (Figure 4A,F). TCF3 is a transcription factor and is known to be a key regulator of BL cell survival and proliferation because it induces cyclin D3-dependent proliferation and promotes oncogenic BCR-driven PI3K survival signals (Figure 4B).11 Mutations in TCF3 and/or its negative regulator ID3 are among the most recurrent mutations in BL, reflecting their functional relevance for BL pathophysiology (Figure 4B).11,25-28 TCF3 essentiality in BL was also observed in our CRISPR-Cas9 screen (Figure 1) and was furthermore confirmed in competitive-growth assays after inducible TCF3 knockdown in BL60 cells (Figure 4C-E). TCF3 knockdown led to a G0/G1 arrest (Figure 4E), which possibly explains the proteomic changes that were observed for the cell cycle–related effectors (Figure 4A; supplemental Figure 4A). TCF3 downregulation upon SHMT2 knockdown was confirmed by western blot analysis showing a reduction of TCF3 protein expression by 36.8% to 81.8% (mean, BL60: P = .02; Ramos: P = .002; BL70: P < .001 in Student t test; Figure 4F; supplemental Figure 4B). In addition, SHMT2 downregulation led to a strong increase in expression of the phosphatase SHP-1 (Figure 4F), a known negative regulator of BCR signaling that has been previously shown to be repressed by TCF3.11 Also in line with previous literature,11 TCF3 knockdown led to reduced phosphorylation of the activator phospho-sites Tyr525/526 of SYK and Ser473 of AKT, indicating that TCF3 controls BCR/PI3K signaling in BL cells (Figure 4G). To demonstrate that TCF3 is a cell survival–regulating effector downstream of SHMT2, we reinforced TCF3 expression in combination with GFP in BL cells after having retrovirally transduced them with an shRNA vector marked by red fluorescent protein (RFP) that targeted SHMT2. Monitoring of RFP/GFP double-positive cells by flow cytometry in a competitive coculture experiment with wild-type cells revealed that 45.3% were double-positive cells (ie, TCF3 reconstituted cells) after 5 days, whereas only 19.1% were double-positive cells when SHMT2 knockdown cells were transduced with an empty vector control construct (P < .05 in Student t test, Figure 4H; supplemental Figure 4C). Hence, downregulation of SHMT2 induces apoptosis at least in part by reducing TCF3 protein levels. Because TCF3 has been described as a key promotor of oncogenic BCR survival signaling (Figure 4B),11 we next performed a quantitative phosphoproteomic analysis to investigate whether BCR signaling is affected by SHMT2 knockdown and the observed subsequent downregulation of TCF3. To this end, we used SILAC-based mass spectrometry to compare tyrosine phosphorylation between control cells expressing an unspecific shRNA and cells inducibly expressing either SHMT2 or CD79A-specific shRNAs, the latter being used to read-out the consequences of disrupted BCR signaling.29 In line with TCF3 function being compromised upon SHMT2 knockdown, the phosphoproteomic analysis showed correlated phosphorylation changes upon interference with SHMT2 function and BCR signaling (R = 0.59; Figure 4I; supplemental Table 2). Several BCR-related signaling effectors, including SYK, PLCγ2, INPP5D, and the PI3K activators CD19 and PIK3AP1, showed concordantly reduced tyrosine phosphorylation under both conditions. These changes were not caused by altered protein expression levels in SHMT2 knockdown cells (Figure 4J; supplemental Figure 4D-E), thus confirming that BCR signaling is disrupted upon SHMT2 knockdown. Next, we investigated whether constitutively activated PI3K signaling can rescue the toxic effect of the SHMT2 knockdown in BL cells. Therefore, we expressed a constitutively active form of the catalytic PI3K subunit P110α (called MP110*)10,30 in BL60 cells. Previous studies have validated this construct and showed that it cooperates with MYC to induce BL in mice.10 In BL60 cells, constitutive PI3K signaling rescued the survival of SHMT2 knockdown cells, indicating that BCR-driven PI3K signaling is a relevant survival pathway downstream of the SHMT2-TCF3 axis (Figure 4K). In contrast to BL, SHMT2 inhibition neither affected TCF3 nor the phosphorylation of SYK in ABC-DLBCL cells (supplemental Figure 4F), suggesting that the identified SHMT2-TCF3-BCR axis is BL specific. Together, these findings indicate that in BL, TCF3 is a critical survival regulator downstream of SHMT2.
![TCF3 expression depends on SHMT2 function. (A) Integration of CRISPR/Cas9 drop out screen results (CSS, CRISPR screen score) in BL60 cells with total proteome changes (SILAC log fold change [LFC]) in inducible SHMT2 knockdown BL60 cells (shSHMT2#2) on day 5 after induction of shRNA expression. Selected gene names/proteins are indicated. (B) Schematic illustration of the functional role of TCF3 in BL adapted from Schmitz et al. Indicated mutation frequencies were obtained from Schmitz et al, 2012; Love et al, 2012; Grande et al, 2019; Bouska et al, 2017; López et al, 2019.11,25-28 (C) Representative western blot shows inducible TCF3 knockdown in BL60 cell line on day 5 after induction of shRNA expression. GAPDH served as loading control (n = 3; Student t test, shCtrl vs shTCF3.1, P = .003; shCtrl vs shTCF3.2, P = .017; shCtrl vs shTCF3.3, P = .005). (D) Coculture experiment with BL60 TCF3 doxycycline-inducible knockdown cells (3 different shRNA constructs) compared with a nontargeting control shRNA (shCtrl). Cells were cocultured with wild-type cells and analyzed by flow cytometry using the GFP reporter of the vector. GFP-positive cells were normalized to day 1 upon induction (n = 3, mean ± SEM; P < .001 in two-way ANOVA). (E) Cell cycle analysis of the BL60 cell line expressing an inducible shRNA against TCF3 (shTCF3#3) or a nontargeting control shRNA (shCtrl; n = 3, mean ± SEM; **P < .01 in Student t test, ***P < .001 in Student t test). (F) Representative western blots of lysates derived from BL60 and Ramos cells at day 8 upon SHMT2 knockdown (n = 3-4; TCF3 [BL60] P = .02 [Ramos] P = .002, SHP-1 [BL60] P = .02 [Ramos] ns). GAPDH served as loading control. (G) Western blots show SYK Tyr525/526 and AKT Ser473 phosphorylation levels upon inducible knockdown (shTCF3#3) in BL60 cell line 48 hours after induction of shRNA expression compared with a nontarget control shRNA (shCtrl). GAPDH served as loading control. Phosphorylation levels were normalized to SYK or AKT expression, respectively (n = 3; P < .04 in Student t test for pSYK). (H) TCF3 reconstitution using a retroviral vector with GFP fluorescent reporter in constitutive SHMT2 knockdown cells (shSHMT2#1) with an RFP fluorescent reporter in BL60 cell line. Double-positive (GFP/RFP) cells were followed up in flow cytometry (n = 4, mean ± SEM; *P < .05 in Student t test). (I) Phosphoproteomic SILAC-based LC-MS analysis in inducible SHMT2 and CD79a knockdown vs control BL60 cells upon doxycycline induction. SILAC ratios are average of 2 replicates. Differentially phosphorylated sites were defined as phosphosites quantified in at least 2 replicates exhibiting an absolute log2 SILAC ratio >0.5. The Pearson’s correlation coefficient is shown. P value is from a Pearson’s correlation test. (J) SILAC log2 fold change (LFC) for BCR effector protein expression and tyrosine phosphorylation of the indicated BCR effectors upon inducible SHMT2 knockdown vs control in BL60 cells on day 5 after induction of shRNA expression. Differentially phosphorylated phosphosites (Benjamini-Hochberg adjusted P value < 1e-3) are colored in red. SILAC ratios are average of 3 and 2 biological replicates for total proteome and phosphoproteome, respectively. (K) Cell viability (XTT assay) of BL60 cells expressing either a constitutively active variant of the catalytic PI3K subunit P110α (MP110*) or the empty vector (control) that were treated with SHIN1 at a final concentration of 2 µM or dimethyl sulfoxide (DMSO) at day 0. Background corrected absorbance is shown (n = 4, mean ± SEM; on day 4 control + DMSO vs control + SHIN1, P < .001; control + DMSO vs MP110* + DMSO, P < .01; MP110* + DMSO vs MP110*+ SHIN1 ns in Bonferroni posttest).](https://ash.silverchair-cdn.com/ash/content_public/journal/blood/139/4/10.1182_blood.2021012081/3/m_bloodbld2021012081f4a.png?Expires=1769084841&Signature=0HN9L-tSKpxqrGxqZkZfxYR3louMdQw8FpEv-6mhhTn4~bOExFJE4fqi6Zux3ddn73AxpVLYzpf9GpHiuxWcIBmmVqgSYvTDKEXZ1KtSnLFVMnvOCYG1Nh8sjKEshvSe~mFgk8ClUZsU0ChbSuDoyHBlf36NzBVbH0H~nH3vGVGqu7s50gi4yS7YZqzvOX1nkgIAtuwoHLZ6zLClaoyUScLWFPkNS1gAzVLqku08smWud5Mb~6Ij7gQoFJivCmHabfO-x7R42KxHmqApoC3KR7rcTC99eS1AfSRFGTsOsKwUesov5rkH2sX~LdIgO739hkxYKbcecBEE1D55PVWIIA__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
![TCF3 expression depends on SHMT2 function. (A) Integration of CRISPR/Cas9 drop out screen results (CSS, CRISPR screen score) in BL60 cells with total proteome changes (SILAC log fold change [LFC]) in inducible SHMT2 knockdown BL60 cells (shSHMT2#2) on day 5 after induction of shRNA expression. Selected gene names/proteins are indicated. (B) Schematic illustration of the functional role of TCF3 in BL adapted from Schmitz et al. Indicated mutation frequencies were obtained from Schmitz et al, 2012; Love et al, 2012; Grande et al, 2019; Bouska et al, 2017; López et al, 2019.11,25-28 (C) Representative western blot shows inducible TCF3 knockdown in BL60 cell line on day 5 after induction of shRNA expression. GAPDH served as loading control (n = 3; Student t test, shCtrl vs shTCF3.1, P = .003; shCtrl vs shTCF3.2, P = .017; shCtrl vs shTCF3.3, P = .005). (D) Coculture experiment with BL60 TCF3 doxycycline-inducible knockdown cells (3 different shRNA constructs) compared with a nontargeting control shRNA (shCtrl). Cells were cocultured with wild-type cells and analyzed by flow cytometry using the GFP reporter of the vector. GFP-positive cells were normalized to day 1 upon induction (n = 3, mean ± SEM; P < .001 in two-way ANOVA). (E) Cell cycle analysis of the BL60 cell line expressing an inducible shRNA against TCF3 (shTCF3#3) or a nontargeting control shRNA (shCtrl; n = 3, mean ± SEM; **P < .01 in Student t test, ***P < .001 in Student t test). (F) Representative western blots of lysates derived from BL60 and Ramos cells at day 8 upon SHMT2 knockdown (n = 3-4; TCF3 [BL60] P = .02 [Ramos] P = .002, SHP-1 [BL60] P = .02 [Ramos] ns). GAPDH served as loading control. (G) Western blots show SYK Tyr525/526 and AKT Ser473 phosphorylation levels upon inducible knockdown (shTCF3#3) in BL60 cell line 48 hours after induction of shRNA expression compared with a nontarget control shRNA (shCtrl). GAPDH served as loading control. Phosphorylation levels were normalized to SYK or AKT expression, respectively (n = 3; P < .04 in Student t test for pSYK). (H) TCF3 reconstitution using a retroviral vector with GFP fluorescent reporter in constitutive SHMT2 knockdown cells (shSHMT2#1) with an RFP fluorescent reporter in BL60 cell line. Double-positive (GFP/RFP) cells were followed up in flow cytometry (n = 4, mean ± SEM; *P < .05 in Student t test). (I) Phosphoproteomic SILAC-based LC-MS analysis in inducible SHMT2 and CD79a knockdown vs control BL60 cells upon doxycycline induction. SILAC ratios are average of 2 replicates. Differentially phosphorylated sites were defined as phosphosites quantified in at least 2 replicates exhibiting an absolute log2 SILAC ratio >0.5. The Pearson’s correlation coefficient is shown. P value is from a Pearson’s correlation test. (J) SILAC log2 fold change (LFC) for BCR effector protein expression and tyrosine phosphorylation of the indicated BCR effectors upon inducible SHMT2 knockdown vs control in BL60 cells on day 5 after induction of shRNA expression. Differentially phosphorylated phosphosites (Benjamini-Hochberg adjusted P value < 1e-3) are colored in red. SILAC ratios are average of 3 and 2 biological replicates for total proteome and phosphoproteome, respectively. (K) Cell viability (XTT assay) of BL60 cells expressing either a constitutively active variant of the catalytic PI3K subunit P110α (MP110*) or the empty vector (control) that were treated with SHIN1 at a final concentration of 2 µM or dimethyl sulfoxide (DMSO) at day 0. Background corrected absorbance is shown (n = 4, mean ± SEM; on day 4 control + DMSO vs control + SHIN1, P < .001; control + DMSO vs MP110* + DMSO, P < .01; MP110* + DMSO vs MP110*+ SHIN1 ns in Bonferroni posttest).](https://ash.silverchair-cdn.com/ash/content_public/journal/blood/139/4/10.1182_blood.2021012081/3/m_bloodbld2021012081f4b.png?Expires=1769084841&Signature=tksiQBYvPuDcickf~LAInSwlNTv7DzfRA0pdoUT-tVdQAlUBJhzU5mSULWh9Gh4pG~yV-tVKjd74j6Z2tW~bJFNOJPmRCMavk3cag1OOvzb6-Sr01Id4k4Fr9YixSWEEAQOBk-1J~8GG~KLA6ehJRvisv6sVZJlLiGLpaktMtvZgPYOUfcrRhDBIu51lHx6xX70ozkba5rcAaGY4rvdl-LEDdry4uez7ZjtRm8K5koZDs8RkTfW7TJT5-icgRRIlii8qTbg9iCs6BXwZ6Is3tmU0LH7geT48e3pRltg-Kz~MqQSAwEoz~EkePoEj39yl8fEghgybtn8EgY50qemebg__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
TCF3 expression depends on SHMT2 function. (A) Integration of CRISPR/Cas9 drop out screen results (CSS, CRISPR screen score) in BL60 cells with total proteome changes (SILAC log fold change [LFC]) in inducible SHMT2 knockdown BL60 cells (shSHMT2#2) on day 5 after induction of shRNA expression. Selected gene names/proteins are indicated. (B) Schematic illustration of the functional role of TCF3 in BL adapted from Schmitz et al. Indicated mutation frequencies were obtained from Schmitz et al, 2012; Love et al, 2012; Grande et al, 2019; Bouska et al, 2017; López et al, 2019.11,25-28 (C) Representative western blot shows inducible TCF3 knockdown in BL60 cell line on day 5 after induction of shRNA expression. GAPDH served as loading control (n = 3; Student t test, shCtrl vs shTCF3.1, P = .003; shCtrl vs shTCF3.2, P = .017; shCtrl vs shTCF3.3, P = .005). (D) Coculture experiment with BL60 TCF3 doxycycline-inducible knockdown cells (3 different shRNA constructs) compared with a nontargeting control shRNA (shCtrl). Cells were cocultured with wild-type cells and analyzed by flow cytometry using the GFP reporter of the vector. GFP-positive cells were normalized to day 1 upon induction (n = 3, mean ± SEM; P < .001 in two-way ANOVA). (E) Cell cycle analysis of the BL60 cell line expressing an inducible shRNA against TCF3 (shTCF3#3) or a nontargeting control shRNA (shCtrl; n = 3, mean ± SEM; **P < .01 in Student t test, ***P < .001 in Student t test). (F) Representative western blots of lysates derived from BL60 and Ramos cells at day 8 upon SHMT2 knockdown (n = 3-4; TCF3 [BL60] P = .02 [Ramos] P = .002, SHP-1 [BL60] P = .02 [Ramos] ns). GAPDH served as loading control. (G) Western blots show SYK Tyr525/526 and AKT Ser473 phosphorylation levels upon inducible knockdown (shTCF3#3) in BL60 cell line 48 hours after induction of shRNA expression compared with a nontarget control shRNA (shCtrl). GAPDH served as loading control. Phosphorylation levels were normalized to SYK or AKT expression, respectively (n = 3; P < .04 in Student t test for pSYK). (H) TCF3 reconstitution using a retroviral vector with GFP fluorescent reporter in constitutive SHMT2 knockdown cells (shSHMT2#1) with an RFP fluorescent reporter in BL60 cell line. Double-positive (GFP/RFP) cells were followed up in flow cytometry (n = 4, mean ± SEM; *P < .05 in Student t test). (I) Phosphoproteomic SILAC-based LC-MS analysis in inducible SHMT2 and CD79a knockdown vs control BL60 cells upon doxycycline induction. SILAC ratios are average of 2 replicates. Differentially phosphorylated sites were defined as phosphosites quantified in at least 2 replicates exhibiting an absolute log2 SILAC ratio >0.5. The Pearson’s correlation coefficient is shown. P value is from a Pearson’s correlation test. (J) SILAC log2 fold change (LFC) for BCR effector protein expression and tyrosine phosphorylation of the indicated BCR effectors upon inducible SHMT2 knockdown vs control in BL60 cells on day 5 after induction of shRNA expression. Differentially phosphorylated phosphosites (Benjamini-Hochberg adjusted P value < 1e-3) are colored in red. SILAC ratios are average of 3 and 2 biological replicates for total proteome and phosphoproteome, respectively. (K) Cell viability (XTT assay) of BL60 cells expressing either a constitutively active variant of the catalytic PI3K subunit P110α (MP110*) or the empty vector (control) that were treated with SHIN1 at a final concentration of 2 µM or dimethyl sulfoxide (DMSO) at day 0. Background corrected absorbance is shown (n = 4, mean ± SEM; on day 4 control + DMSO vs control + SHIN1, P < .001; control + DMSO vs MP110* + DMSO, P < .01; MP110* + DMSO vs MP110*+ SHIN1 ns in Bonferroni posttest).
TCF3 expression depends on SHMT2 function. (A) Integration of CRISPR/Cas9 drop out screen results (CSS, CRISPR screen score) in BL60 cells with total proteome changes (SILAC log fold change [LFC]) in inducible SHMT2 knockdown BL60 cells (shSHMT2#2) on day 5 after induction of shRNA expression. Selected gene names/proteins are indicated. (B) Schematic illustration of the functional role of TCF3 in BL adapted from Schmitz et al. Indicated mutation frequencies were obtained from Schmitz et al, 2012; Love et al, 2012; Grande et al, 2019; Bouska et al, 2017; López et al, 2019.11,25-28 (C) Representative western blot shows inducible TCF3 knockdown in BL60 cell line on day 5 after induction of shRNA expression. GAPDH served as loading control (n = 3; Student t test, shCtrl vs shTCF3.1, P = .003; shCtrl vs shTCF3.2, P = .017; shCtrl vs shTCF3.3, P = .005). (D) Coculture experiment with BL60 TCF3 doxycycline-inducible knockdown cells (3 different shRNA constructs) compared with a nontargeting control shRNA (shCtrl). Cells were cocultured with wild-type cells and analyzed by flow cytometry using the GFP reporter of the vector. GFP-positive cells were normalized to day 1 upon induction (n = 3, mean ± SEM; P < .001 in two-way ANOVA). (E) Cell cycle analysis of the BL60 cell line expressing an inducible shRNA against TCF3 (shTCF3#3) or a nontargeting control shRNA (shCtrl; n = 3, mean ± SEM; **P < .01 in Student t test, ***P < .001 in Student t test). (F) Representative western blots of lysates derived from BL60 and Ramos cells at day 8 upon SHMT2 knockdown (n = 3-4; TCF3 [BL60] P = .02 [Ramos] P = .002, SHP-1 [BL60] P = .02 [Ramos] ns). GAPDH served as loading control. (G) Western blots show SYK Tyr525/526 and AKT Ser473 phosphorylation levels upon inducible knockdown (shTCF3#3) in BL60 cell line 48 hours after induction of shRNA expression compared with a nontarget control shRNA (shCtrl). GAPDH served as loading control. Phosphorylation levels were normalized to SYK or AKT expression, respectively (n = 3; P < .04 in Student t test for pSYK). (H) TCF3 reconstitution using a retroviral vector with GFP fluorescent reporter in constitutive SHMT2 knockdown cells (shSHMT2#1) with an RFP fluorescent reporter in BL60 cell line. Double-positive (GFP/RFP) cells were followed up in flow cytometry (n = 4, mean ± SEM; *P < .05 in Student t test). (I) Phosphoproteomic SILAC-based LC-MS analysis in inducible SHMT2 and CD79a knockdown vs control BL60 cells upon doxycycline induction. SILAC ratios are average of 2 replicates. Differentially phosphorylated sites were defined as phosphosites quantified in at least 2 replicates exhibiting an absolute log2 SILAC ratio >0.5. The Pearson’s correlation coefficient is shown. P value is from a Pearson’s correlation test. (J) SILAC log2 fold change (LFC) for BCR effector protein expression and tyrosine phosphorylation of the indicated BCR effectors upon inducible SHMT2 knockdown vs control in BL60 cells on day 5 after induction of shRNA expression. Differentially phosphorylated phosphosites (Benjamini-Hochberg adjusted P value < 1e-3) are colored in red. SILAC ratios are average of 3 and 2 biological replicates for total proteome and phosphoproteome, respectively. (K) Cell viability (XTT assay) of BL60 cells expressing either a constitutively active variant of the catalytic PI3K subunit P110α (MP110*) or the empty vector (control) that were treated with SHIN1 at a final concentration of 2 µM or dimethyl sulfoxide (DMSO) at day 0. Background corrected absorbance is shown (n = 4, mean ± SEM; on day 4 control + DMSO vs control + SHIN1, P < .001; control + DMSO vs MP110* + DMSO, P < .01; MP110* + DMSO vs MP110*+ SHIN1 ns in Bonferroni posttest).
SHMT2 inhibition triggers an autophagic degradation of TCF3 through activation of ULK1
Next, we investigated the molecular link between SHMT2 and TCF3, addressing particularly the question of how TCF3 is downregulated upon SHMT2 knockdown. RNA-sequencing (RNA-seq) revealed that TCF3 mRNA was not significantly downregulated upon SHMT2 knockdown, implicating a posttranscriptional mechanism (supplemental Figure 5A). mRNA translation blockade by cycloheximide treatment did not rescue TCF3 protein expression in SHMT2 knockdown cells, ruling out that mRNA translation is affected in these cells (supplemental Figure 5B). Also, proteasomal degradation was ruled out as relevant for TCF3 downregulation because the proteasome inhibitor MG132 did not rescue TCF3 protein expression in SHMT2 knockdown cells (supplemental Figure 5C-D). Finally, we investigated whether autophagy as an alternative mechanism controlling protein expression in a posttranscriptional manner could be involved in TCF3 regulation. To this end, we found that SHMT2 knockdown led to an induction of autophagy measured by a significant increase of LC3-II, the lipidated form of LC3 that is used as a marker for autophagy (Figure 5A; supplemental Figure 5E).31,32 To confirm this finding, we employed 2 additional widely used autophagy assays. First, we used a tandem-fluorescent LC3 (tfLC3) autophagic flux reporter in which the amino terminus of LC3 is fused to 2 fluorescent protein tags: RFP and GFP (supplemental Figure 5F).33 Upon fusion of the autophagosome with the lysosome, GFP fluorescence is selectively quenched because of the decrease in pH. This leads to a reduction in the GFP/RFP fluorescence ratio (supplemental Figure 5F), which is used to quantify autophagy. Treatment of BL60 tfLC3 reporter cells with the SHMT inhibitor SHIN1 leads to a reduction in the GFP/RFP fluorescence ratio (ie, increased autophagic flux) comparable to the reduction observed in cells treated with the mTOR inhibitors Torin1 and AZD2014, which are known autophagy activators.32,34 Importantly, this effect was reversed by blocking the autophagic flux with Bafilomycin A1 (Figure 5B), clearly indicating that inhibition of SHMT2 induces autophagic flux in BL cells. Similar results were obtained for Ramos tfLC3 reporter cells (supplemental Figure 5G). In addition, we labeled the autophagy marker LC3 with an immunofluorescent staining to quantify the number of intracellular LC3 punctae as a read-out for autophagy. In accordance with the previous assays, it revealed that an increase of the autophagic flux occurs upon SHMT inhibition (Figure 5C-D). To follow up on this, we next investigated the mechanistic link between SHMT2 and autophagy. Autophagy is known to be regulated by the nutrient sensor mTOR, which under nutrient-rich conditions inhibits ULK kinases through phosphorylation of inhibitory serine residues. mTOR inhibition (eg, mediated by starvation) induces autophagy through dephosphorylation of these inhibitory residues within ULK, thereby inducing a complex cascade involving ULK activation and LC3 lipidation, finally leading to the formation of autophagosomes.35 In addition to nutrient depletion, reactive oxygen species have been described as potent activators of autophagy.36 However, we detected no ROS induction upon SHMT2 inhibition in BL cells (supplemental Figure 5H-I). Because we found that SHMT2 inhibition leads to reduced levels of intracellular glycine and formate and because supplementation of both rescued the toxic effect of the SHMT inhibitor, we tested whether the supplementation of glycine and formate has an impact on autophagy. Indeed, supplementation of glycine and formate abrogated LC3 lipidation (autophagy) upon SHMT inhibition (Figure 5E; supplemental Figure 5J). Similarly and in line with these results, ULK1 phosphorylation of the inhibitory serine at position 757 was rescued by glycine/formate supplementation in BL cells treated with an SHMT inhibitor, while we clearly observed reduced inhibitory ULK1 phosphorylation upon SHMT inhibition in the absence of glycine and formate supplementation (Figure 5F). These results indicate that autophagy was triggered by SHMT inhibition through the mTOR-ULK axis. Moreover, and in line with our previous results, glycine/formate supplementation also restored TCF3 levels (and subsequently led to SHP-1 repression) in SHMT inhibitor–treated BL cells (Figure 5G; supplemental Figure 5K). To further confirm the involvement of autophagy in TCF3 degradation, we knocked out ATG5, a central effector protein for autophagic vesicle formation, in BL60 cells and monitored TCF3 levels after treatment of the cells with the SHMT inhibitor SHIN1. ATG5 is essential for autophagy induction because it facilitates the lipidation of LC3, as we confirmed in BL60 cells (supplemental Figure 5L). TCF3 protein expression was largely restored in ATG5 knockout cells after SHIN1 treatment (Figure 5H), showing that TCF3 expression is controlled by autophagy. Finally, we employed a proximity ligation assay (PLA)18,37 to visualize the intracellular LC3-TCF3 interaction. Association of TCF3 and LC3 in cells treated with SHIN1 in regular medium was increased, but it was blunted in medium supplemented with glycine and formate, in which the autophagic response is inhibited (Figure 5I-J; supplemental Figure 5M-P). In summary, we found that SHMT inhibition triggered autophagy through reduction of glycine/formate and subsequent ULK1 activation, finally leading to autophagic degradation of TCF3.
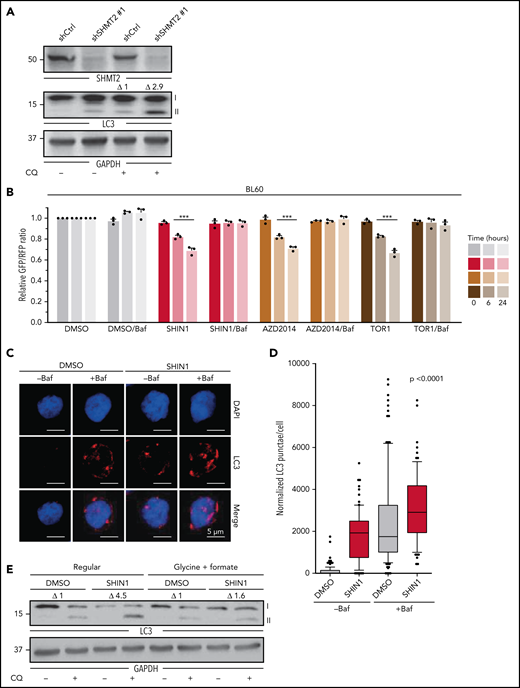
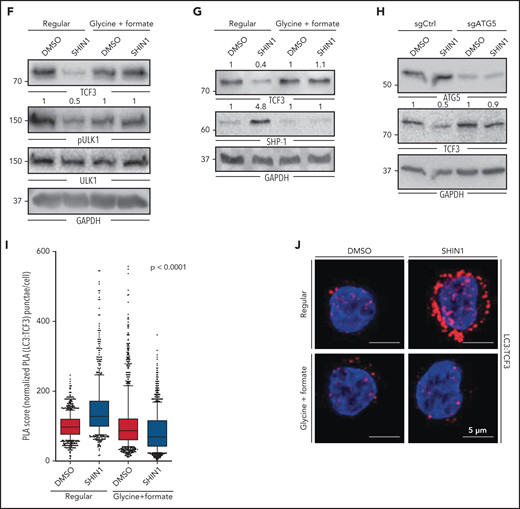
SHMT2 inhibition induces autophagic degradation of TCF3. (A) Representative western blot analysis showing LC3 levels in BL60 cells expressing either SHMT2-specific shRNA or nonspecific control shRNA that were treated with chloroquine at a concentration of 100 µM for 4 hours as indicated. GAPDH served as loading control. (n = 4; P = .01 in Student t test). (B) GFP/RFP ratio of BL60 cells transduced with tfLC3 reporter upon treatment with SHIN1 at a concentration of 2.5 µM, AZD2014 at a concentration of 200 nM, and Torin 1 (TOR1) at a concentration of 500 nM for indicated durations. A reduced ratio represents an increased level of autophagy. Bafilomycin A1 (Baf) treatment at 50 nM was used to inhibit autophagy (n = 3, mean ± SEM; P < .0001 in Tukey’s multiple comparison test for TOR1, AZD2014, and SHIN1 compared with DMSO after 6 hours and 24 hours; P = ns for rescue with Baf after 6 hours and 24 hours). (C) Representative confocal images of LC3 immunofluorescence staining. BL60 cells were treated for 24 hours with 2.5 µM SHIN1 or DMSO as well as for the last 6 hours with 50 nM of Bafilomycin or DMSO and stained for LC3 and DAPI. LC3 was stained with Alexa Fluor 647 (red) and nuclei were counterstained with DAPI (blue). Representative images display the overlay max intensity of the 41 z-stacks of the 647 channel and the average intensity of the z-stacks of the DAPI signal. (D) Quantification of LC3 punctae from microscopic images of BL60 cells treated as described in Figure 5C. Data were normalized to control and reported as percentage (n = 2, with n ≥ 31 single cells per condition). Box plots represent the median and 25th to 75th percentiles, whiskers display 10th to 90th percentiles, and outliers are displayed as dots. P < .0001 according to a Kruskal-Wallis test. (E) Representative western blot analysis showing LC3 levels in BL60 cells upon SHIN1 treatment at 2.5 µM for 48 hours in regular medium and upon supplementation with formate and glycine. Chloroquine treatment was applied at a concentration of 100 µM for 4 hours. GAPDH served as loading control (n = 3; P = .03 in Student t test for ΔLC3-II in SHIN1 vs DMSO in regular medium). (F-G) Representative western blots in BL60 cells showing ULK1 Ser757 phosphorylation, ULK1, TCF3, and SHP-1 after treatment of BL60 cells with 2.5 µM SHIN1 for 48 hours in regular medium and upon supplementation with glycine and formate (3.3 mM and 2 mM, respectively). pULK1 and ULK1 were probed on different membranes, but samples were derived from the same experiment and blots were processed in parallel. GAPDH served as loading control (n = 3; P = .02 in Student t test for pULK1 levels in SHIN1 vs DMSO in regular medium; P = .003 in Student t test for TCF3 levels in regular medium vs glycine/formate supplementation; and P = .008 in Student t test for SHP-1 levels in regular medium vs glycine/formate supplementation). (H) Representative western blots showing TCF3 levels in ATG5 KO compared with control-sgRNA in BL60 cell line upon induction of knockout with 250 ng/mL of doxycycline and 48 hours of SHIN1 treatment at a concentration of 2.5 µM vs DMSO control. GAPDH served as loading control (n = 3; P = .037 in paired Student t test for TCF3 levels in SHIN1 vs DMSO in samples with sgCtrl; P = ns in paired Student t test for TCF3 levels in SHIN1 vs DMSO in samples with sgATG5). (I) PLA score is shown for PLA of TCF3 and LC3 in SHIN1-treated BL60 cells at a concentration of 2.5 µM for 18 hours compared with DMSO control, in regular medium as well as upon supplementation of glycine/formate at a concentration of 3.3 mM and 2 mM, respectively (n = 4; n ≥ 105 single cells per condition). Box plots represent the median and 25th to 75th percentiles, whiskers display 10th to 90th percentiles, and outliers are displayed as dots (P < .001 in Kruskal-Wallis test). (J) Representative confocal images from PLA for TCF3 and LC3 in BL60 cell line, as described in Figure 5I. Merged images represent the composite images of the PLA of TCF3 and LC3 (red) and the DAPI signal (blue).
SHMT2 inhibition induces autophagic degradation of TCF3. (A) Representative western blot analysis showing LC3 levels in BL60 cells expressing either SHMT2-specific shRNA or nonspecific control shRNA that were treated with chloroquine at a concentration of 100 µM for 4 hours as indicated. GAPDH served as loading control. (n = 4; P = .01 in Student t test). (B) GFP/RFP ratio of BL60 cells transduced with tfLC3 reporter upon treatment with SHIN1 at a concentration of 2.5 µM, AZD2014 at a concentration of 200 nM, and Torin 1 (TOR1) at a concentration of 500 nM for indicated durations. A reduced ratio represents an increased level of autophagy. Bafilomycin A1 (Baf) treatment at 50 nM was used to inhibit autophagy (n = 3, mean ± SEM; P < .0001 in Tukey’s multiple comparison test for TOR1, AZD2014, and SHIN1 compared with DMSO after 6 hours and 24 hours; P = ns for rescue with Baf after 6 hours and 24 hours). (C) Representative confocal images of LC3 immunofluorescence staining. BL60 cells were treated for 24 hours with 2.5 µM SHIN1 or DMSO as well as for the last 6 hours with 50 nM of Bafilomycin or DMSO and stained for LC3 and DAPI. LC3 was stained with Alexa Fluor 647 (red) and nuclei were counterstained with DAPI (blue). Representative images display the overlay max intensity of the 41 z-stacks of the 647 channel and the average intensity of the z-stacks of the DAPI signal. (D) Quantification of LC3 punctae from microscopic images of BL60 cells treated as described in Figure 5C. Data were normalized to control and reported as percentage (n = 2, with n ≥ 31 single cells per condition). Box plots represent the median and 25th to 75th percentiles, whiskers display 10th to 90th percentiles, and outliers are displayed as dots. P < .0001 according to a Kruskal-Wallis test. (E) Representative western blot analysis showing LC3 levels in BL60 cells upon SHIN1 treatment at 2.5 µM for 48 hours in regular medium and upon supplementation with formate and glycine. Chloroquine treatment was applied at a concentration of 100 µM for 4 hours. GAPDH served as loading control (n = 3; P = .03 in Student t test for ΔLC3-II in SHIN1 vs DMSO in regular medium). (F-G) Representative western blots in BL60 cells showing ULK1 Ser757 phosphorylation, ULK1, TCF3, and SHP-1 after treatment of BL60 cells with 2.5 µM SHIN1 for 48 hours in regular medium and upon supplementation with glycine and formate (3.3 mM and 2 mM, respectively). pULK1 and ULK1 were probed on different membranes, but samples were derived from the same experiment and blots were processed in parallel. GAPDH served as loading control (n = 3; P = .02 in Student t test for pULK1 levels in SHIN1 vs DMSO in regular medium; P = .003 in Student t test for TCF3 levels in regular medium vs glycine/formate supplementation; and P = .008 in Student t test for SHP-1 levels in regular medium vs glycine/formate supplementation). (H) Representative western blots showing TCF3 levels in ATG5 KO compared with control-sgRNA in BL60 cell line upon induction of knockout with 250 ng/mL of doxycycline and 48 hours of SHIN1 treatment at a concentration of 2.5 µM vs DMSO control. GAPDH served as loading control (n = 3; P = .037 in paired Student t test for TCF3 levels in SHIN1 vs DMSO in samples with sgCtrl; P = ns in paired Student t test for TCF3 levels in SHIN1 vs DMSO in samples with sgATG5). (I) PLA score is shown for PLA of TCF3 and LC3 in SHIN1-treated BL60 cells at a concentration of 2.5 µM for 18 hours compared with DMSO control, in regular medium as well as upon supplementation of glycine/formate at a concentration of 3.3 mM and 2 mM, respectively (n = 4; n ≥ 105 single cells per condition). Box plots represent the median and 25th to 75th percentiles, whiskers display 10th to 90th percentiles, and outliers are displayed as dots (P < .001 in Kruskal-Wallis test). (J) Representative confocal images from PLA for TCF3 and LC3 in BL60 cell line, as described in Figure 5I. Merged images represent the composite images of the PLA of TCF3 and LC3 (red) and the DAPI signal (blue).
High-throughput screens identify drugs synergizing with SHMT2 inhibitors in BL
With this insight into the mechanisms by which SHMT2 controls BL cell survival, we next wished to investigate the translational potential of our findings. To this end, we first tested the efficacy of the small molecule inhibitor SHIN1 in 8 BL cell lines.15 Response to SHMT inhibition tracked with the TCF3/ID3 mutation status. Cell lines with validated oncogenic mutations in these genes showed higher sensitivity, which fits well with our mechanistic results (Figure 6A; supplemental Table 3).
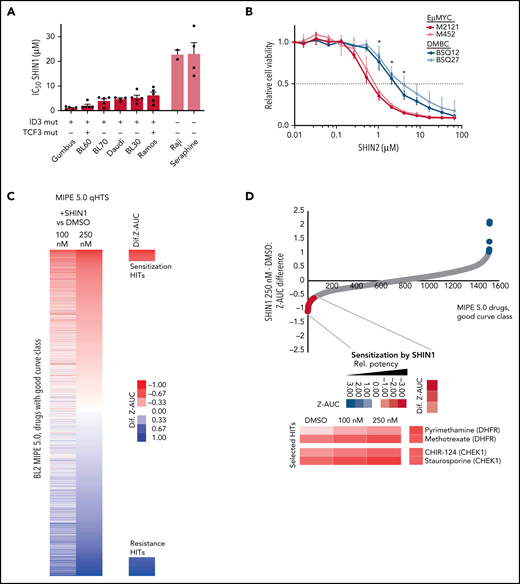
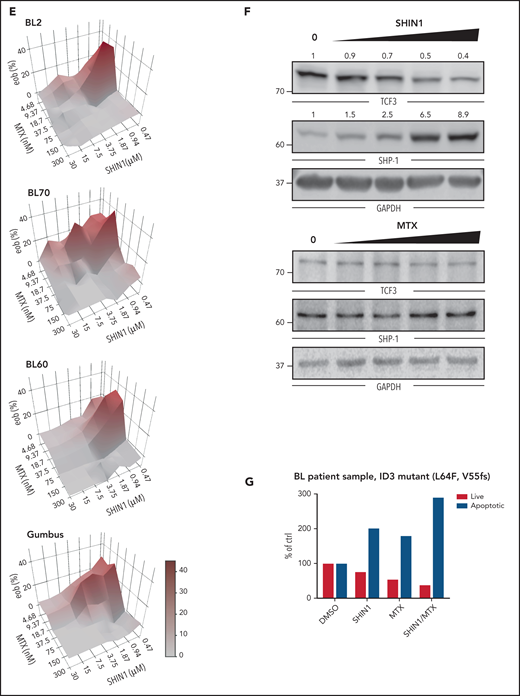
Identification of drugs acting synergistically with an SHMT inhibitor. (A) IC50 of SHIN1 in different BL cell lines measured by MTT assay. Cells were treated for 4 days (n = 4; mean ± SEM). The presence/absence of reported oncogenic ID3 and TCF3 mutations for the individual cell lines is indicated by + or −. (B) Stable cell lines isolated from murine Eµ:Myc (M2121, M452) and DMBC (BSQ12, BSQ27) tumors were treated with varying doses of SHIN2 for 72 hours, and viability was measured by CellTiterGlo assay (n = 3). Error bars represent standard deviation (SD). For each concentration, both Eµ:Myc lines were compared with both DMBC lines by Welch t test. *P < .05 for all comparisons at that concentration. (C) Results from “spiked-in,” quantitative high-throughput drug screening utilizing a mechanistically annotated library (MIPE 5.0), either synergizing or antagonizing SHIN1 treatment. (D) Selected hits from “spiked-in,” quantitative high-throughput drug screening that are synergistic with SHIN1 treatment. (E) Combined treatment of SHIN1 and methotrexate (MTX) shows synergy in MTT assay (n = 3; 1 representative analysis is shown). eob, excess over bliss. (F) Western blot of BL60 wild-type cells treated with either SHIN1 (concentrations ranging from 0.5-5 µM) or MTX (10-100 nM) for 3 days. GAPDH served as loading control. Cropped blots of representative experiments are shown with quantification for SHIN1-treated cells (n = 3-5; P = .002 in Student t test for altered TCF3 levels in 2.5 µM concentration and P = .003 for SHP-1 levels, respectively). (G) Annexin V staining in CD19+ cells derived from bone marrow of a 27-year-old patient with BL after in vitro treatment with SHIN1 (5 µM) and MTX (20 nM) for 96 hours. Cells were normalized to DMSO control. ID3 mutations (L64F, V55fs) were detected by exome sequencing (supplemental Figure 7A; supplemental Table 4). F, phenylalanine; fs, frame shift; L, leucin; V, valine.
Identification of drugs acting synergistically with an SHMT inhibitor. (A) IC50 of SHIN1 in different BL cell lines measured by MTT assay. Cells were treated for 4 days (n = 4; mean ± SEM). The presence/absence of reported oncogenic ID3 and TCF3 mutations for the individual cell lines is indicated by + or −. (B) Stable cell lines isolated from murine Eµ:Myc (M2121, M452) and DMBC (BSQ12, BSQ27) tumors were treated with varying doses of SHIN2 for 72 hours, and viability was measured by CellTiterGlo assay (n = 3). Error bars represent standard deviation (SD). For each concentration, both Eµ:Myc lines were compared with both DMBC lines by Welch t test. *P < .05 for all comparisons at that concentration. (C) Results from “spiked-in,” quantitative high-throughput drug screening utilizing a mechanistically annotated library (MIPE 5.0), either synergizing or antagonizing SHIN1 treatment. (D) Selected hits from “spiked-in,” quantitative high-throughput drug screening that are synergistic with SHIN1 treatment. (E) Combined treatment of SHIN1 and methotrexate (MTX) shows synergy in MTT assay (n = 3; 1 representative analysis is shown). eob, excess over bliss. (F) Western blot of BL60 wild-type cells treated with either SHIN1 (concentrations ranging from 0.5-5 µM) or MTX (10-100 nM) for 3 days. GAPDH served as loading control. Cropped blots of representative experiments are shown with quantification for SHIN1-treated cells (n = 3-5; P = .002 in Student t test for altered TCF3 levels in 2.5 µM concentration and P = .003 for SHP-1 levels, respectively). (G) Annexin V staining in CD19+ cells derived from bone marrow of a 27-year-old patient with BL after in vitro treatment with SHIN1 (5 µM) and MTX (20 nM) for 96 hours. Cells were normalized to DMSO control. ID3 mutations (L64F, V55fs) were detected by exome sequencing (supplemental Figure 7A; supplemental Table 4). F, phenylalanine; fs, frame shift; L, leucin; V, valine.
Next, we investigated the in vivo efficacy of SHMT inhibition. Because SHIN1 was shown to lack bioavailability,15 we used the recently developed bioavailable SHMT inhibitor SHIN2.20 To assess the in vivo activity of SHIN2 in a strictly Myc-driven neoplasm, we investigated the therapeutic efficacy of SHIN2 in the well-established Eμ:Myc model.38-40 For the purpose of our experiments, we initially isolated 2 distinct lymphoma cell lines from the Eμ:Myc model (M2121, M452) which both expressed BCR at their cell surface (supplemental Figure 6A). In a first approach, we compared the in vitro sensitivity of these cell lines to SHIN2 with the response of 2 distinct murine diffuse large B-cell lymphoma cell lines (BSQ12, BSQ27) that were isolated from Cd79bcond.p.Y195H/wt, Myd88cond.p.L252P/wt, Rosa26LSL.BCL2.IRES.GFP/wt, and Cd19Cre/wt (DMBC) mice, which are known to develop a DLBCL-like disease (Figure 6B; supplemental Figure 6A-B).41 The Eμ:Myc-derived lymphoma cells (M2121, M452) displayed a significantly lower GI50 than the DMBC-derived cells (BSQ12, BSQ27) when treated with SHIN2 (Figure 6B). We next injected 107 M2121 cells intraperitoneally into Rag1−/− recipients, allowed lymphomas to develop for 7 days, and then initiated treatment with SHIN2 (200 mg/kg administered via intraperitoneal injection (IP, twice daily) or vehicle control. While vehicle-treated animals succumbed to lymphoma within 10 days after transplantation (median overall survival 8.5 ± 0.8 days), SHIN2-treated mice derived a significant (P < .0001) survival benefit (median overall survival 11.0 ± 0.8 days) in this highly aggressive MYC-driven lymphoma model (supplemental Figure 6C). Although we found only a moderate survival benefit, we believe this to be relevant in the context of our highly aggressive BL transplant model. Also, the lack of ID3/TFC3 mutations in this model may have contributed to this aspect.
Thereafter, we identified drugs that synergize with SHIN1 because treatment with single drugs usually leads to rapidly evolving resistance to therapy in patients. To this end, we performed a “spiked-in,” quantitative, high-throughput drug screening using a mechanistically annotated library (MIPE 5.0) of 2480 approved and investigational drugs, targeting >860 distinct mechanisms-of-action and strongly focusing on oncogenic targets. Briefly, each drug was tested at 11 different concentrations (spanning a dose range from 45 μM to 0.7 nM) either alone or in the presence of sublethal amount of the SHMT inhibitor SHIN1 (100 and 250 nM) in BL cells.15 Interestingly, drugs that became more potent in the presence of SHIN1 included agents known to be efficacious in BL and thus relevant in the context of BL treatment, such as the DHFR inhibitors MTX and pyrimethamine, which were found to synergize with SHIN1 at nanomolar concentrations (Figure 6C-D; supplemental Figure 6D-F). Moreover, other drugs with clinical potential showed increased potency when combined with SHIN1; including, for example, inhibitors of CHEK1. The synergistic effect of combined MTX/SHIN1 was confirmed in MTT assays (Figure 6E; for MTX efficacy, compare supplemental Figure 6D,F). In line with our results from SHMT2 knockdown models, TCF3 levels were found to be depleted in a dose-dependent manner after SHIN1 treatment. Also, SHP-1 levels were increased after SHIN1 treatment as a downstream effect of reduced TCF3 activity (P < .01 in Student t test). Interestingly, however, we did not observe these effects after treatment with MTX, possibly providing a molecular rationale to explain their synergistic activity (Figure 6F). Finally, we were able to obtain primary BL cells from bone marrow of a 27-year-old patient with BL and cocultured these cells using irradiated YK6-CD40Lg-IL21 feeder cells.42 Importantly, in addition to genomic alterations in MYC, CCND3 and ARID1A, these BL cells harbored pathogenic ID3 mutations (p.55fs and p.L64F) that are known to enhance the oncogenic TCF3 pathway (supplemental Figure 7A; supplemental Table 4; compare Schmitz et al 2012).11 Upon treatment with SHIN1 alone and in combination with MTX, we detected an increased apoptotic rate in CD19-positive BL cells using Annexin-V staining (Figure 6G; supplemental Figure 7B), providing a future perspective for targeting the SHMT2 axis in BL.
Discussion
In this study, we elucidated the dependency landscape of BL by genome-scale CRISPR screens and identified SHMT2 as a therapeutic target.
Mechanistically, we found a molecular link between SHMT2 and TCF3 as the main reason for hypersensitivity of BL cells to SHMT2 inhibition (see also visual abstract). SHMT2 depletion or its pharmacological inhibition resulted in reduced intracellular glycine and formate levels leading to autophagosomal degradation of TCF3 protein. Hence, SHMT2 inhibition interfered with the oncogenic TCF3 transcriptional program, which is essential for BL cell survival because it critically controls important survival pathways, including BCR signaling.11,12,25-28 Our study provides an example of how metabolic pathways are functionally connected with oncogenic transcriptional programs that are currently considered untargetable. Hence, therapeutic exploitation of such an unexpected molecular link seems warranted, making SHMT2 a particularly interesting drug target in BL. Because of the aggressive nature of BL, however, SHMT2 inhibitors should be combined with other potent drugs to exert synergistic antitumoral activity. We found that SHMT inhibitors synergize with MTX, a dihydrofolate reductase inhibitor that also targets one-carbon metabolism. Interestingly, MTX, in contrast to SHMT inhibitors, did not affect TCF3 expression, pointing toward a more complex wiring of one-carbon metabolism and providing a molecular explanation for their synergistic activity. The newly identified SHMT2-autophagy-TCF3 axis seems to be highly specific for BL cells. In contrast to BL cells, SHMT2 inhibition in ABC-DLBCL cells had no effect on TCF3 expression. Also, the majority of ABC-DLBCL are not TCF3 dependent, and TCF3 mutations occur only in ∼1% of ABCs (in contrast to up to 30% of BL cells). Probably because of their similar cell of origin, the dependency profiles between GCB-DLBCL and BL are more similar to each other.18 Both BL and GCB-DLBCL depend on BCR expression, PI3K and mTOR signaling, and SHMT2 and TCF3. However, there are also specific differences. For example, BL cells depend on IRF4 whereas GCB-DLBCL cells depend more on the apoptosis regulator BCL2 (supplemental Figure 8). These findings clearly highlight the lymphoma-type specific wiring of oncogenic processes, including the interplay of metabolic and transcriptional programs. Taken together, we have identified SHMT2 as a target in BL and discovered a functional link between SHMT2 and TCF3 that may be amenable to therapeutic exploitation as SHMT2 inhibitors critically interfere with the oncogenic TCF3 transcription program.
Acknowledgments
The authors thank the Microarray and High Throughput Sequencing unit of the DKFZ Genomics Core Facility and the Whitehead Institute Metabolite Profiling Core Facility for their services. The authors also thank Adam Friedman, Feng Zhang, Johannes Zuber, and Didier Trono for providing reagents and Silvia Münch, Uwe Plessmann, Monika Raabe, Martine Pape, Samira Hitschler, and Jennifer Appelhans for their technical support. The authors thank the GMALL study group for providing patient samples.
This work was supported in part by research funding from the Else-Kroener-Fresenius Foundation (A.C.W.); grants from the German Research Foundation (Deutsche Forschungsgemeinschaft [DFG]) (SCHE 2120/1-1 [S.S.]), (Project-ID 259130777 – SFB 1177 [T.O.]) (RE 2246/13-1 [H.C.R.]), and (GO 2688/1-1 | SCHM 1633/11-1, SCHM 1633/9-1 [C.A.S.]); the German Cancer Aid (Deutsche Krebshilfe) (grant no. 70113536 [T.O.]), (grant nos. 1117240, 70113041, 70114055 [H.C.R.]), (grant no. 7011377629 [C.A.S.]); from the German-Israeli Foundation for Research and Development (I-65-412.20-2016) and the Deutsche Jose Carreras Leukämie Stiftung (R12/08) (H.C.R.); the German Ministry of Education and Research (BMBF) (e:Med 01ZX1303A [H.C.R.]) and (e:Med, project SeneSys, 031L0189A [C.A.S.]); and faculty scholars award from HHMI, SU2C, the MIT Center for Precision Cancer Medicine, the Ludwig Center at MIT, and the National Cancer Institute grant no. R35CA242379 [M.G.V.H.]. Y.P. is supported by National Cancer Institute K08 CA222684 and Hyundai Hope on Wheels grant.
Authorship
Contribution: T.O. conceived, designed and supervised the study; A.C.W., C.D., and A.Z. performed most of the experiments; K.S.L., S.A.R., M.C., J.Q.W., Y.P., D.J., C.S., S.S., F.A.T., H.B., P. Stauder, M.S., S.W., K.B., M.E., F.K., and G.K. acquired or contributed to data acquisition; A.C.W., C.D., A.Z., K.S.L., S.A.R., M.C., F.C., J.D.P., J.Q.W., Y.P., B.H., C.S., S.S., H.B., M.S., Z.A.C., C.A.L., S.W., K.B., B.C., M.E., G.K., T.H.D., R.S., H.C.R., T.Z., and T.O. analyzed and interpreted data; T.O., A.C.W., C.D., S.A.R., F.C., and M.C. wrote the manuscript and all other coauthors reviewed it; and F.S., X.X., B.C., C.B., P. Stroebel, K.S., H.K., D.J.H., R.S., H.U., H.S., C.A.S., J.D.R., M.G.V.H., C.T., L.M.S., and T.Z. provided administrative and technical support.
Conflict-of-interest disclosure: F.C. is co-founder of enGene Statistics GmbH. M.G.V.H. is a consultant and scientific advisory board member for Agios Pharmaceuticals, Aeglea Biotherapeutics, Auron Therapeutics, and iTeos Therapeutics. H.K is a founder, director, and stockholder of Farber Partners and inventor of patents held by Princeton University. H.C.R. received consulting and lecture fees from Abbvie, Astra-Zeneca, Vertex, and and Merck, received research funding from GILEAD, and is a co-founder of CDL Therapeutics GmbH. J.D.R. is an advisor and stockholder in Kadmon Pharmaceuticals, Colorado Research Partners, L.E.A.F. Pharmaceuticals, Bantam Pharmaceuticals, Barer Institute, and Rafael Pharmaceuticals, is a paid consultant for Pfizer, is a founder, director, and stockholder of Farber Partners, Serien Therapeutics, and Sofro Pharmaceuticals, is a founder and stockholder in Toran Therapeutics, is inventor of patents held by Princeton University, and is a director of the Princeton University–PKU Shenzhen collaboration. T.O. is a consultant for Merck KGaA and KronosBio and receives research funding for projects unrelated to this study from Merck KGaA and GILEAD. K.S. has consulted for KronosBio, AstraZeneca, Bristol Myers Squibb, and Auron Therapeutics, has stock options with Auron Therapeutics, and received grant funding from Novartis on topics unrelated to this manuscript. The remaining authors declare no competing financial interests.
Correspondence: Thomas Oellerich, Department of Medicine II, Hematology/Oncology, Goethe University, Theodor-Stern-Kai 7, 60590 Frankfurt, Germany; e-mail: thomas.oellerich@kgu.de.
CRISPR Cas9 screen analysis raw data (Figure 1; supplemental Figure 1A-F; Figure 4A; supplemental Figure 4A) are reported in supplemental Table 1. RNA-seq raw data (supplemental Figure 5A) have been deposited to the Sequence Read Archive (SRA)68 and are available under SRA accession number PRJNA623692. The mass spectrometry proteomics data (Figure 4A,I-J; supplemental Figure 4A,D-E) have been deposited to the ProteomeXchange Consortium via the PRIDE69 partner repository with the dataset identifier PXD018961. Results from the mass spectrometry phosphoproteomic data are also reported in supplemental Table 2. The metabolomic analysis (Figure 3A-B; supplemental Figure 3A-C) has been made available at Metabolights (https://www.ebi.ac.uk/metabolights/MTBLS1702). Code will be made available upon request. CRISPR screen analysis and RNAi screen analysis are publicly available on https://www.depmap.org/portal/.
The online version of this article contains a data supplement.
There is a Blood Commentary on this article in this issue.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal