

Key Points
SOX9 plays an oncogenic role in germinal center B-cell type, IGH-BCL2+ DLBCL, by promoting cell proliferation and inhibiting apoptosis.
SOX9 drives lymphomagenesis through upregulation of DHCR24, the key final enzyme in the cholesterol biosynthesis pathway.

Abstract
Although oncogenicity of the stem cell regulator SOX9 has been implicated in many solid tumors, its role in lymphomagenesis remains largely unknown. In this study, SOX9 was overexpressed preferentially in a subset of diffuse large B-cell lymphomas (DLBCLs) that harbor IGH-BCL2 translocations. SOX9 positivity in DLBCL correlated with an advanced stage of disease. Silencing of SOX9 decreased cell proliferation, induced G1/S arrest, and increased apoptosis of DLBCL cells, both in vitro and in vivo. Whole-transcriptome analysis and chromatin immunoprecipitation–sequencing assays identified DHCR24, a terminal enzyme in cholesterol biosynthesis, as a direct target of SOX9, which promotes cholesterol synthesis by increasing DHCR24 expression. Enforced expression of DHCR24 was capable of rescuing the phenotypes associated with SOX9 knockdown in DLBCL cells. In models of DLBCL cell line xenografts, SOX9 knockdown resulted in a lower DHCR24 level, reduced cholesterol content, and decreased tumor load. Pharmacological inhibition of cholesterol synthesis also inhibited DLBCL xenograft tumorigenesis, the reduction of which is more pronounced in DLBCL cell lines with higher SOX9 expression, suggesting that it may be addicted to cholesterol. In summary, our study demonstrated that SOX9 can drive lymphomagenesis through DHCR24 and the cholesterol biosynthesis pathway. This SOX9-DHCR24-cholesterol biosynthesis axis may serve as a novel treatment target for DLBCLs.
Introduction
Sex-determining region Y box 9 protein (SOX9) is a member of the SOX family of transcription factors, which are developmental regulators that possess high-mobility group (HMG) box DNA binding and transactivation domains.1 SOX9 plays a critical role in cell fate determination,2 cell differentiation,3 maintenance of the stem cell pool,4 and tissue homeostasis5 in a variety of developing and adult tissues, the most studied of which are cartilage6,7 and testis.8,9 The SOX proteins bind to ATTGTT consensus or related sequence motifs through their high-mobility group domain, and SOX9 induces significant bending at the consensus-binding motif (A/TA/TCAAA/TG) by forming an L-shaped complex in the minor groove of DNA.1,10 Apart from its normal role as a cell fate and stem-cell regulator, SOX9 has been implicated in human diseases, including cancer. Most studies have supported an oncogenic role for SOX9, but a tumor suppressor function for SOX9 has also been suggested.11 SOX9 is overexpressed in many solid tumors, including hepatocellular carcinoma and breast, bladder, gastric, prostate, pancreatic, and colorectal cancers.12,13 Clinically, SOX9 overexpression in these cancers appear to be associated with worse prognosis. SOX9 mutations, most of which are missense, are infrequent and are detected in ∼1% of tumors overall (https://cancer.sanger.ac.uk/cosmic/gene/analysis?ln=SOX9). However, SOX9 mutations are enriched in colorectal cancers and are present in ∼11% of these tumors.14 More than 80% of these mutations are frameshift or nonsense mutations that are associated with mutated KRAS and wild-type TP53 status.14
There is also emerging evidence that SOX9 can regulate diverse cellular processes related to tumorigenesis and tumor progression, including cell proliferation, apoptosis, migration, invasion, chemoresistance, cancer stem cell maintenance, autophagy, angiogenesis, immune escape, and metastasis.12,15 These protumoral activities of SOX9 are effected through regulation of downstream genes and multiple signaling pathways. In triple-negative breast cancer, SOX9 is essential for cancer cell growth and metastasis and has been found to suppress the expression of apoptosis-related genes and increase expression of genes involved in epithelial-mesenchymal transition by binding to the respective promoters.16 In gastric cancer, glioblastoma, and pancreatic cancer, SOX9 promotes cancer cell proliferation and survival, as well as tumor growth, by upregulation of BMI1 expression, which consequently inhibits the level of the tumor suppressor p21.17 It has also been shown that SOX9 drives prostate cancer by activating WNT/β-catenin signaling through positively regulating genes within the WNT pathway, including those encoding WNT receptors and the β-catenin effector TCF4.18 SOX9 is also a stem-cell marker in hepatocellular carcinoma, regulating the Wnt/β-catenin pathway and its downstream target osteopontin.19 SOX9 mediates chemoresistance in non–small-cell lung cancer cells by transcriptionally activating aldehyde dehydrogenase A1 and promoting their stemlike properties.20 As a pivotal molecule with multifaceted functions, SOX9 may serve as a potential therapeutic target in many cancers.
Although the role of SOX9 in the development and progression of solid tumors is well established, little is known about SOX9 in lymphomagenesis. In this study, we defined a novel role of SOX9 in modulating DHCR24-mediated cholesterol synthesis, and targeting of this axis inhibited lymphomagenesis.
Methods
Ethics approval
This study was conducted in accordance with the Declaration of Helsinki regulations for the protocols approved by the Institutional Review Board of Weill Cornell Medicine (approval 0107004999). Written consent for the use of the human tissue samples for research was obtained from patients or their guardians. All experiments were performed under an Institutional Animal Care and Use Committee–approved protocol of Shanghai Jiaotong University School of Medicine, and guidelines for the proper use of animals in research and animal welfare were followed.
Patient tissue samples
All tissue samples were diagnosed according to World Health Organization classification criteria by attending hematopathologists at New York Presbyterian/Weill Cornell Medical Center, and clinical information was obtained from electronic clinical records. DLBCLs were subclassified into the germinal center B-cell (GCB) and non-GCB subtypes based on the Hans’ immunohistochemistry algorithm.21
Drug compounds and antibodies
Simvastatin, triparanol, and JQ1 were purchased from Med Chem Express (Monmouth Junction, NJ), Santa Cruz Biotechnology (Dallas, TX), and Selleckchem (Houston, TX), respectively. All compounds, except for those used in in vivo experiments, were reconstituted in dimethyl sulfoxide, stored in 100-mM stock concentrations in −80°C and used at the indicated doses, as suggested by the vendor. A CellTrace Violet Cell Proliferation kit for flow cytometry was obtained from ThermoFisher Scientific (Waltham, MA). Flow cytometry antibodies, BV421 mouse anti-Ki67, and Alexa Fluor 647 rabbit anti-active caspase 3 were purchased from BD Pharmingen (San Jose, CA); immunoblot antibodies, SOX9, and DHCR24 from Abcam (London, United Kingdom); Bax, Mcl-1, Bcl2, Bcl-XL, and Bad from Cell Signaling Technology (Danvers, MA); and tubulin antibody from Proteintech (Rosemont, IL).
Descriptions of cell lines, culturing, and assays; animal studies; and statistical analyses are provided in the supplemental Methods (available on the Blood Web site). Primer sequences are all available in supplemental Table 2.
Transcriptome RNA sequencing and data processing
Total RNA was extracted from SOX9 short hairpin RNA#1 (shRNA#1), SOX9 shRNA#2, and scrambled control lentiviral–transduced Karpas-422 cells. Messenger RNA (mRNA) was isolated from total RNA by using the Next Poly(A) mRNA Magnetic Isolation Module (New England BioLabs, Ipswich, MA). The RNA library was prepared by using the KAPA Stranded RNA-SEQ Library Prep Kit (Illumina, San Diego, CA), according to an instruction manual before it was subjected to sequencing on an Illumina NovaSeq 6000. Raw sequencing data were quality-control qualified, and trimmed data were aligned with reference genome/transcriptome (GRCh37). Differentially expressed genes or transcripts were subjected to either pathway or Gene Ontology analysis. A Venn graph and heat map were generated in R language.
Results
SOX9 is highly expressed in GCB subtypes of DLBCL accompanied by IGH-BCL2 mutations
To determine the expression of SOX9 protein in DLBCL, we performed immunohistochemistry by using microarrays on tissue from a cohort of patients with DLBCL (n = 114, see supplemental Table 1 for patients’ clinical information). The SOX9+ DLBCL cases showed variable staining, whereas SOX9 was absent in the normal GCB cells (Figure 1A). Overall, 11 of the 114 DLBCL samples (∼10%) were positive for SOX9, among which 90.9% (10 of 11) were of the germinal center B-cell–like (GCB) subtype, according to Hans’ classifier (P = .008). In 93 cases, cytogenetic data were available. Although only 2 of 73 (2.7%) of IGH-BCL2− DLBCL samples expressed SOX9, 7 of 20 (35%) of IGH-BCL2+ samples were positive for SOX9 (P < .001; see supplemental Table 3). Thus, most of the SOX9+ DLBCLs likely belong to the recently proposed EZB genetic subtype,22 given that IGH-BCL2 fusion is highly specific for this subtype of DLBCL. Mutation data for 2 of these 7 IGH-BCL2+, SOX9+ DLBCLs were also available. One of the 2 cases harbored an EZH2 p.A692V pathogenic mutation, which provided additional supporting evidence for the EZB genetic subtype. The other was associated with a TP53 p.T253P hotspot mutation. TP53 mutations, though most frequently seen in the A53 genetic subtype, are not specific among the genetic subtypes and can be present in ∼38% of the DLBCL of the EZB subtype. We attempted to look for SOX9 mutations in SOX9+DLBCL by Sanger performing Sanger sequencing on 7 cases. We did not identify SOX9 mutations but the sequencing was limited by the poor quality of the DNA extracted from archived paraffin blocks (see supplemental Data). We also tried to correlate SOX9 expression with different clinicopathologic parameters in DLBCL and found that SOX9 positivity in DLBCL was significantly associated with advanced stage disease (supplemental Table 2; P < .011).
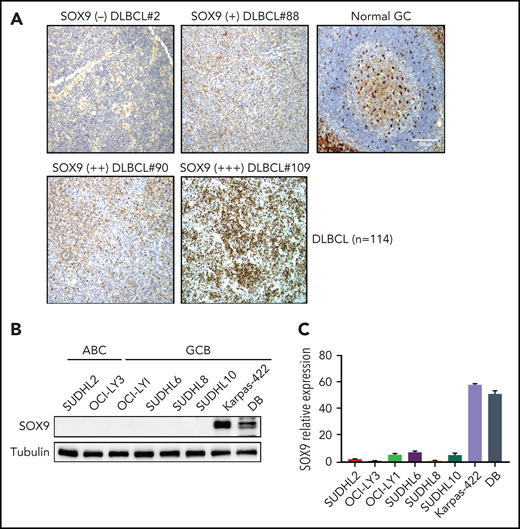
IGH-BCL2+DLBCLs manifest high levels of endogenous SOX9. (A) immunohistochemical staining of SOX9 protein in tissue samples from a DLBCL cohort (n = 114). Original magnification ×200. Bar represents 100 μm. SOX9 immunostainings are denoted as (+), (++), and (+++) to indicate the expression levels of SOX9. Negative staining is denoted as (−). Normal tonsil tissue was used as the control. SOX9 highlighted scattered follicular dendritic cells and showed only background staining in the germinal centers. Results of immunoblot (B) and qRT-PCR (C) analysis of endogenous SOX9 protein or SOX9 mRNA levels in DLBCL cell lines. (B-C) Data represent the mean ± standard deviation (SD) from technical triplicates.
IGH-BCL2+DLBCLs manifest high levels of endogenous SOX9. (A) immunohistochemical staining of SOX9 protein in tissue samples from a DLBCL cohort (n = 114). Original magnification ×200. Bar represents 100 μm. SOX9 immunostainings are denoted as (+), (++), and (+++) to indicate the expression levels of SOX9. Negative staining is denoted as (−). Normal tonsil tissue was used as the control. SOX9 highlighted scattered follicular dendritic cells and showed only background staining in the germinal centers. Results of immunoblot (B) and qRT-PCR (C) analysis of endogenous SOX9 protein or SOX9 mRNA levels in DLBCL cell lines. (B-C) Data represent the mean ± standard deviation (SD) from technical triplicates.
To investigate SOX9 expression in DLBCL cell lines, we performed immunoblot analysis and quantitative reverse transcription polymerase chain reaction (qRT-PCR) analysis in 8 DLBCL cell lines, including Karpas-422 and DB, which are of the GCB subtype and harbor IGH-BCL2 translocation (Figure 1B-C). In concordance with the protein level, the SOX9 mRNA level was also high in Karpas-422 and DB cells, but relatively low in the other cell lines (Figure 1C). In light of the tumorigenic role of SOX9 in many solid tumors, these studies suggest that SOX9 participates in the pathogenesis of DLBCLs, particularly those associated with IGH-BCL2 translocation.
Inhibition of SOX9 in IGH-BCL2+ DLBCL cells impairs cell survival and proliferation
To further explore the biological effects of SOX9 on DLBCL, lentivirus encoding shRNAs against SOX9 (shSOX9#1, shSOX9#2) or overexpressing SOX09 (pCDH-SOX9) were transduced to Karpas-422/DB cells, or OCI-LY1/SUDHL-6 cells, respectively. SOX9 protein was significantly reduced in shSOX9#1- or shSOX9#2-transduced Karpas-422 cells (2.5- or 3.3-fold reduction relative to the scrambled control; Figure 2A; supplemental Figure 6A) or DB cells (2.2- or 2-fold reduction relative to scrambled control, Figure 2A; supplemental Figure 6B), but was significantly increased in OCI-LY1 or SUDHL6 cells carrying pCDH-SOX9 compared with the vector control (supplemental Figures 1A and 6C-D). Both shSOX9-transduced cell lines showed a reduction in SOX9 mRNA, consistent with the decrease in SOX9 protein levels (supplemental Figure 2A). We then performed the CellTrace carboxyfluorescein diacetate succinimidyl ester cell proliferation assay to determine whether inhibition or overexpression of SOX9 affects DLBCL cell proliferation. shSOX9#1- or shSOX9#2-transduced Karpas-422 or DB cells, or pCDH-SOX9–transduced OCI-LY1 or SUDHL6 cells were seeded, harvested, and labeled with CellTrace violet solution per the manufacturer’s instruction manual. Cells were then subcultured at 37°C for another 3 days, followed by flow cytometry analysis of cell proliferation at day 0 (proliferation control) and day 3. As demonstrated in Figure 2B, the knockdown of SOX9 significantly inhibited cell proliferation when compared with its scrambled control counterpart. In contrast, overexpression of SOX9 had no major impact on DLBCL cell proliferation (supplemental Figure 1B). To explore whether inhibition or overexpression of SOX9 affected DLBCL cell growth, shSOX9#1- or shSOX9#2-transduced Karpas-422 or DB cells or pCDH-SOX9-–transduced OCI-LY1 or SUDHL6 cells were seeded in 96-well plates, and the number of cells was counted manually on a hemocytometer at the indicated time points (Figure 2C). Knockdown of SOX9 significantly inhibited an increase in the number of cells over time when compared with its scrambled control counterpart (Figure 2C). In contrast, overexpression of SOX9 had no major impact on the number of DLBCL cells over time (supplemental Figure 1C). A cell-cycle analysis was performed by exposing shSOX9#1, shSOX9#2, or scrambled control-transduced Karpas-422 or DB cells to BrdU for 4 hours. Inhibition of SOX9 promoted G1/S phase arrest, resulting in a significant increase in the percentage of cells in G1 phase from 51% (scrambled control) to 62.7% (shSOX9#1) or 61% (shSOX9#2) in Karpas-422 cells, or from 47.9% (scrambled control) to 65% (shSOX9#1) or 64.2% (shSOX9#2) in DB cells. Meanwhile, the percentage of cells in S phase decreased from 39.5% (scrambled control) to 28.4% (shSOX9#1) or 28.5% (shSOX9#2) in Karpas-422 cells, or from 40.4% (scrambled control) to 26.7% (shSOX9#1) or 26.5% (shSOX9#2) in DB cells (Figure 2D). However, overexpression of SOX9 had no significant impact on OCI-LY1 (supplemental Figure 1D, top left and right) and SUDH6 cell cycle distributions (supplemental Figure 1D, bottom left and right). To investigate whether inhibition of SOX9 modulates the survival of DLBCL cells, an annexin V/7ADD apoptosis assay was performed, and caspase 3 activity was determined in the aforementioned cells. Inhibition of SOX9 significantly increased the percentage of V+/7AAD+ cells (supplemental Figure 2B) and induced caspase 3 activity in SOX9-silenced Karpas-422 (supplemental Figure 2C) and DB (supplemental Figure 2D) cells compared with the scrambled control. In contrast, overexpression of SOX9 showed no significant effect on the percentage of either annexin V+/7AAD+ (supplemental Figure 1E) or caspase 3 activity (supplemental Figure 1G-H).
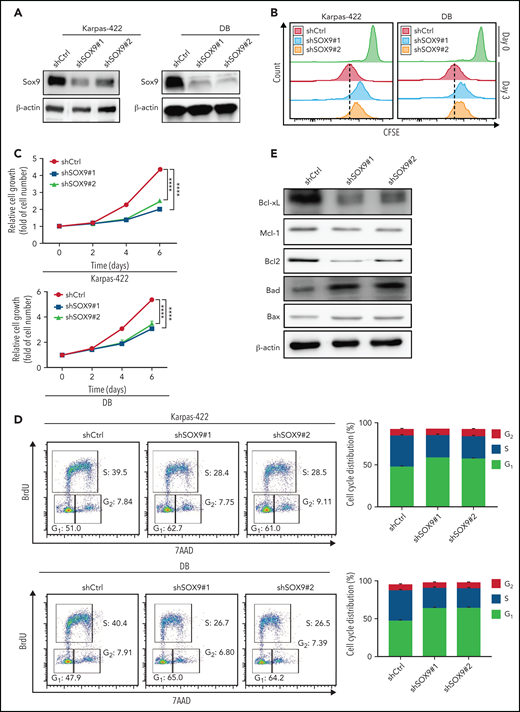
Inhibition of SOX9 in IGH-BCL2+ DLBCL cells impairs cell survival and proliferation. Immunoblot (A) and CFSE proliferation (B) assays of SOX9− Karpas-422 and DB cells. (C) Hemocytometer counts of the number of SOX9− Karpas-422 and DB cells. (D) SOX9− Karpas-422 and DB cells were exposed to BrdU for 4 hours before flow cytometry analysis. Quantitation of the percentage distribution of cell cycle phases was determined by gating of live cells in the G1 (light gray bar), S (dark gray bar), or G2 (white bar) phases. (E) Immunoblot assay of expression of Bcl-xL, Mcl-1, Bcl-2, Bad, and Bax. β-Actin was included as the control for equal loading. (C-D) Data represent the mean ± SD of technical triplicates. ****P < .005. CFSE, carboxyfluorescein diacetate succinimidyl ester.
Inhibition of SOX9 in IGH-BCL2+ DLBCL cells impairs cell survival and proliferation. Immunoblot (A) and CFSE proliferation (B) assays of SOX9− Karpas-422 and DB cells. (C) Hemocytometer counts of the number of SOX9− Karpas-422 and DB cells. (D) SOX9− Karpas-422 and DB cells were exposed to BrdU for 4 hours before flow cytometry analysis. Quantitation of the percentage distribution of cell cycle phases was determined by gating of live cells in the G1 (light gray bar), S (dark gray bar), or G2 (white bar) phases. (E) Immunoblot assay of expression of Bcl-xL, Mcl-1, Bcl-2, Bad, and Bax. β-Actin was included as the control for equal loading. (C-D) Data represent the mean ± SD of technical triplicates. ****P < .005. CFSE, carboxyfluorescein diacetate succinimidyl ester.
To explore possible mechanisms of SOX9-mediated cell survival, immunoblot analysis was performed to determine the expression of antiapoptotic proteins, such as Bcl-xL, Mcl-1, and Bcl-2, or proapoptotic proteins, such as Bad and Bax. Inhibition of SOX9 downregulated Bcl-xL, Mcl-1, and Bcl-2 expression, but upregulated Bad and Bax expression (Figure 2E; supplemental Figure 7A-E). Overexpression of SOX9 only mildly elevated Bcl-2 expression, but did not result in significant changes in Bcl-xL and Mcl-1 expression. However, overexpression of SOX9 downregulated Bad and Bax expression (supplemental Figures 1F and 9A-E). Taken together, these results suggest that SOX9 may play an oncogenic role in GCB-type, IGH-BCL2 DLBCL by promoting cell proliferation and inhibiting apoptosis.
SOX9 regulates expression of cholesterol metabolism–related genes
We explored the molecular mechanisms underlying the SOX9-induced biological consequences in DLBCL. RNA extracted from shSOX9#1, shSOX9#2, and scrambled control–transduced Karpas-422 cells were subjected to whole-transcriptome RNA-sequencing (RNA-seq) analysis (GSE180051; see the footnote). A small subset of genes (n = 135) were downregulated in both shSOX9#1- and shSOX9#2-transduced cells (Figure 3A). Kyoto Encyclopedia of Genes and Genomes analysis revealed that the steroid biosynthesis signaling pathway was the most significantly altered pathway in SOX9− cells (Figure 3A-B). This result was further confirmed by Gene Set Enrichment Analysis showing a downregulation of gene sets involved in steroid biosynthesis relative to the scrambled control (Figure 3C). We also observed a concordance in the modulation of a small subset of genes involved in steroid biosynthesis by constructing a heat map of both shSOX9#1- and shSOX9#2-transduced Karpas-422 cells (supplemental Figure 3A). Downregulation of the expression of these genes was confirmed by qRT-PCR in Karpas-422 and DB cells transduced with shSOX9#1, shSOX9#2 (Figure 3D), or SUDHL6 and OCI-LY1 cells transduced with pCDH-SOX9 (Figure 3E). To further evaluate whether expression of steroid biosynthesis–related genes are dependent on SOX9, the RNA-seq raw data were analyzed, and the expression of these gene was compared between 2 IGH-BCL2+ DLBCL cell lines expressing high levels of SOX9 and 6 IGH-BCL2− DLBCL cell lines manifesting low levels of SOX9 (Figure 3F), and the results were validated by qRT-PCR (supplemental Figure 3B-F). These analyses demonstrate that, except for FDFT1, the expression of the steroid synthesis–related genes tested including CYP51A1, DHCR24, MSMO1, and NSDHL1 were significantly higher in DLBCL cells with higher SOX9 expression. These data support the notion that SOX9 may regulate cholesterol metabolism in DLBCL.
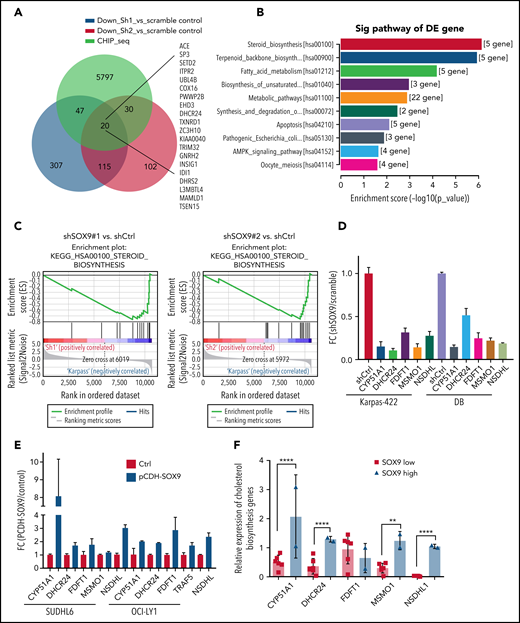
SOX9 regulates expression of cholesterol biosynthesis–related genes. (A) A Venn diagram of a small subset of overlapping SOX9-responsive genes downregulated in RNA-seq and showing a SOX9 peak in ChIP-seq. Green, blue, and red overlaid region (n = 20). Genes are listed to the right. (B) KEGG analysis of SOX9-responsive genes categorized according to the top 10 signaling pathways. (C) Downregulation of the steroid biosynthesis pathway in SOX9− Karpas-422 cells compared with the control, based on Gene Set Enrichment Analysis of SOX9-responsive genes. (D) Real-time PCR validation of steroid biosynthesized gene expression in SOX9− Karpas-422 cells and DB cells. (E) qRT-PCR determination of steroid biosynthesized gene expression in SUDHL6 and OCI-LY1 cells transduced with pCDH-SOX9 lentivirus. (F) Real-time PCR assay determined the relative expression of steroid biosynthesized genes in DLBCL cells with high (n = 2) and low (n = 6) levels of SOX9. (D-F) Data represent the mean ± SD of technical triplicates. **P < .01; ****P < .005. KEGG, Kyoto Encyclopedia of Genes and Genomes.
SOX9 regulates expression of cholesterol biosynthesis–related genes. (A) A Venn diagram of a small subset of overlapping SOX9-responsive genes downregulated in RNA-seq and showing a SOX9 peak in ChIP-seq. Green, blue, and red overlaid region (n = 20). Genes are listed to the right. (B) KEGG analysis of SOX9-responsive genes categorized according to the top 10 signaling pathways. (C) Downregulation of the steroid biosynthesis pathway in SOX9− Karpas-422 cells compared with the control, based on Gene Set Enrichment Analysis of SOX9-responsive genes. (D) Real-time PCR validation of steroid biosynthesized gene expression in SOX9− Karpas-422 cells and DB cells. (E) qRT-PCR determination of steroid biosynthesized gene expression in SUDHL6 and OCI-LY1 cells transduced with pCDH-SOX9 lentivirus. (F) Real-time PCR assay determined the relative expression of steroid biosynthesized genes in DLBCL cells with high (n = 2) and low (n = 6) levels of SOX9. (D-F) Data represent the mean ± SD of technical triplicates. **P < .01; ****P < .005. KEGG, Kyoto Encyclopedia of Genes and Genomes.
SOX9 directly targets and transcriptionally activates DHCR24 via direct binding to the SOX9 binding site
Chromatin immunoprecipitation–sequencing (ChIP-seq) was performed in the Karpas-422 cell line to identify the genes directly targeted by SOX9. Peak calling by the model-based analysis for the ChIP-seq algorithm revealed 9922 transcription sites bound directly to SOX9 (GSE179960, see footnote for ChIP-seq data deposition and supplemental Table 4 for peak calling). Interestingly, when we compared the 135 commonly downregulated genes identified by RNA-seq and the genes with SOX9-bound transcription sites detected by ChIP-seq, we identified 20 overlapping genes, among which DHCR24 showed the highest enrichment of bound SOX9 within its promoter and a significant change in RNA expression (Figure 3A; supplemental Table 5), in addition to being one of the genes in the steroid biosynthesis pathway. The 800-bp region upstream of the transcription start site of DHCR24 was highly enriched for SOX9 binding (Figure 4A). To validate the ChIP-seq results, the ChIP assay was performed in the Karpas-422 DLBCL cells. We identified a putative SOX9 binding site with the previously published consensus SOX9 binding motif (ACAATG)23 at position −576 of DHCR24. Three pairs of PCR primers were used to amplify 3 separate segments of the DHCR24 promoter. An amplification product was seen in 2 short DNA fragments, corresponding to −599 to −348 nt or −636 to −452 nt of the DHCR24 promoter, which harbor the SOX9 consensus motif (ACAATG; Figure 4B). However, no band was observed in the negative control DNA fragment corresponding to −251 to −78 nt of DHCR24. These results suggest direct binding of SOX9 to the DHCR24 promoter at the putative SOX9 binding site. To further confirm that DHCR24 is indeed a functional target of SOX9, we constructed luciferase reporters containing either the wild-type DHCR24 promoter encompassing a consensus binding motif of SOX9 or the DHCR24 promoter harboring a mutated SOX9 consensus motif (Figure 4B, top). The wild-type or mutant DHCR24 (encompassing 5 point mutations of SOX9 consensus motif; Figure 4C, top) reporter plasmids were cotransfected with the pCDH-SOX9, shSOX9, or empty vector lentiviral plasmids into 293T cells, and their luciferase activities were measured at 48 hours. Interestingly, overexpression of SOX9 significantly increased wild-type DHCR24 luciferase reporter activity compared with either empty vector or SOX9− counterparts (Figure 4C, bottom). In contrast, overexpressing SOX9 had no effect on the luciferase activity of the mutant DHCR24 promoter (Figure 4C, bottom). These results indicate that SOX9 directly targets and transcriptionally activates DHCR24 via direct binding to the SOX9 binding site.
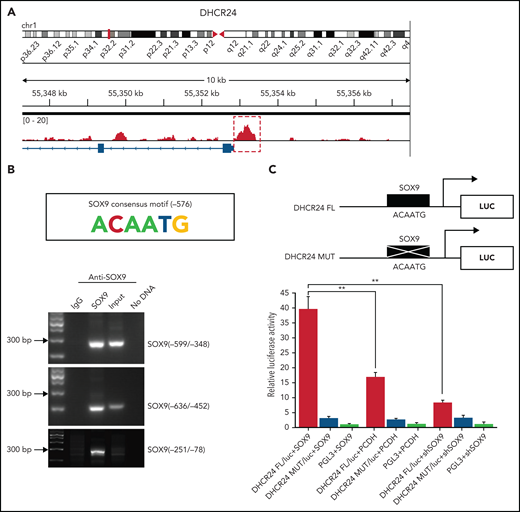
SOX9 binds directly to the cholesterol biosynthesized gene DHCR24. (A) Binding profiles and peak calling records of SOX9 in the DHCR24 promoter. Significant peak (P < 3. 3 × 10−11) is highlighted by a red box with dotted line. (B) ChIP assay validation of the SOX9 binding site in the promoter of DHCR24. The SOX9 consensus binding motif from −576 to −571 on the promoter of DHCR24 is schematically represented. (C) Luciferase reporter plasmids carrying wild-type DHCR24 promoter or DHCR24 promoter with mutated SOX9 binding site was cotransfected with pCDH-SOX9, shSOX9, or vector control lentiviral plasmids in 293T cells. Luciferase activities were measured 48 hours after transfection, and normalized against firefly luciferase activities. Schematic representation of DHCR24 FL and DHCR24 mutant luciferase reporter plasmids is shown. (C) Data represent the mean ± SD of technical triplicates. **P < .01.
SOX9 binds directly to the cholesterol biosynthesized gene DHCR24. (A) Binding profiles and peak calling records of SOX9 in the DHCR24 promoter. Significant peak (P < 3. 3 × 10−11) is highlighted by a red box with dotted line. (B) ChIP assay validation of the SOX9 binding site in the promoter of DHCR24. The SOX9 consensus binding motif from −576 to −571 on the promoter of DHCR24 is schematically represented. (C) Luciferase reporter plasmids carrying wild-type DHCR24 promoter or DHCR24 promoter with mutated SOX9 binding site was cotransfected with pCDH-SOX9, shSOX9, or vector control lentiviral plasmids in 293T cells. Luciferase activities were measured 48 hours after transfection, and normalized against firefly luciferase activities. Schematic representation of DHCR24 FL and DHCR24 mutant luciferase reporter plasmids is shown. (C) Data represent the mean ± SD of technical triplicates. **P < .01.
SOX9 regulates cholesterol biosynthesis via DHCR24
To determine whether steroid biosynthesis can be modulated in a SOX9-dependent manner in DLBCL cells, cholesterol content was measured in DLBCL expressing either a high (Karpas-422 and DB cell lines) or a low (OCI-LY1 and SUDHL6 cell lines) level of SOX9. Total cholesterol and cholesterol ester levels were present at significantly higher levels in the high SOX9-expressing Karpas-422 and DB cells (Figure 5A). Cholesterol levels were also determined in Karpas-422 and DB cells transduced with shSOX9#1, shSOX9#2, or in OCI-LY1 cells transduced with pCDH-SOX9 lentivirus. Inhibition of SOX9 significantly decreased total cholesterol or cholesterol ester contents in Karpas-422 and DB cells (Figure 5B-C). Vice versa, induction of SOX9 increased total cholesterol and cholesterol ester contents in OCI-LY1 cells (Figure 5D). Because DHCR24 was reported to be a pivotal enzyme in cholesterol biosynthesis that catalyzes the transformation of desmosterol to cholesterol, we explored whether SOX9 modulates cholesterol biosynthesis through regulation of DHCR24. Indeed, inhibition of SOX9 downregulated the expression of DHCR24 by 1.3-fold (shSOX9#1) or 2.5-fold (shSOX9#2) in Karpas-422 cells, or by 2.5-fold (shSOX9#1) or 3.3-fold (shSOX9#2) in DB cells (Figure 5E, top and bottom). Moreover, inhibition of DHCR24 by triparanol, a DHCR24 inhibitor, partially counteracted the SOX9-induced increase in total cholesterol and cholesterol ester contents (Figure 5F). Collectively, these data suggest that SOX9 is a positive regulator of cholesterol biosynthesis via increasing DHCR24 expression.
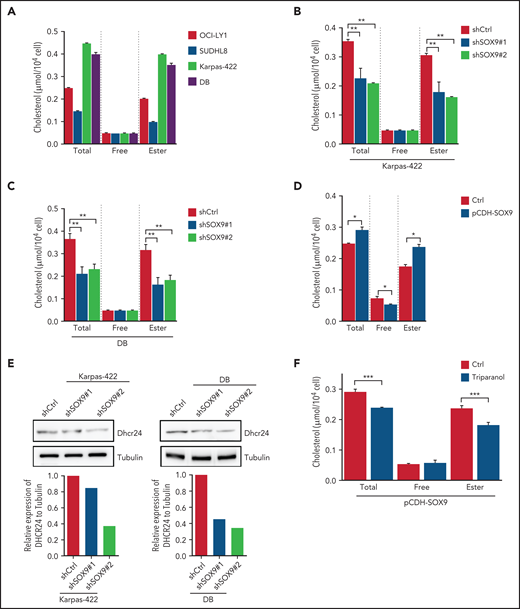
SOX9 modulates cholesterol biosynthesis via DHCR24. (A) Total and free cholesterol levels were measured in DLBCL cells with high (Karpas-422 and DB) or low (OCI-LY1 and SUDHL8) levels of SOX9. A total of 1 × 106 cells were used for each measurement. (B-D) Total and free cholesterol levels were measured in SOX9− Karpas-422 cells and DB cells and in SOX9-overexpressing OCI-LY1 cells. The level of cholesterol ester was calculated: total cholesterol − free cholesterol content. (E) Immunoblot assay and densitometry quantitation of DHCR24 protein in SOX9− Karpas-422 and DB cells. (F) Karpas-422 or DB cells were treated with triparanol for 48 hours or left untreated before they were subjected to cholesterol content measurement. (A-D, F) Data represent the mean ± SD of technical triplicates. *P < .05; **P < .01; ***P < .001.
SOX9 modulates cholesterol biosynthesis via DHCR24. (A) Total and free cholesterol levels were measured in DLBCL cells with high (Karpas-422 and DB) or low (OCI-LY1 and SUDHL8) levels of SOX9. A total of 1 × 106 cells were used for each measurement. (B-D) Total and free cholesterol levels were measured in SOX9− Karpas-422 cells and DB cells and in SOX9-overexpressing OCI-LY1 cells. The level of cholesterol ester was calculated: total cholesterol − free cholesterol content. (E) Immunoblot assay and densitometry quantitation of DHCR24 protein in SOX9− Karpas-422 and DB cells. (F) Karpas-422 or DB cells were treated with triparanol for 48 hours or left untreated before they were subjected to cholesterol content measurement. (A-D, F) Data represent the mean ± SD of technical triplicates. *P < .05; **P < .01; ***P < .001.
DHCR24 rescues the SOX9 knockdown phenotype
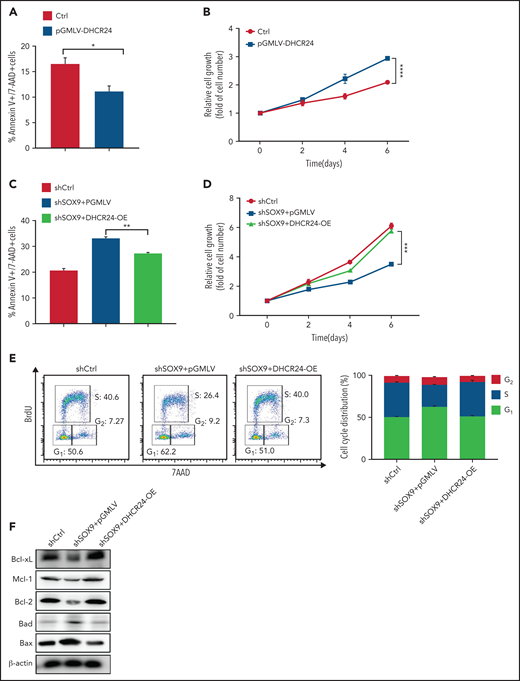
To investigate whether DHCR24 has a role in modulating the potentially oncogenic functions of SOX9, a lentiviral vector encoding a DHCR24-overexpressing plasmid (pGMLV-DHCR24) was constructed and transduced into SOX9-expressing Karpas-422 cells. Overexpression of pGMLV-DHCR24 significantly decreased DLBCL cell apoptosis and accelerated DLBCL cell growth (Figure 6A-B). To further test whether overexpression of DHCR24 rescues the phenotype associated with SOX9 knockdown, pGMLV-DHCR24 lentivirus was transduced to SOX9− Karpas-422 cells and incubated for 48 hours. Short hairpin (sh) scrambled control-overexpressing Karpas-422 cells and vector control-transduced SOX9− Karpas-422 cells were used as experimental controls. Elevated DHCR24 expression was capable of rescuing the decrease in DLBCL cell survival and cell growth associated with SOX9 knockdown (Figure 6C-D). Moreover, overexpression of DHCR24 rescued SOX9 shRNA-induced G1/S arrest (Figure 6E, left and right). Furthermore, overexpression of DHCR24 overcame the downregulation of Bcl-xL, Mcl-1, and Bcl2 and the upregulation of Bad and Bax in Karpas-422 cells after SOX9 knockdown (Figure 6F; supplemental Figure 8A-E). Taken together, our findings provide supportive evidence of a pivotal role of DCHR24 in modulating SOX9 functions.
SOX9 modulates cholesterol biosynthesis via DHCR24. (A) Total and free cholesterol levels were measured in DLBCL cells with high (Karpas-422 and DB) or low (OCI-LY1 and SUDHL8) levels of SOX9. A total of 1 × 106 cells were used for each measurement. (B-D) Total and free cholesterol levels were measured in SOX9− Karpas-422 cells and DB cells and in SOX9-overexpressing OCI-LY1 cells. The level of cholesterol ester was calculated: total cholesterol − free cholesterol content. (E) Immunoblot assay and densitometry quantitation of DHCR24 protein in SOX9− Karpas-422 and DB cells. (F) Karpas-422 or DB cells were treated with triparanol for 48 hours or left untreated before they were subjected to cholesterol content measurement. (A-D, F) Data represent the mean ± SD of technical triplicates. *P < .05; **P < .01; ***P < .001; ****P < 0.0005.
SOX9 modulates cholesterol biosynthesis via DHCR24. (A) Total and free cholesterol levels were measured in DLBCL cells with high (Karpas-422 and DB) or low (OCI-LY1 and SUDHL8) levels of SOX9. A total of 1 × 106 cells were used for each measurement. (B-D) Total and free cholesterol levels were measured in SOX9− Karpas-422 cells and DB cells and in SOX9-overexpressing OCI-LY1 cells. The level of cholesterol ester was calculated: total cholesterol − free cholesterol content. (E) Immunoblot assay and densitometry quantitation of DHCR24 protein in SOX9− Karpas-422 and DB cells. (F) Karpas-422 or DB cells were treated with triparanol for 48 hours or left untreated before they were subjected to cholesterol content measurement. (A-D, F) Data represent the mean ± SD of technical triplicates. *P < .05; **P < .01; ***P < .001; ****P < 0.0005.
Inhibition of SOX9-dependent cholesterol biosynthesis reduces tumor growth in vivo
To explore the biological significance of inhibiting SOX9 in vivo, 10 million shSOX9 lentivirus-transduced Karpas-422 cells were subcutaneously injected into each nude mouse (n = 5). Three weeks after transplantation, the DLBCL-recipient mice were euthanized, and tumorigenesis-related phenotypes were measured. The tumor size and volume were dramatically reduced in recipients of shSOX9-transduced Karpas-422–xenografts when compared with the controls (Figure 7A-B). Moreover, the increased cell death and the decreased proliferation rate of the DLBCL cells were observed in recipients of SOX9− Karpas-422 xenografts compared with scrambled control recipients (Figure 7C-D). Moreover, total cholesterol and cholesterol ester levels were significantly reduced in SOX9− Karpas-422–recipient mice (Figure 7E). Furthermore, DLBCL cell morphology was confirmed by hematoxylin-eosin staining in mice with SOX9− Karpas-422 xenografts and scrambled control–recipient mice. (Figure 7F). We also generated mice with DB xenografts mice, and results similar to those of Karpas-422–recipients were obtained (supplemental Figure 4B-F).
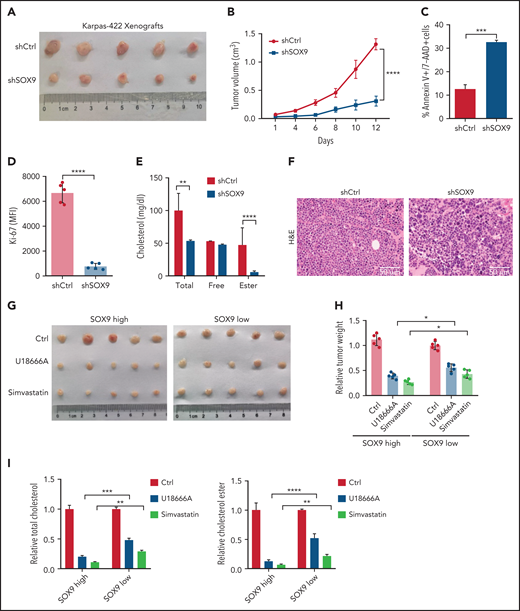
Inhibition of SOX9 and cholesterol synthesis deaccelerates tumor growth in vivo. (A) Ten million SOX9− Karpas-422 cells were subcutaneously injected into each nude mouse (n = 5). Three weeks after transplantation, the mice were euthanized and tumor size (A), tumor volume (B), apoptosis (C), proliferation (D), and cholesterol contents (E) were measured, and DLBCL morphology (F) was examined in the mice. Ten million DLBCL cells harboring a high level (Karpas-422) or a low level (SUDHL8) of SOX9 were subcutaneously injected into each nude mouse (n = 5). Meanwhile, 10 mg/kg U18666A (every 2 days) or 50 mg/kg simvastatin (every 2 days) were either orally administered or intraperitoneal injected into the DLBCL xenograft-recipient mice for a week. Bar represents 50 μm. Three weeks after transplantation and drug administration, the mice were euthanized and tumor size (G), relative tumor weight (H), and relative cholesterol contents (I) were measured. (B-I) Data represent the mean ± SD from technical triplicates. *P < .05; **P < .01; ***P < .001; ****P < .005.
Inhibition of SOX9 and cholesterol synthesis deaccelerates tumor growth in vivo. (A) Ten million SOX9− Karpas-422 cells were subcutaneously injected into each nude mouse (n = 5). Three weeks after transplantation, the mice were euthanized and tumor size (A), tumor volume (B), apoptosis (C), proliferation (D), and cholesterol contents (E) were measured, and DLBCL morphology (F) was examined in the mice. Ten million DLBCL cells harboring a high level (Karpas-422) or a low level (SUDHL8) of SOX9 were subcutaneously injected into each nude mouse (n = 5). Meanwhile, 10 mg/kg U18666A (every 2 days) or 50 mg/kg simvastatin (every 2 days) were either orally administered or intraperitoneal injected into the DLBCL xenograft-recipient mice for a week. Bar represents 50 μm. Three weeks after transplantation and drug administration, the mice were euthanized and tumor size (G), relative tumor weight (H), and relative cholesterol contents (I) were measured. (B-I) Data represent the mean ± SD from technical triplicates. *P < .05; **P < .01; ***P < .001; ****P < .005.
To further investigate whether inhibition of the SOX9-DCHR24-choelsterol biosynthesis axis represses lymphomagenesis, 10 million Karpas-422 cells expressing high levels of SOX9 or SUDHL8 cells exhibiting low levels of SOX9 were transplanted into nude mice. Three weeks after transplantation, the xenograft-recipient mice were randomized and administered simvastatin (oral) or U18666A (intraperitoneal injection) at the indicated doses and time intervals (supplemental Figure 4A) or were left untreated. Although tumor size and weight were significantly reduced in both recipients of Karpas-422 or SUDHL8 xenografts, the degree of reduction was higher in the former after treatment (Figure 7G-H). Moreover, an approximately three- to fourfold reduction in tumor weight was observed in Karpas-422 xenograft–recipient mice that had been treated with simvastatin or U18666A, respectively, when compared with a 1.8- or 2.8-fold reduction in mice receiving SUDHL8 xenografts after having the same treatments (Figure 7H). Furthermore, the relative fold reductions of total and cholesterol ester contents were more pronounced in the Karpas-422 recipients (P < .001) than in the SUDHL8 (P < .01) recipients (Figure 7I). In summary, our in vivo data support an oncogenic role of SOX9 in DLBCL via the SOX9-DHCR24-cholesterol biosynthesis axis, which appears to confer increased vulnerability of the DLBCL cells to inhibition of cholesterol synthesis.
To explore whether SOX9 can be a promising therapeutic target in DLBCL, SOX9high (Karpas-422) and SOX9low (SUDHL6) cell lines and SOX9-overexpressing OCI-LY1 cells were treated in vitro with 0.5 μM JQ1, a BET inhibitor reported to inhibit SOX9 transcription and protein stability.24 In addition, Karpas-422– or SUDHL6-recipient mice were injected with JQ1 (50 mg/kg, intraperitoneally). JQ1 inhibited cholesterol synthesis in a SOX9-dependent manner in vitro (supplemental Figure 5A-D). Importantly, JQ1 inhibited lymphomagenesis and cholesterol biosynthesis in a SOX9-dependent manner in vivo (supplemental Figure 5E-H), suggesting the potential of therapeutic targeting of SOX9 in management of DLBCL.
Discussion
In our study, SOX9 was overexpressed in a subset of DLBCLs, particularly those harboring IGH-BCL2 translocations. Limited Sanger sequencing results indicated that the overexpressed SOX9 is most likely wild-type, which is similar to the mutation profiling results of Schmitz et al, who rarely found SOX9 mutations in a large cohort of patients with DLBCL.25 Our data also suggest that SOX9 expression in DLBCL is associated with advanced clinical stage, which must be confirmed in a larger, independent cohort.
The molecular mechanism behind the association of SOX9 overexpression and the presence of IGH-BCL2 is unknown. We speculate that certain signaling pathways (eg, NF-κB signaling) are responsible for inducing SOX9 expression. Lymphoma cells with the t(14;18) translocation showed high levels of nuclear NF-κB, which may induce BCL2 expression in these cells as a result of NF-κB activation in the setting of global epigenetic dysregulation.26-28 Further investigation is necessary to delineate the precise mechanisms of SOX9 overexpression in these lymphoma cells.
Knockdown of SOX9 in SOX9high cell lines decreased cell proliferation and enhanced survival of DLBCL cells in vitro and in mouse xenograft recipients, in line with the published data regarding SOX9 function in promoting cell proliferation and inhibiting apoptosis. SOX9 has been shown to facilitate cell proliferation and differentiation of normal tissues or stem cells, such as sebocytes,29 human lung fibroblast cells,30 and undifferentiated rat mesenchymal stem cells.31 In addition, SOX9 was found to exert its oncogenic role by modulating cell proliferation and apoptosis and promoting tumor progression and chemoresistance in various solid tumor models including gastric,17,32 hepatocellular,33,34 and pancreatic35 cancers and in glioma,36 chordoma,37 and chondrosarcoma,38 among others.
Screening for a potential SOX9-regulated signaling pathway revealed that genes differentially expressed after SOX9 knockdown were enriched in the cholesterol biosynthesis signaling cascade. In combination with our ChIP-seq data, we identified DHCR24, a terminal enzyme in cholesterol biosynthesis that catalyzes the conversion of desmosterol to cholesterol, as a SOX9 target for transcription activation. Enforced expression of DHCR24 rescued the SOX9-silencing phenotype. SOX9 overexpression elevated the DHCR24 level, resulting in promoted cholesterol synthesis and lymphomagenesis. We showed that cholesterol content was higher in DLBCL cells with high levels of SOX9. Inhibitors targeting cholesterol synthesis induced apoptosis and reduced tumorigenesis in DLBCL cells expressing high levels of SOX9, suggesting that these cells are more addicted to cholesterol. The link between a transcription factor and addiction to cholesterol biosynthesis has been demonstrated previously. Multiple myeloma cells have been shown to be addicted to an abnormal, pleiotropic regulatory network controlled by the transcriptional factor interferon regulatory factor-4.39 Among the various molecules and cellular processes regulated by interferon regulatory factor-4 are many enzymes and regulators of sterol and lipid synthesis, including squalene monooxygenase and stearoyl-CoA desaturase, which encode rate-limiting enzymes in these pathways.39
Accumulating evidence suggests that unbalanced cholesterol homeostasis and dysregulation of cholesterol metabolism contributes to carcinogenesis.40,41 Interestingly, the incidence of coronary diseases is higher in patients with DLBCL than in the general population,42,43 suggesting a link between de novo cholesterol synthesis in lymphoma development and maintenance. Our study showed that pharmacological inhibition of cholesterol biosynthesis using simvastatin dramatically decreased growth of DLBCL cells and lymphomagenesis in DLBCL xenografts. This pharmacological approach is especially effective in DLBCL cells expressing high levels of SOX9 because of their addiction to the cholesterol biosynthesis pathway, which supports an important role of de novo cholesterol synthesis in lymphoma development and maintenance. The role of simvastatin in preventively managing and treating patients with atherosclerotic cardiovascular disease,44 heart failure,45 and stroke46 via inhibition of low-density lipoprotein cholesterol synthesis has been well established. Repurposing of this common drug to treat DLBCLs with high SOX9 expression may represent a potentially effective therapeutic regimen for this subset of DLBCLs. Other studies have identified potential targets for lowering cholesterol content in lymphoma cells. For instance, high-density lipoprotein nanoparticles targeting scavenger receptor type B1 reduce cholesterol uptake and induce ferroptosislike cell death, thus inhibiting lymphomagenesis.47-49 Moreover, cholesterol auxotrophy, because of loss of squalene monooxygenase expression, may present a targetable liability in anaplastic large cell lymphoma. Furthermore, inhibition of the SYK/PI3K pathway can interfere with multiple components of the cholesterol synthesis pathway in DLBCL.50
Cholesterol metabolism contributes to tumorigenesis via regulation of signaling pathways, and it can be regulated by signaling pathways. For example, cholesterol enhances liver carcinogenesis by activating the MAPK signaling pathway.51 Moreover, the EGFR/SRC/extracellular signal–regulated kinase–signaling cascade stabilized YTHDF2 and promoted cholesterol dysregulation and growth of glioblastoma.52 Our study illustrates SOX9-mediated regulation of de novo cholesterol synthesis in DLBCL by a novel pathway involving DHCR24, which has been shown to promote tumorigenesis through its positive regulation of cholesterol synthesis in a variety of cancers including breast cancer stem cells,53 endometrial carcinoma,54 and hepatocellular carcinoma cells.55
In summary, our study demonstrates, for the first time, the oncogenicity of SOX9 in DLBCL and identifies a novel role for the SOX9-DHCR24-cholesterol biosynthesis axis in lymphomagenesis. It is likely that a subset of DLBCLs, in particular those with IGH-BCL2 rearrangement, are addicted to this axis. Pharmacological targeting of SOX9 with selective SOX9 inhibitor, nanoparticle delivery, or long noncoding RNA may be promising treatment strategies for DLBCL.
Acknowledgments
The authors thank Hongliang Zong for valuable research suggestions and technical support and the Research Program of the Department of Pathology and Laboratory Medicine at Weill Cornell Medicine for assistance with the immunohistochemistry.
This projected was funded by grants from the Innovative Research Team of high-level local universities in Shanghai, China; the National Key Research and Development Program of China (2020YFA0803603) (J.C.) and the National Natural Science Foundation of China (81700134), the Pujiang Talent Program in Shanghai (17PJ1405500), the Science Foundation of Shanghai (17ZR1415600), and the Youth Eastern Scholar of Shanghai 1730000034) (all to J.M.). Work related to patient lymphoma samples described in this study was supported by a Specialized Center of Research (SCOR) grant (7012-16) from the Leukemia and Lymphoma Society (United States).
Authorship
Contribution: Y.S. performed the experiments, analyzed the data, and wrote the manuscript; J.Z., S.C., and Z.C. analyzed the data; K.N. performed the experiments and analyzed the data; W. Wang, W. Wei, D.J., Z.P., Y.R., and Q.F. performed the experiments; K.L.R. collected the clinical data; Y.Q. made intellectual contributions; J.C. conceptualized the project and made intellectual contributions; W.T. conceptualized the project, provided critical clinical cases, analyzed the data, performed experiments, and wrote the manuscript; and J.M. conceptualized and directed the project, designed the experiments, and wrote the manuscript.
Conflict-of-interest disclosure: The authors report no competing financial interests.
Kristy L. Richards died on 30 March 2019.
Correspondence: Jiao Ma, Department of Biochemistry and Molecular Cell Biology, Shanghai Jiaotong University School of Medicine, No. 280 South Chongqing Rd, West Building 7, Room 408, Shanghai 200025, China; e-mail: drjiaoma@shsmu.edu.cn; Wayne Tam, Division of Hematopathology, Department of Pathology and Laboratory Medicine, Weill Cornell Medicine, 525 East 68th Street, F544C, New York, NY 10065; e-mail: wtam@med.cornell.edu; and Jinke Cheng, Department of Biochemistry and Molecular Cell Biology, Shanghai Jiaotong University School of Medicine, No. 280 South Chongqing Rd, West Building 7, Room 408, Shanghai 200025, China; e-mail: jkcheng@shsmu.edu.cn.
ChIP-Seq data have been deposited in the Gene Expression Omnibus (GEO) database (accession number GSE179960;_https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE179960/ private access token: ilafewewlvqdnit). RNA-Seq data from gene expression profiles in SOX9 shRNAs and scrambled control–transduced Karpas-422 cells have been deposited in the GEO database (accession number GSE180051; https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE180051/ private access token: ujwfmusijfwvnqx).
The online version of this article contains a data supplement.
There is a Blood Commentary on this article in this issue.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal