

Key Points
14-3-3ζ synergizes c-Src to β3-integrin, and forms the 14-3-3ζ–c-Src–integrin-β3 complex during platelet activation.
Interference with the formation of the complex abolishes platelet outside-in signaling and suppresses thrombosis without causing bleeding.

Abstract
Several adaptor molecules bind to cytoplasmic tails of β-integrins and facilitate bidirectional signaling, which is critical in thrombosis and hemostasis. Interfering with integrin-adaptor interactions spatially or temporally to inhibit thrombosis without affecting hemostasis is an attractive strategy for the development of safe antithrombotic drugs. We show for the first time that the 14-3-3ζ–c-Src–integrin-β3 complex is formed during platelet activation. 14-3-3ζ–c-Src interaction is mediated by the -PIRLGLALNFSVFYYE- fragment (PE16) on the 14-3-3ζ and SH2-domain on c-Src, whereas the 14-3-3ζ–integrin-β3 interaction is mediated by the -ESKVFYLKMKGDYYRYL- fragment (EL17) on the 14-3-3ζ and -KEATSTF- fragment (KF7) on the β3-integrin cytoplasmic tail. The EL17-motif inhibitor, or KF7 peptide, interferes with the formation of the 14-3-3ζ–c-Src–integrin-β3 complex and selectively inhibits β3 outside-in signaling without affecting the integrin-fibrinogen interaction, which suppresses thrombosis without causing significant bleeding. This study characterized a previously unidentified 14-3-3ζ–c-Src–integrin-β3 complex in platelets and provided a novel strategy for the development of safe and effective antithrombotic treatments.
Introduction
Bleeding is the major risk associated with all antithrombotic drugs, including anticoagulant and antiplatelet agents.1,2 Several integrins have been identified in platelets that play key roles in platelet functions, such as collagen receptor α2β1, fibronectin receptor α5β1, laminin receptor α6β1, and vitronectin receptor αvβ3.3-5 However, αIIbβ3 is the most abundant6 and is crucial to platelet function, because it is required for stable platelet adhesion to the vascular wall and for platelet aggregation.7-9 Through interactions with its ligands (eg, fibrinogen, von Willebrand factor [VWF], fibronectin, and vitronectin)10-13 and the integrin-β3 tail-binding adaptor proteins (eg, talin, kindlin, 14-3-3ζ, and c-Src family kinase),14,15 αIIbβ3 can transmit signals bidirectionally in a process known as outside-in and inside-out signaling.16,17 Platelet activation orchestrated by these complementary signaling pathways play critical roles in hemostasis and thrombosis.15,18 Inside-out signaling, upon agonist stimulation, activates integrins to mediate stable platelet adhesion and aggregation.19,20 Upon integrin ligation, outside-in signaling amplifies platelet activation and thrombus size.21,22 Accordingly, selectively attenuating αIIbβ3 outside-in signaling without interfering with inside-out signaling and integrin ligation would interfere with thrombus growth with minimal effects on hemostasis,18 making this an ideal strategy for development of antithrombotic agents. Modification of adaptor binding to integrin-β3, adaptor composition, and coadaptor-mediated binding are possible means of disrupting outside-in signaling.
Several adaptors are now known to interact with the cytoplasmic tails (CTs) of β-integrins, and more are still being uncovered.14 The adaptor molecules assemble into focal adhesions at the integrin-β3 adhesome to regulate platelet signaling. In this study, we investigate the scaffold of focal adhesion at the integrin-β3 tail and identify novel interactions among the adaptor molecules with the aim of developing strategies and novel agents against thrombosis without causing significant bleeding.
Methods
Platelet preparation and aggregation
Platelets were prepared from human blood or from wild-type (WT), GPIbα-deficient (GPIbα−/−), IL4Rα/GPIbα-transgenic,23 and β3-integrin–deficient (β3−/−) mice. Aggregation experiments were performed as previously described.24 In brief, washed platelets were resuspended in Tyrode’s buffer, with or without an addition of 100 μg/mL fibrinogen (F3879; MilliporeSigma, Burlington MA). After a 5-minute incubation with candidate compounds at 37°C, platelet aggregation was initiated by addition of agonist and measured in an aggregometer (LBY-NJ4; Techlink, Beijing, China). Detailed information is in the supplemental Materials, available on the Blood Web site.
Platelet spreading, immunofluorescence staining, and confocal microscopy
Washed platelets were added to 100 μg/mL fibrinogen-coated coverslips and incubated for 40 minutes at 37°C. Platelets were fixed, permeabilized, and blocked, followed by further staining with anti-β3, -14-3-3ζ, and -c-Src antibodies and a related fluorescence-conjugated secondary antibody. For the spreading assay, platelets were stained with fluorescence isothiocyanate (FITC)–labeled phalloidin (40735ES75; Yeasen) and visualized by confocal microscope (A1 MP+; Nikon, Tokyo, Japan). Detailed information is in the supplemental Materials.
Immunoprecipitation and western blot analysis
Platelets or transfected HEK293T cells were solubilized in cold NP-40 lysis buffer for immunoprecipitation or in RIPA lysis buffer (R0278; MilliporeSigma) for western blot analysis. Protein A–conjugated magnetic beads (10002D; Thermo Fisher Scientific, Waltham, MA) were used for coimmunoprecipitation, per the manufacturer’s instructions. Lysates from transfected HEK293T cells were coimmunoprecipitated and immunoblotted with hemagglutinin or Myc-tagged antibody. Platelet lysate was coimmunoprecipitated with integrin-β3 antibody or isotype-matched control IgG and immunoblotted with integrin-β3, 14-3-3ζ, c-Src, or Src pTyr416. Detailed information is in the supplemental Materials.
14-3-3ζ Binding compound screening
The workflow for screening for the 14-3-3ζ binding compounds in our in-house library (LR-NP1792; Mendeley Data doi:10.17632/2kbjft8jd3.1) is illustrated in supplemental Figure 3A. In brief, the 14-3-3ζ crystal structure (Protein Data Bank ID: 2V7D) was obtained online (https://www.rcsb.org/structure/2V7D). For virtual screening, the protein structure was subjected to a brief molecular dynamics simulation (10 ps) for further refinement with Studio software (version 3.1; Biovia, San Diego, CA). LR-NP1792 was docked to the structure of 14-3-3ζ in the site sphere (x:37.594256, y:19.966698, z:23.903805, radius:6.7) by LibDock.25 The CHARMm minimization was performed to optimize the docked poses. The binding capacity of the top 4% LibDock score (supplemental Table 3) compounds to 14-3-3ζ was further verified by surface plasmon resonance (SPR) and the potential effect of these candidates on 0.05-U/mL thrombin-induced platelet aggregation was detected at the concentration of 30 μM.
Peptide precipitations
Binding of recombinant c-Src and 14-3-3ζ to synthetic biotin-labeled integrin-β3 CT was performed with biotin-streptavidin. Detailed information is in the supplemental Materials.
Platelet-fibrinogen binding and P-selectin detection
Detection of fibrinogen binding26 and P-selectin expression27,28 were performed with modifications according published methods. In brief, washed platelets were suspended in Tyrode’s buffer B containing candidate compounds and 100 μg/mL FITC-conjugated fibrinogen or anti-mouse P-selectin antibody (RB40.34, BD Pharmingen), followed by incubation, with or without agonists (200 μM PAR4AP or 1 μM anti-αIIb transmembrane [TM] peptide) at 37°C in the dark for 40 minutes. FITC-conjugated fibrinogen bound platelets, and P-selectin expression were quantified with flow cytometry (LSR Fortessa; BD, Franklin Lakes, NJ).
SPR analysis
SPR was performed as previously described, with modifications.29 In brief, recombinant 14-3-3ζ was immobilized on the activated Sensor Chip CM-5 by amine coupling, candidate compounds in HBS-EP+ running buffer were applied to the immobilized ligand with a flow rate of 10 μL/min, and the real-time binding signal was recorded with BIAcore 3000 software (GE Healthcare, Milwaukee, WI). The 6xHis-tagged EL17 or PE16 immobilized Sensor Chip NTA was used to analyze binding with KF7 and 3′,4′,7′-trihydroxyisoflavone (THO). Detailed information is in the supplemental Materials.
Carrageenan-induced tail thrombosis in mice
Tail thrombosis in mice was induced by carrageenan according to a modified reported method.24 In brief, 30 minutes after oral gavage of candidate compounds in Swiss mice (Kunming mice) of either sex (18-22 g), the mice were injected IP with 100 μL (0.8% w/v) carrageenan (type I; MilliporeSigma) dissolved in saline to induce thrombosis. The length of the thrombus in the tail was measured 24 hours after treatment. All animal experimental protocols were approved by the Animal Care and Use Committee at Kunming Institute of Zoology, Chinese Academy of Sciences (SMKX2017023).
FeCl3-induced thrombosis
C57BL/6J mice (male, 7-8 weeks old) were anesthetized by isoflurane inhalation with and anesthesia respirator (R540IP; RWD Life Science, Shenzhen, China) 2 hours after oral gavage of 100 mg/kg candidate compounds. Carotid arterial thrombosis was induced with a filter paper disc (diameter = 2 mm) soaked with 10% FeCl3, and blood flow was monitored with a laser-speckle blood flow imaging system (RFLSI Pro; RWD Life Science).
Bleeding assays
C57BL/6J mice of either sex (7-8 weeks old) were gavaged orally with the candidate compound and the control (100 μL normal saline containing 2 μL dimethyl sulfoxide [DMSO]). A 5-mm tail-tip transection was made to evaluate tail-bleeding times, or a calibrated section (4.92 ± 0.26 mg) was removed from the liver lobe to evaluate liver bleeding. For cerebral bleeding, the skull of anesthetized mice was drilled (diameter = 2 mm) laterally to the bregma and a 4-mm-deep injury was made with a needle.30 Detailed information is in the supplemental Materials.
Clot retraction
Platelets (500 × 109/L) were resuspended in platelet-depleted human plasma and incubated with our candidate compound or control at 37°C for 5 minutes. α-Thrombin (0.05 U/mL) and CaCl2 (10 mM) were added to initiate the coagulation. Images of clots were acquired at various time points. The size of the retracted clots was quantified using ImageJ 1.35h software (National Institutes of Health, Bethesda, MD).
Statistical analysis
Statistical significance was assessed by Student t test. Analyses were performed with Prism 6.1 software (GraphPad, La Jolla CA). Results were reported as means ± standard deviation (SD), with significance set at P < .05.
Results
14-3-3ζ–c-Src–integrin-β3 forms a complex in platelets during activation
Tyrosine kinase c-Src is known to bind to the cytoplasmic tail of the integrin-β3 subunit via its SH3 domain.26,31 As a integrin scaffolding adaptor, 14-3-3ζ also has been reported to bind to integrin-β2.32,33 In this study, confocal immunofluorescence microscopy results revealed that integrin-β3, c-Src, and 14-3-3ζ were highly colocalized in platelets adhering to immobilized fibrinogen (Figure 1A), especially at the joint between the filopodia and platelet body, suggesting that the 14-3-3ζ–c-Src–integrin-β3 complex forms during platelet activation. Meanwhile, overlapping yellow regions indicated a possible interaction of c-Src with 14-3-3ζ (merged image; Figure 1A). The deletion of a conserved region (-165PIRLGLALNFSVFYYE180-; PE16) on 14-3-3ζ (Figure 1B) and residues 200 to 246 on c-Src (Figure 1C) abolished c-Src–14-3-3ζ interaction.
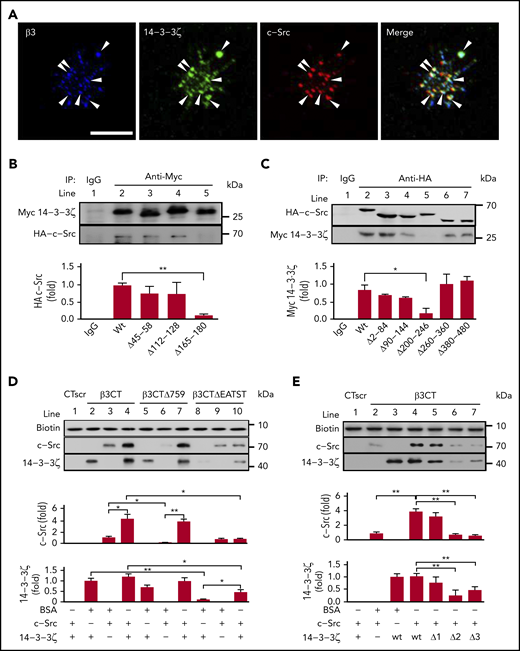
The 14-3-3ζ–c-Src–integrin-β3 complex in platelets. (A) Washed human platelets were plated on fibrinogen-coated coverslips at 37°C for 20 minutes. Confocal microscopy of permeabilized platelets stained with antibodies against integrin-β3 (blue), 14-3-3ζ (green), and c-Src (red). Arrowheads indicate 14-3-3ζ+, c-Src+, and integrin-β3+ deposits. Scale bar, 10 μm. (B) Pull-down analysis of the interaction between 14-3-3ζ or the 14-3-3ζ mutant and WT c-Src. (C) Pull-down analysis of the interaction between c-Src or the c-Src mutant and WT 14-3-3ζ. (D) Western blot analysis of recombinant c-Src or 14-3-3ζ protein precipitated by biotin-conjugated WT β3CT, β3CT lacking amino acids 759 to 761 (β3CTΔ759) or β3CT lacking amino acids 749 to 753 (β3CTΔEATST). (E) Western blot analysis of recombinant c-Src or 14-3-3ζ mutant proteins precipitated by biotin-conjugated β3CT. Δ1, Δ2, and Δ3 refer to recombinant 14-3-3ζ proteins lacking amino acids 45 to 58, 112 to 128, and 165 to 180, respectively. Data are shown as means ± SD (n = 3). Statistical significance was determined by Student t test. *P < .05, **P < .01.
The 14-3-3ζ–c-Src–integrin-β3 complex in platelets. (A) Washed human platelets were plated on fibrinogen-coated coverslips at 37°C for 20 minutes. Confocal microscopy of permeabilized platelets stained with antibodies against integrin-β3 (blue), 14-3-3ζ (green), and c-Src (red). Arrowheads indicate 14-3-3ζ+, c-Src+, and integrin-β3+ deposits. Scale bar, 10 μm. (B) Pull-down analysis of the interaction between 14-3-3ζ or the 14-3-3ζ mutant and WT c-Src. (C) Pull-down analysis of the interaction between c-Src or the c-Src mutant and WT 14-3-3ζ. (D) Western blot analysis of recombinant c-Src or 14-3-3ζ protein precipitated by biotin-conjugated WT β3CT, β3CT lacking amino acids 759 to 761 (β3CTΔ759) or β3CT lacking amino acids 749 to 753 (β3CTΔEATST). (E) Western blot analysis of recombinant c-Src or 14-3-3ζ mutant proteins precipitated by biotin-conjugated β3CT. Δ1, Δ2, and Δ3 refer to recombinant 14-3-3ζ proteins lacking amino acids 45 to 58, 112 to 128, and 165 to 180, respectively. Data are shown as means ± SD (n = 3). Statistical significance was determined by Student t test. *P < .05, **P < .01.
In vitro interactions of 14-3-3ζ, c-Src, and integrin-β3 were further characterized by peptide-bait experiments (Figure 1D-E) with biotinylated peptides, including β3CT (biotin-NNPLYKEATSTFTNITYRGT, containing WT residues 743-762 of the β3CT), β3CTΔ759 (biotin-NNPLYKEATSTFTNITY, lacking CT residues 760-762), β3CTΔEATST (biotin-NNPLYKFTNITYRGT, lacking CT residues 749-753), and β3CT scrambled peptide (biotin-TNTNGPRLYSYTKIETNAFT; CTscr), recombinant 14-3-3ζ, and c-Src. Recombinant 14-3-3ζ showed a direct interaction with recombinant c-Src (supplemental Figure 1). 14-3-3ζ (lane 2; Figure 1D) or c-Src (lane 3; Figure 1D) alone was pulled down by β3CT to form 14-3-3ζ–β3CT or c-Src–β3CT complexes. Interestingly, when both 14-3-3ζ and c-Src were present, more 14-3-3ζ and c-Src were pulled down by β3CT (lane 4, Figure 1D), which likely resulted, not only from 14-3-3ζ–β3CT or c-Src–β3CT, but also from 14-3-3ζ–β3CT–c-Src complex formation. Meanwhile, β3CTΔ759, which lacks binding sites for c-Src,34 pulled down only 14-3-3ζ (lane 5, Figure 1D). However, β3CTΔEATST, which lacks -749EATST753- on β3CT, did not bind to 14-3-3ζ (lane 8; Figure 1D) but retained the c-Src-β3CT interaction (lane 9; Figure 1D), suggesting that -749EATST753- is essential for 14-3-3 ζ–integrin-β3 interaction. Deletion of EL17 (Δ2, -112ESKVFYLKMKGDYYRYL128-) from 14-3-3ζ inhibited the 14-3-3ζ–integrin-β3 interaction, suggesting that EL17 on 14-3-3ζ mediates binding to integrin-β3 (lane 6; Figure 1E). Consistent with the data highlighting the importance of PE16 in c-Src–14-3-3ζ coimmunoprecipitation (lane 5; Figure 1B), deletion of PE16 (Δ3) inhibited 14-3-3ζ binding to c-Src (lane 7; Figure 1E). Notably, 14-3-3ζ dimerization may not be essential for 14-3-3ζ–c-Src–integrin-β3 complex formation, given that complex formation was normal with 14-3-3ζ lacking the critical residues 45 to 58 for 14-3-3ζ dimerization35 (Δ1, lane 5; Figure 1E).
The 14-3-3ζ–integrin-β3 interaction is mediated by the EL17 fragment on 14-3-3ζ and the KF7 fragment on integrin-β3
14-3-3ζ has been reported to bind to the phosphorylated TTT motif of integrin-β2.32 It is also capable of interacting with nonphosphorylated ligands.36 Because our findings implied a critical role for -749EATST753- on integrin-β3 for 14-3-3ζ binding (Figure 1D), we synthesized a TST motif containing the β3 peptide KEATSTF (KF7) to characterize the site on β3 that mediates 14-3-3ζ–integrin-β3 interactions. SPR measurements indicated that the association (Ka) and dissociation (Kd) rate constants, as well as the Rmax and equilibrium dissociation constant (KD) of the interaction between KF7 and recombinant 14-3-3ζ was 8.6 × 102 M−1s−1, 1.8 × 10−2 s−1, 581 resonance units (RU), and 2.1 × 10−5 M, respectively (Figure 2A). KF7pT1 (KEApTSTF) and KF7pT2 (KEATSpTF) also bound to 14-3-3ζ. The estimated Ka, Kd, Rmax, and KD for KF7pT1–14-3-3ζ interaction was 1.1 × 103 M−1s−1, 9.6 × 10−3 s−1, 681 RU, and 8.7 × 10−6 M, whereas the parameter for KF7pT2 was 3.7 × 103 M−1s−1), 1.3 × 10−2 s−1, 629 RU, and 3.4 × 10−6 M, respectively (supplemental Figure 2A-B). Further investigation found that KF7 interacted with the fragment EL17 on 14-3-3ζ, with an estimated Ka, Kd, Rmax, and KD of 8.2 × 102 M−1s−1, 9.8 × 10−3 s−1, 314 RU, and 1.2 × 10−5 M (Figure 2B). Scrambled KF7 peptide (TKSETFA, KF7scr) did not bind to immobilized 14-3-3ζ or EL17 (supplemental Figure 2C-D).
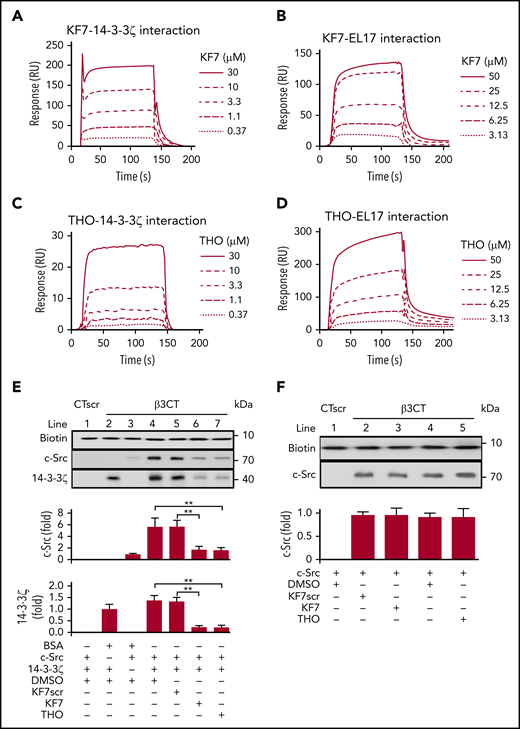
Interference with the 14-3-3ζ–c-Src–integrin-β3 complex by KF7 and THO. (A-D) SPR analysis of the interaction between KF7 or THO and 14-3-3ζ immobilized on Sensor Chip CM-5 and EL17 immobilized on Sensor Chip NTA. SPR sensorgrams of KF7 binding to 14-3-3ζ (A) and EL17 (B) or THO binding to 14-3-3ζ (C) and EL17 (D). (E) Western blot analysis of recombinant c-Src or 14-3-3ζ protein precipitated by biotin-conjugated β3CT in the presence of 0.1% DMSO, 30 μM KF7, 30 μM KF7 scrambled peptide (KF7scr), and 30 μM THO. (F) Western blot analysis of recombinant c-Src protein precipitated by biotin-conjugated WT β3CT in the presence of 100 μM KF7, KF7scr, THO or 0.1% DMSO. Data are means ± SD (n = 3). Statistical significance was determined by Student t test. **P < .01.
Interference with the 14-3-3ζ–c-Src–integrin-β3 complex by KF7 and THO. (A-D) SPR analysis of the interaction between KF7 or THO and 14-3-3ζ immobilized on Sensor Chip CM-5 and EL17 immobilized on Sensor Chip NTA. SPR sensorgrams of KF7 binding to 14-3-3ζ (A) and EL17 (B) or THO binding to 14-3-3ζ (C) and EL17 (D). (E) Western blot analysis of recombinant c-Src or 14-3-3ζ protein precipitated by biotin-conjugated β3CT in the presence of 0.1% DMSO, 30 μM KF7, 30 μM KF7 scrambled peptide (KF7scr), and 30 μM THO. (F) Western blot analysis of recombinant c-Src protein precipitated by biotin-conjugated WT β3CT in the presence of 100 μM KF7, KF7scr, THO or 0.1% DMSO. Data are means ± SD (n = 3). Statistical significance was determined by Student t test. **P < .01.
In the experimental screening from the library (Mendeley Data doi:10.17632/2kbjft8jd3.1) of ∼2000 small molecules (supplemental Figure 3; supplemental Table 3), we found that THO interacted with 14-3-3ζ with an estimated Ka, Kd, Rmax, and KD of 8.1 × 102 M−1s−1, 2.2 × 10−2 s−1, 123 RU, and 2.7 × 10−5 M, respectively, whereas that for THO-EL17 interaction was 4.3 × 102 M−1s−1, 3.5 × 10−2 s−1, 490 RU, and 8.2 × 10−5 M, respectively (Figure 2C-D). Neither KF7 nor THO directly interacted with immobilized Gα13, talin-1, PE16 peptide, integrin αIIb, or β3 cytoplasmic peptide (supplemental Figure 2E-H). We next tested the effects of KF7 and THO on the interaction of integrin β3CT with 14-3-3ζ or c-Src. In vitro peptide precipitation experiments showed that both KF7 and THO inhibited binding of 14-3-3ζ to integrin-β3CT at a concentration of 30 μM (Figure 2E). Baseline c-Src-β3CT interaction, which reflects the weak binding of c-Src with β3CT in unstimulated platelets,37 was not affected by KF7 or THO (Figure 2F).
Interference with the 14-3-3ζ–c-Src–integrin-β3 complex inhibits platelet aggregation and spreading without affecting fibrinogen binding
c-Src is primed for activation by direct interaction with an integrin-β tail,31 and upon activation of integrin, C-terminal–associated c-Src is critical for early outside-in signaling transduction and platelet aggregation.15 Because both KF7 and THO inhibited 14-3-3ζ–integrin-β3 interaction (Figure 2E), we next examined the effect of myristoylated-KF7 (myr-KF7) and THO on platelet aggregation and spreading. Both myr-KF7 (Figure 3A-C) and THO (Figure 3D-F), as well as myr-KF7pT1 and myr-KF7pT2 (supplemental Figure 4), dose dependently inhibited the platelet aggregation induced by 0.03 U/mL thrombin, 1 μg/mL collagen, and 1 μM anti-αIIb TM peptide (in the presence of 100 μg/mL fibrinogen). The anti-αIIb TM designer peptide directly targets the TM region of integrin αIIb, switches αIIbβ3 to its high-affinity state, and initiates αIIbβ3-mediated outside-in signaling.38,39 Anti-αIIb TM–induced platelet aggregation was suppressed by myr-KF7 and THO, but was not affected by the TXA2 receptor antagonist SQ29548 and the adenosine diphosphatase apyrase in the presence of fibrinogen. However, SQ29548 and apyrase suppressed anti-αIIb TM–induced platelet aggregation in the absence of fibrinogen (supplemental Figure 5). These results indicate that the TXA2 receptor and ADP-P2Y12 signaling are responsible for platelet secretion, whereas myr-KF7 and THO may act on αIIbβ3 outside-in signaling. Furthermore, THO and myr-KF7 did not significantly affect P-selectin expression on β3−/− platelets, although they significantly reduced P-selectin expression on WT platelets (supplemental Figure 6). This finding suggests that the observed myr-KF7 and THO inhibitory effects on platelet α-granule release are induced by αIIbβ3 outside-in signaling.27

Effects of myr-KF7 and THO on platelet’s aggregation, binding, and spreading. (A-C) Aggregation of washed human platelets (300 × 109/L) stimulated with thrombin (0.03 U/mL; A), collagen (1 μg/mL; B), or anti-αIIb TM peptide (1 μM; C) in the presence of myr-KF7 and its scrambled peptide myr-KF7scr (50 μM). (D-F) Aggregation of washed human platelets (300 × 109/L) stimulated with thrombin (0.03 U/mL; D), collagen (1 μg/mL; E), or anti-αIIb TM peptide (1 μM; F) in the presence of THO and 0.1% DMSO. (G) The effect of myr-KF7 and THO on platelet spreading. Washed human platelets (100 × 109/L) were preincubated with myr-KF7, myr-KF7scr, and THO at 50 μM; SQ29548 at 4 μM; apyrase at 0.5 U/mL; or eptifibatide (10 μM). The platelets were allowed to spread on immobilized fibrinogen at 37°C for 40 minutes and stained with FITC-conjugated phalloidin for immunofluorescence microscopy. The bar represents 10 μm. (H) ImageJ software was used to quantify the platelets and area of spread. *P < .05; ***P < .001. (I) The effect of myr-KF7 and THO on soluble fibrinogen binding to platelets. Washed human platelets (300 × 109/L) were preincubated with myr-KF7, myr-KF7scr, and THO at 50 μM. Binding of FITC-conjugated fibrinogen (100 μg/mL) to platelets was measured by flow cytometry. (J-L) The effect of 100 μM myr-KF7 and THO on 100 μM for both PAR4AP- and collagen-induced aggregation of GPIbα−/− platelets. (M) Effect of 100 μM myr-KF7 and THO on GPIbα−/− spreading of platelets on immobilized fibrinogen.Scale bar, 10 μm.
Effects of myr-KF7 and THO on platelet’s aggregation, binding, and spreading. (A-C) Aggregation of washed human platelets (300 × 109/L) stimulated with thrombin (0.03 U/mL; A), collagen (1 μg/mL; B), or anti-αIIb TM peptide (1 μM; C) in the presence of myr-KF7 and its scrambled peptide myr-KF7scr (50 μM). (D-F) Aggregation of washed human platelets (300 × 109/L) stimulated with thrombin (0.03 U/mL; D), collagen (1 μg/mL; E), or anti-αIIb TM peptide (1 μM; F) in the presence of THO and 0.1% DMSO. (G) The effect of myr-KF7 and THO on platelet spreading. Washed human platelets (100 × 109/L) were preincubated with myr-KF7, myr-KF7scr, and THO at 50 μM; SQ29548 at 4 μM; apyrase at 0.5 U/mL; or eptifibatide (10 μM). The platelets were allowed to spread on immobilized fibrinogen at 37°C for 40 minutes and stained with FITC-conjugated phalloidin for immunofluorescence microscopy. The bar represents 10 μm. (H) ImageJ software was used to quantify the platelets and area of spread. *P < .05; ***P < .001. (I) The effect of myr-KF7 and THO on soluble fibrinogen binding to platelets. Washed human platelets (300 × 109/L) were preincubated with myr-KF7, myr-KF7scr, and THO at 50 μM. Binding of FITC-conjugated fibrinogen (100 μg/mL) to platelets was measured by flow cytometry. (J-L) The effect of 100 μM myr-KF7 and THO on 100 μM for both PAR4AP- and collagen-induced aggregation of GPIbα−/− platelets. (M) Effect of 100 μM myr-KF7 and THO on GPIbα−/− spreading of platelets on immobilized fibrinogen.Scale bar, 10 μm.
Myr-KF7 and THO also significantly inhibited platelet spreading on immobilized fibrinogen (Figure 3G-H), suggesting that the 14-3-3ζ–c-Src–integrin-β3 complex plays a vital role in platelet lamellipodia formation and spreading. However, the number of platelets attached to the coverslips was not altered by myr-KF7 and THO, whereas SQ29548 and apyrase reduced the attachment significantly (Figure 3G-H). To investigate whether interference with the 14-3-3ζ–c-Src–integrin-β3 complex affects integrin inside-out signaling, we examined the effects of myr-KF7 and THO on soluble fibrinogen binding to platelets. Flow cytometry results showed no significant inhibitory effect of myr-KF7 and THO on soluble fibrinogen binding, with or without added agonists (Figure 3I; supplemental Figure 7).
It has been widely accepted that 14-3-3ζ associates with the GPIb-IX complex40,41 and plays an important role in VWF-mediated GPIb-IX signaling transduction.42-44 Indeed, we found that a high dose of THO (200 μM) inhibited 1.25 mg/mL ristocetin-induced platelet agglutination/aggregation in human platelet-rich plasma, and this inhibitory effect was further enhanced in the presence of 1 mM eptifibatide (supplemental Figure 8). However, myr-KF7 or THO also exhibited an inhibitory effect on both GPIbα−/− (Figure 3J-L) and IL4Rα/GPIbα-transgenic (N-terminal domain of GPIbα replaced with interleukin-4 receptor-α) platelet aggregation (supplemental Figure 9). Furthermore, myr-KF7 and THO suppressed GPIbα−/− platelet spreading on immobilized fibrinogen (Figure 3M), indicating that these inhibitors affect αIIbβ3 outside-in signaling, independent of GPIb-IX.
Myr-KF7 and THO alters the platelet integrin-β3 outside-in signaling pathway
c-Src activation is critical for early outside-in signaling transduction. To assess whether the 14-3-3ζ–c-Src–integrin-β3 complex mediates integrin-induced activation of c-Src, we measured the phosphorylation of c-Src at Tyr416. The expected elevated Tyr phosphorylation of c-Src after platelet attachment to fibrinogen was attenuated when the platelets were treated with myr-KF7 and THO (Figure 4A-B), suggesting inhibition of integrin-dependent c-Src activation. Myr-KF7 and THO also inhibited the c-Src activation induced by anti-αIIb TM in the presence of shear forces, which was unaffected by SQ29548 and apyrase (supplemental Figure 10A-B). These findings indicate that 14-3-3ζ plays important roles in regulating integrin-dependent c-Src activation. The ρ family of small GTPases, such as RhoA and Rac1, are critical in regulating integrin-induced cytoskeletal reorganization.45 Because c-Src activation negatively regulates RhoA activation in platelets,15 we examined activation of RhoA and Rac1 during platelet spreading on immobilized fibrinogen in the presence or absence of myr-KF7 and THO. Myr-KF7 and THO significantly accelerated platelet RhoA (Figure 4C-D) and Rac1 (supplemental Figure 11) activation during spreading. Akt phosphorylation, an important downstream effector of platelet integrin outside-in signaling transduction,46 was also inhibited dose dependently by myr-KF7 and THO (Figure 4E-J). Thus, 14-3-3ζ–integrin-β3 interaction appears to be critical for mediating integrin signaling to c-Src, RhoA, and Rac1, sequentially regulating Akt phosphorylation and platelet activation and aggregation.

Effects of myr-KF7 and THO on platelet c-Src, RhoA, and AKT activation. (A-B) Washed human platelets pretreated with 50 μM myr-KF7, myr-KF7scr, THO, and 0.1% DMSO were allowed to adhere to immobilized fibrinogen and then solubilized at the indicated time points. Proteins from lysates were immunoblotted with antibodies to Src pY416 and c-Src. (C-D) GTP-bound RhoA was measured by association with the RhoA-binding domain of Rhotekin (GST-RBD). Washed human platelets (500 × 109/L) pretreated with myr-KF7 (E-F) or THO (G-H) were activated by 0.03 U/mL thrombin or 1 μg/mL collagen, and the lysates were immunoblotted with AKT1 pS473 or AKT1 antibody. (I-J) Platelets pretreated with myr-KF7 or THO were activated by anti-αIIb TM peptide (1 μM), and the lysates were immunoblotted with AKT1 pS473 or AKT1 antibody. All data are expressed as means ± SD (n = 3). Statistical significance was determined by Student t test. *P < .05; **P < .01.
Effects of myr-KF7 and THO on platelet c-Src, RhoA, and AKT activation. (A-B) Washed human platelets pretreated with 50 μM myr-KF7, myr-KF7scr, THO, and 0.1% DMSO were allowed to adhere to immobilized fibrinogen and then solubilized at the indicated time points. Proteins from lysates were immunoblotted with antibodies to Src pY416 and c-Src. (C-D) GTP-bound RhoA was measured by association with the RhoA-binding domain of Rhotekin (GST-RBD). Washed human platelets (500 × 109/L) pretreated with myr-KF7 (E-F) or THO (G-H) were activated by 0.03 U/mL thrombin or 1 μg/mL collagen, and the lysates were immunoblotted with AKT1 pS473 or AKT1 antibody. (I-J) Platelets pretreated with myr-KF7 or THO were activated by anti-αIIb TM peptide (1 μM), and the lysates were immunoblotted with AKT1 pS473 or AKT1 antibody. All data are expressed as means ± SD (n = 3). Statistical significance was determined by Student t test. *P < .05; **P < .01.
Myr-EL17 inhibits the destabilizing effect of myr-KF7 and THO on the 14-3-3ζ–c-Src–integrin-β3 complex
The EL17 fragment on 14-3-3ζ mediated the 14-3-3ζ–integrin-β3 interaction and was the cognate target of KF7 and THO (Figure 2B,D). Blocking EL17 interfered with the 14-3-3ζ–c-Src–integrin-β3 complex and was shown to be an effective method of preventing integrin outside-in signaling and platelet aggregation (Figures 3 and 4). We further investigated whether the 14-3-3ζ–c-Src–integrin-β3 complex destabilized by KF7 or THO could be restored by EL17. Myristoylated-EL17 (myr-EL17), but not the myristoylated-scrambled peptide (myr-EL17scr), dose dependently interfered with the inhibitory effect of myr-KF7 (Figure 5A-C) and THO (Figure 5D-F) on platelet aggregation. In addition, pretreatment of platelets with myr-KF7 or THO restored spreading on immobilized fibrinogen in the presence of myr-EL17 (Figure 5G-H). The agonist-induced phosphorylation of c-Src Tyr416 was reduced in the presence of myr-KF7 and THO but was rescued when myr-EL17 was added simultaneously (Figure 5I). In vitro peptide precipitation results also showed that binding of 14-3-3ζ or c-Src to integrin-β3CT returned to levels comparable with those in the control groups in the presence of EL17 (Figure 5J). However, myr-EL17 itself had no effect on agonist-induced platelet aggregation (supplemental Figure 12A) or 14-3-3ζ and c-Src binding to integrin-β3CT in vitro (supplemental Figure 13), whereas myristoylated-PE16 (myr-PE16) inhibited platelet aggregation at high concentrations (supplemental Figure 12B). Immunoprecipitation demonstrated that the 14-3-3ζ–c-Src–integrin-β3 complex, destabilized in platelets by myr-KF7 and THO, was restored by myr-EL17 (supplemental Figure 14).

Myr-EL17 restored the platelet function affected by myr-KF7 and THO. (A-F) Washed human platelets (300 × 109/L) were preincubated with myr-KF7 (A-C) or THO (D-F) with various concentrations of myr-EL17. Aggregations were elicited by thrombin (0.03 U/mL), collagen (1 μg/mL), or anti-αIIb TM peptide (1 μM). (G) Washed human platelets (100 × 109/L) were preincubated with 50 μM myr-KF7 or THO in the presence of 100 μM myr-EL17 or myr-EL17scr (both 100 μM). After 40 minutes of spreading on immobilized fibrinogen at 37°C, the platelets were stained with FITC-conjugated phalloidin for immunofluorescence microscopy. myr-EL17 and myr-EL17scr (both 100 μM) and 10 μM eptifibatide were used as the controls. The bar represents 10 μm. (H) Image J software was used to quantify the number of platelets and area of spread. (I) Platelets preincubated with 30 μM myr-KF7 and THO in the presence of 90 μM myr-EL17 or myr-EL17scr were activated by thrombin (0.03 U/mL), collagen (1 μg/mL), or anti-αIIb TM peptide (1 μM), and the cell lysates were immunoblotted with Src pY416 and c-Src antibodies. (J) Western blot analysis of the recombinant c-Src and 14-3-3ζ precipitated by biotin-conjugated β3CT in the presence of 30 μM KF7 or THO, with or without 90 μM EL17. All data are expressed as means ± SD (n = 3). *P < .05; **P < .01; ***P < .001.
Myr-EL17 restored the platelet function affected by myr-KF7 and THO. (A-F) Washed human platelets (300 × 109/L) were preincubated with myr-KF7 (A-C) or THO (D-F) with various concentrations of myr-EL17. Aggregations were elicited by thrombin (0.03 U/mL), collagen (1 μg/mL), or anti-αIIb TM peptide (1 μM). (G) Washed human platelets (100 × 109/L) were preincubated with 50 μM myr-KF7 or THO in the presence of 100 μM myr-EL17 or myr-EL17scr (both 100 μM). After 40 minutes of spreading on immobilized fibrinogen at 37°C, the platelets were stained with FITC-conjugated phalloidin for immunofluorescence microscopy. myr-EL17 and myr-EL17scr (both 100 μM) and 10 μM eptifibatide were used as the controls. The bar represents 10 μm. (H) Image J software was used to quantify the number of platelets and area of spread. (I) Platelets preincubated with 30 μM myr-KF7 and THO in the presence of 90 μM myr-EL17 or myr-EL17scr were activated by thrombin (0.03 U/mL), collagen (1 μg/mL), or anti-αIIb TM peptide (1 μM), and the cell lysates were immunoblotted with Src pY416 and c-Src antibodies. (J) Western blot analysis of the recombinant c-Src and 14-3-3ζ precipitated by biotin-conjugated β3CT in the presence of 30 μM KF7 or THO, with or without 90 μM EL17. All data are expressed as means ± SD (n = 3). *P < .05; **P < .01; ***P < .001.
Dynamics of c-Src and 14-3-3ζ binding to platelet integrin-β3
It has been reported that binding of c-Src to integrin-β3 increases progressively after αIIbβ3 activation.37 However, the underlying mechanism remains unknown. We performed platelet lysate coimmunoprecipitation to investigate the temporal progression of 14-3-3ζ–c-Src–integrin-β3 complex formation during platelet aggregation. We found that c-Src and 14-3-3ζ coimmunoprecipitated with integrin-β3 by the β3 antibody but not by control IgG (Figure 6A). Binding of c-Src and 14-3-3ζ to integrin-β3 in anti-αIIb TM–activated platelets increased significantly compared with resting platelets, suggesting that constitutive binding of c-Src with integrin-β3 is not sufficient to trigger integrin outside-in signaling and initiate platelet activation and aggregation. Similar to in vitro experiments, myr-KF7 and THO interfered with the formation of the 14-3-3ζ–c-Src–integrin-β3 complex in human platelets (Figure 6A-B) and the 14-3-3ζ–integrin-β3 complex in GPIbα−/− platelets (supplemental Figure 15). We also found that, during platelet aggregation, binding of both c-Src and activated c-Src to integrin-β3 initially increased, then subsequently decreased, with continuous stimulation. The binding of 14-3-3ζ to integrin-β3 showed a similar trend. However, the addition of THO significantly decreased the binding of both 14-3-3ζ and c-Src to integrin-β3 (Figure 6C-D). Kindlin-3 is essential for integrin activation and platelet aggregation, and mutations of the membrane-distal NxxY motif of the integrin-β3 tails (Y759A) and ST752/753AA abolished kindlin-3 but not talin binding.47 THO (supplemental Figure 16A) and myr-KF7 (supplemental Figure 16B) did not affect kindlin-3 binding to integrin-β3 during platelet activation.
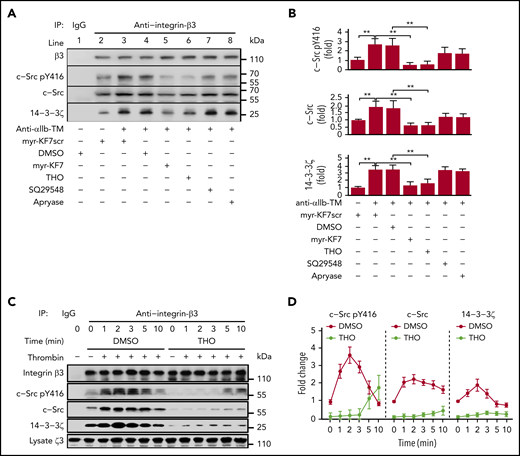
Dynamics of 14-3-3ζ and c-Src binding to integrin-β3 and the inhibiting effect of myr-KF7 and THO. (A-B) Washed human platelets (1000 × 109/L) were preincubated with myr-KF7 or myr-KF7scr at 50 μM, THO at 100 μM, SQ29548 at 5 μM, and apyrase at 1 U/mL in the presence of 100 μg/mL fibrinogen, and the platelets were activated by 1 μM anti-αIIb TM. Platelet lysates were harvested in 30 seconds and immunoprecipitated with integrin-β3 antibody, and the immunoprecipitates were blotted with integrin-β3, c-Src pY416, c-Src, and 14-3-3ζ antibodies. (C-D) Washed human platelets (1000 × 109/L), preincubated with or without 100 μM THO, were activated by thrombin (0.03 U/mL). Then, the lysates were harvested at various time points, immunoprecipitated with integrin-β3 antibody and blotted with integrin-β3, Src pTyr416, c-Src, and 14-3-3ζ antibodies. All data are expressed as means ± SD (n = 3). Statistical significance was determined by Student t test. *P < .05; **P < .01.
Dynamics of 14-3-3ζ and c-Src binding to integrin-β3 and the inhibiting effect of myr-KF7 and THO. (A-B) Washed human platelets (1000 × 109/L) were preincubated with myr-KF7 or myr-KF7scr at 50 μM, THO at 100 μM, SQ29548 at 5 μM, and apyrase at 1 U/mL in the presence of 100 μg/mL fibrinogen, and the platelets were activated by 1 μM anti-αIIb TM. Platelet lysates were harvested in 30 seconds and immunoprecipitated with integrin-β3 antibody, and the immunoprecipitates were blotted with integrin-β3, c-Src pY416, c-Src, and 14-3-3ζ antibodies. (C-D) Washed human platelets (1000 × 109/L), preincubated with or without 100 μM THO, were activated by thrombin (0.03 U/mL). Then, the lysates were harvested at various time points, immunoprecipitated with integrin-β3 antibody and blotted with integrin-β3, Src pTyr416, c-Src, and 14-3-3ζ antibodies. All data are expressed as means ± SD (n = 3). Statistical significance was determined by Student t test. *P < .05; **P < .01.
Interference with the formation of the 14-3-3ζ–c-Src–integrin-β3 complex inhibits thrombus growth without impairing hemostasis
To investigate the antithrombotic potential and bleeding risk of THO, mice were orally administered THO 2 hours before the experiments that followed. THO gavage (100 mg/kg) significantly prolonged FeCl3-induced carotid artery thrombus occlusion time from 322 ± 25 to 653 ± 46 seconds (Figure 7A). Tail thrombi induced by 40 mg/kg IP-injected carrageenan were markedly reduced by THO. Compared with the thrombus length in the control (3.62 ± 0.37 cm), the thrombus length in the THO-treated mice was significantly reduced, with 33 mg/kg THO (2.37 ± 0.33 cm), 100 mg/kg THO (1.14 ± 0.39 cm), and 100 mg/kg clopidogrel (1.42 ± 0.44 cm; Figure 7B). Three, 6, and 12 hours after the gavage, tail-bleeding time in the THO group (∼100 seconds) was not affected compared with that in the control group (Figure 7C), whereas clopidogrel administration at 100 mg/kg increased time to ∼800, 600, and 500 seconds, respectively. We further explored the effect of THO on liver (Figure 7D) and brain bleeding (Figure 7E-F). The number of red blood cells (RBCs) in the peritoneal lavage of the mice did not change significantly, compared with the counts in the THO and control groups (∼1 × 105/μL; n = 8). Whereas the amount of RBCs increased to ∼1.2 × 106/μL, 1.1 × 106/μL, and 1.0 × 106/μL after administration of 10 mg/kg clopidogrel for 3, 6, and 12 hours, respectively (Figure 7D). To investigate the effect on brain hemostasis, we used a model of needle-induced intracerebral hemorrhage.30 Compared with vehicle-treated mice, the cerebral hematoma area was significantly increased (by 4.5-fold) in the mice that received 10 mg/kg clopidogrel, whereas the mice that received 100 mg/kg THO exhibited no significant difference (Figure 7E-F). Phosphorylation of c-Src at Tyr416 represents the early stage of integrin outside-in signaling, which mediates platelet spreading.15 Interference with the formation of the 14-3-3ζ–c-Src–integrin-β3 complex by myr-KF7 and THO inhibited integrin-dependent activation of c-Src (Figure 4A-B) and increased activation of RhoA (Figure 4C-D) and Rac1 (supplemental Figure 11). As this small-GTPase–mediated signaling is important for cell retraction and primary hemostasis,34,45,48,49 we next investigated whether myr-KF7 and THO can affect platelet clot retraction. Interestingly, myr-KF7 and THO did not inhibit, but slightly accelerated thrombin (Figure 7G-H), and U46619 (supplemental Figure 17) induced clot retraction within 30 minutes. The results suggest that interference with formation of the 14-3-3ζ–c-Src–integrin-β3 complex by THO suppresses thrombosis but has no significant bleeding side effects.

Interference in the formation of the 14-3-3ζ–c-Src–integrin-β3 complex in thrombosis without affecting hemostasis. (A) Oral administration of THO (100 mg/kg) alleviated 10% of the FeCl3-induced occlusive carotid artery thrombosis in mice. (B) Oral administration of THO (100 mg/kg) reduced the mouse tail thrombus induced by 40 mg/kg carrageenan. (C) Tail transection bleeding times of mice orally receiving THO (100 mg/kg) or clopidogrel (100 mg/kg) at 3, 6, and 12 hours after administration. (D) The number of RBCs in peritoneal lavage after a calibrated injury of the liver in mice treated with THO (100 mg/kg) or clopidogrel (10 mg/kg) at 3, 6, and 12 hours after administration. (E-F) Effect of oral administration of THO (100 mg/kg) or clopidogrel (100 mg/kg) on bleeding in the mouse brain. Photographs (E) and relative quantification (F) of the areas of brain bleeding. (G-H) In vitro effects of 100 μM THO, myr-KF7, and their relative controls on clot retraction of human platelet-rich plasma in the presence of 0.03 U/mL thrombin. Data are expressed as means ± SD (n = 3). *P < .05; **P < .01.
Interference in the formation of the 14-3-3ζ–c-Src–integrin-β3 complex in thrombosis without affecting hemostasis. (A) Oral administration of THO (100 mg/kg) alleviated 10% of the FeCl3-induced occlusive carotid artery thrombosis in mice. (B) Oral administration of THO (100 mg/kg) reduced the mouse tail thrombus induced by 40 mg/kg carrageenan. (C) Tail transection bleeding times of mice orally receiving THO (100 mg/kg) or clopidogrel (100 mg/kg) at 3, 6, and 12 hours after administration. (D) The number of RBCs in peritoneal lavage after a calibrated injury of the liver in mice treated with THO (100 mg/kg) or clopidogrel (10 mg/kg) at 3, 6, and 12 hours after administration. (E-F) Effect of oral administration of THO (100 mg/kg) or clopidogrel (100 mg/kg) on bleeding in the mouse brain. Photographs (E) and relative quantification (F) of the areas of brain bleeding. (G-H) In vitro effects of 100 μM THO, myr-KF7, and their relative controls on clot retraction of human platelet-rich plasma in the presence of 0.03 U/mL thrombin. Data are expressed as means ± SD (n = 3). *P < .05; **P < .01.
Discussion
In this study, we identified a direct interaction between 2 adaptor molecules (14-3-3ζ and c-Src) and a direct interaction between 14-3-3ζ and integrin-β3. We demonstrated that 14-3-3ζ plays a critical role in regulating integrin-dependent c-Src activation and that the 14-3-3ζ–c-Src–integrin-β3 complex is critical in platelet outside-in signaling. Notably, through compound library screening, we identified THO as a novel inhibitor of 14-3-3ζ that interferes with the formation of the 14-3-3ζ–c-Src–integrin-β3 complex and inhibits thrombosis without causing bleeding. Thus, interference with the formation of the 14-3-3ζ–c-Src–integrin-β3 complex may be a promising antithrombotic strategy.
The integrin adhesome comprises a group of proteins that form focal adhesions, in which protein components directly or indirectly interact with β-integrin tails.50 At least 28 adaptor proteins, including 6 structural adaptors, 14 scaffolding adaptors, and 8 catalytic adaptors, have been identified to bind to integrin-β3CT.14 The adaptor-adaptor interactions are interconnected to regulate the assembly of the complex structural and signaling platform of the focal adhesion. Previous reports have demonstrated that 14-3-3ζ binds to integrin-β2.32,33,51 We found that 14-3-3ζ also bound to integrin-β3 via the interaction mediated by the -112ESKVFYLKMKGDYYRYL128- fragment on 14-3-3ζ and the -749EATST753- fragment on integrin-β3. Unexpectedly, 14-3-3ζ was found to interact directly with c-Src, a catalytic adaptor of integrin, and the interaction was mediated by both the -165PIRLGLALNFSVFYYE180- fragment on 14-3-3ζ and the SH2 domain on c-Src. c-Src is known to bind to integrin-β3CT via the interaction mediated by both the SH3 domain on c-Src and the -759YRGT762- fragment on integrin-β3.32,52 The interconnectedness of 14-3-3ζ–integrin-β3, integrin-β3–c-Src, and c-Src–14-3-3ζ interactions forms the scaffold for the 14-3-3ζ–c-Src–integrin-β3 complex, as reported herein (Figure 1). Previous reports have shown that adaptor molecules assemble into focal adhesion sites in a sequential manner.53-56 It appears that 14-3-3ζ synergizes with c-Src to bind integrin-β3 (Figures 1D-E and 2E) and promote c-Src activation (Figure 6). It is possible that 14-3-3ζ forms a bridge from c-Src to integrin-β3 to allow continued platelet spreading and inhibit clot retraction, despite calpain-mediated integrin-β3 C-terminal cleavage (at Tyr759).34,57
The 14-3-3 family of 27- to 32-kDa conserved acidic proteins has 7 isotypes (α/β, γ, τ/θ, ε, η, σ, and ζ/δ) that are expressed in the cytoplasm of eukaryotic cells.36,58 They are crucial regulators of intracellular signaling pathways.43 Platelets contain high levels of the ζ, β, and γ isoforms and lower levels of the ε and η isoforms.59,60 14-3-3ζ has been reported to bind GPIb and regulate GPIb-IX signaling.42,44 The sequestration of 14-3-3ζ by GPIb-IX may affect integrin-induced cytoskeletal reorganization.60 In our experiments, GPIb-IX–mediated platelet aggregation was attenuated by 14-3-3ζ EL17 motif-based inhibitor (THO; supplemental Figure 8), suggesting that 14-3-3ζ mediated GPIb-IX signaling was affected. However, KF7 or THO also suppressed the aggregation (Figure 3J-L) and integrin-dependent spreading (Figure 3M) of GPIb-deficient platelets. This finding establishes the direct and integral role of 14-3-3ζ in platelet integrin biology. Disruption of 14-3-3ζ–integrin-β3 interaction, by either KF7 or THO, inhibited formation of the 14-3-3ζ–c-Src–integrin-β3 complex, thus attenuating outside-in signaling transduction and platelet aggregation (Figures 3, 4, and 6). The EL17 fragment in 14-3-3ζ is highly conserved among all the 14-3-3 isotypes in platelets, and therefore 14-3-3 or its EL17 fragment may be a promising target for antiplatelet drugs and thrombotic therapy.
c-Src family kinases (SFKs) play a central role in mediating the response of platelets to vascular injury by transmitting activation signals from platelet surface receptors, which is crucial for thrombus growth and stability.61 The most abundant SFK in human platelets is c-Src,62 which is an important positive regulator of β3-integrin signaling.63 c-Src binds to the C-terminal region of β3-integrin and is thought to be important in integrin outside-in signaling.34,52,64 The SFK inhibitor dasatinib suppresses platelet activation and causes ubiquitous bleeding in both rodent models and humans.61,65-68 In unstimulated platelets, c-Src may weakly bind to integrin-β3 and exist in a moderately activated state37 (Figure 6), potentially priming the platelet to respond to vascular injury. However, after platelet activation, c-Src is rapidly mobilized to the integrin-β337 (Figure 6A-D). The mechanism behind this phenomenon remains unclear, but further elucidation of this process may lead to novel agents that block specific platelet functions that are essential for thrombus growth but cause few adverse bleeding effects.61 Herein, we identified 14-3-3ζ as a novel interaction partner of c-Src. Importantly, 14-3-3ζ synergizes c-Src to integrin-β3CT and thus regulates c-Src activation (Figure 1). Considering that c-Src initiates and propagates signals from integrin-β3 to play a central role in platelet functions, interference with the interaction of 14-3-3ζ–integrin-β3, thus inhibiting the formation and stabilization of the 14-3-3ζ–c-Src–integrin-β3 complex, may provide a unique opportunity to specifically suppress outside-in signaling without affecting integrin ligation and without impairing hemostasis. Indeed, disruption of the 14-3-3ζ–integrin-β3 interaction, by either the KF7 or EL17 competitor compound (THO) selectively suppressed outside-in signaling to inhibit platelet spreading, aggregation, and thrombus formation without affecting integrin ligation, clot retraction, and bleeding.
VWF deficiency may lead to a bleeding disorder.69,70 However, in certain concentrations, inhibitors that block the interaction between VWF and GPIb-IX may inhibit thrombus formation without significantly prolonging bleeding time.71-73 According to previous studies, bioavailability of isoflavones in the blood after oral administration is low,74,75 a single dose of 20 mg/kg isoflavone (similar structure with THO) showed an apparent peak plasma concentration of 5 to 11 μM.76 It is conceivable that the peak plasma concentration of THO after oral administration (100 mg/kg) would also be relatively low. The plasma concentration of THO may therefore be sufficient to inhibit thrombosis but not to reach the threshold that causes significant bleeding disorders. This concept is also consistent with an early study that showed that an antithrombotic effect could be achieved before bleeding occurs.77 Interfering with αIIbβ3 early outside-in signaling may exert an antithrombotic effect without causing a significant bleeding side effect.18 The ρ family of small GTPases, such as RhoA and Rac1, which is activated in late αIIbβ3 outside-in signaling, plays an important role in thrombosis and hemostasis.49,78 However, in our experiments, clot retraction was slightly accelerated in the presence of THO (Figure 7G), which was probably related to preserved fibrinogen binding ability (Figure 3I; supplemental Figure 7) and increased RhoA (Figure 4)/Rac1 (supplemental Figure 11) activation. This accelerated clot retraction may have partially compensated for the inhibitory effect of THO on GPIb and αIIbβ3, and thus no significant bleeding occurred.
In summary, our study provides a conceptual advance by revealing a novel c-Src partner, 14-3-3ζ, and the presence of a 14-3-3ζ–c-Src–integrin-β3 complex scaffold in platelets that regulates integrin signaling. We demonstrated that 14-3-3ζ synergizes c-Src to integrin-β3, induces massive mobilization of c-Src to integrin-β3 during platelet activation, and likely regulates c-Src activation. The discovery of the 14-3-3ζ–c-Src–integrin-β3 association may form a conceptual basis for selectively inhibiting outside-in signaling without perturbing the ligand binding of integrins. Based on this new concept, we found novel antithrombotic agents that contain potent ability to inhibit thrombosis without the adverse effect of bleeding and that provide a novel avenue for the development of antithrombotic therapies.
Original data may be obtained by e-mail request to the corresponding authors: nih@smh.ca and rlai@kiz.ac.cn.
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Acknowledgments
This work was supported by grants from the Chinese Academy of Sciences (XDB31000000, Y802B81201, and XDA12020334); the Youth Innovation Promotion Association (QYZDJ-SSW-SMC012); the National Science Foundation of China (331372208, 31640071, 31770835 and 31801975); Yunnan Province (2015HA023); the Biological Resources Programme, Chinese Academy of Sciences (KFJ-BRP-008); the Heart and Stroke Foundation of Ontario, Canada (G-17-0018663); and the Canadian Institutes of Health Research Foundation (389035).
Authorship
Contribution: C.S. performed the experiments, created the figures, analyzed the data, and wrote the paper; M.L., R.X., and G.W. performed the experiments, created the figures, analyzed the data, and provided valuable suggestions; J.L., P.C., J.M., Q.L., and Z.D. performed experiments and provided valuable suggestions; W.M., F.Z.D., D.T.M., and Z.Z. provided valuable suggestions and helped in writing the manuscript; and H.N. and R.L. designed the research, analyzed the data and wrote the manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Heyu Ni, LKSKI-Keenan Research Centre, 209 Victoria St, Toronto, ON M5B 1W8, Canada; e-mail: nih@smh.ca; and Ren Lai, Kunming Institute of Zoology, Chinese Academy of Sciences, 32 Jiaochang Donglu, Kunming 650223, Yunnan, China; e-mail: rlai@mail.kiz.ac.cn.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal