

Key Points
The mutational landscape of DTFL is highly related to nodal FL but harbors fewer multiple/biallelic mutations in KMT2D.
The immune microenvironment of DTFL is distinct from nodal FL and characterized by a chronic inflammation gene signature.

Abstract
Duodenal-type follicular lymphoma (DTFL) is a rare and highly indolent follicular lymphoma (FL) variant. It is morphologically and immunophenotypically indistinguishable from typical FL, characterized by restricted involvement of intestinal mucosa, and lacks extraintestinal manifestations. The molecular determinants of this distinct clinical behavior are largely unknown. Thirty-eight diagnostic biopsies from patients with DTFL were evaluated. The 10-year overall survival rate was 100% in clinically evaluable patients (n = 19). We compared the targeted mutation profile of DTFL (n = 31), limited-stage typical FL (LSTFL; n = 17), and advanced-stage typical FL (ASTFL; n = 241). The mutation frequencies of recurrently mutated genes, including CREBBP, TNFRSF14/HVEM, and EZH2 were not significantly different. However, KMT2D was less commonly mutated in DTFL (52%) and LSTFL (24%) as compared with ASTFL (79%). In ASTFL, 41% of KMT2D-mutated cases harbored multiple mutations in KMT2D, as compared with only 12% in LSTFL (P = .019) and 0% in DTFL (P < .0001). Whole exome and targeted sequencing of DTFL revealed high mutation frequencies of EEF1A1 (35%) and HVCN1 (22%). We compared the immune microenvironment gene expression signatures of DTFL (n = 8) and LSTFL (n = 7). DTFL clearly separated from LSTFL by unsupervised, hierarchical clustering of 147 chemokines and cytokines and was enriched for a chronic inflammation signature. In conclusion, the mutational landscape of DTFL is highly related to typical FL. The lower frequency of multiple mutations in KMT2D in DTFL and LSTFL indicates an increasing selection pressure for complete KMT2D loss in ASTFL pathogenesis. The highly dissimilar immune microenvironment of DTFL suggests a central role in the biology of this disease.
Introduction
Follicular lymphoma (FL) comprises a heterogeneous group of diseases with variable clinical behavior. Typical FL most commonly presents with widespread lymphadenopathy and eventually secondary involvement of extranodal sites (ie, advanced-stage disease). While advanced-stage typical FL (ASTFL) is still considered an incurable disease,1 a smaller subset of patients with typical FL present with limited-stage disease (limited-stage typical FL [LSTFL]) and may be cured with radiotherapy (RT). The updated 2016 World Health Organization classification also recognizes rare nontypical FL variants as definitive entities, such as pediatric-type follicular lymphoma, which has invariably benign clinical behavior2 and is molecularly distinguished by lack of BCL2 rearrangements,3 absence of mutations in epigenetic modifier genes, but high frequency of distinct gene mutations that activate the MAPK pathway.4,5
Duodenal-type FL (DTFL) is another rare variant of FL, characterized by its restriction to the intestine (often the duodenum) and a remarkably low frequency of progression and dissemination.6,7 Despite its unique clinical presentation and highly indolent behavior, the tumor architecture and cellular features of DTFL are indistinguishable from those of typical FL by routine histology and immunohistochemistry alone.2 Molecularly, DTFL is also well known to harbor BCL2 translocations as seen in typical FL. To identify the determinants of this distinct disease biology, we performed a comprehensive analysis of FL cell-intrinsic alterations (ie, the mutational landscape) and cell-extrinsic perturbations (ie, the immune microenvironment) in a larger series of DTFL.
Methods
Patients
The DTFL cohort: We stringently defined DTFL as FL occurring in any part of the gastrointestinal tract without evidence of extraintestinal manifestation. Cases with infiltration beyond the submucosa or involvement of mesenteric lymph nodes (LNs) were excluded. A total of 39 intestinal FL samples were identified through the Kiel Lymph Node Registry (n = 20) and the Dana-Farber Cancer Institute/Massachusetts General Hospital (MGH) (n = 19). One case was excluded from the DTFL cohort because of nodal involvement by histopathological review (n = 1). Clinical follow-up data were available for 19/38 patients of the final DTFL cohort (supplemental Figure 1; available on the Blood Web site).
The LSTFL cohort: A total of 17 samples were available from Dana-Farber Cancer Institute/MGH, all confirmed to be Ann Arbor stage I or II at the time of initial diagnosis by complete clinical staging including positron emission tomography/computed tomography. Clinical follow-up was available for 15 patients.
The ASTFL cohort: We included 241 cases with available targeted DNA sequencing data from our previous study,8 including patients from the GLSG2000 trial (n = 137) and a population-based registry of British Columbia (n = 104). All patients had nodal FL, symptomatic, advanced stage disease, and received rituximab-containing immunochemotherapy as first-line treatment.
All diagnostic lymphoma biopsies were reviewed by expert hematopathologists (A.L., R.D.G., and W.K.). All research described herein was approved by the respective research ethics boards.
Pathology
Next-generation sequencing
Deep sequencing using customized hybrid capture was performed as previously described.8,10 DTFL cases from the Kiel Lymph Node Registry and ASTFL cases were sequenced with a previously reported bait set covering 108 genes/regions.8 DTFL and LSTFL cases from MGH were sequenced using a modified bait set covering 104 genes. The overlap between both bait sets consisted of 82 coding genes (supplemental Table 1). Whole-exome sequencing (WES) libraries of tumor/germ-line pairs of DTFL were prepared, sequenced, and analyzed at the Broad Institute as previously described.4 A variant allele frequency cutoff of 5% was used to call nonsilent variants.
Profiling of the immune microenvironment
Digital multiplexed gene expression profiling of formalin-fixed and paraffin-embedded biopsy specimens was performed as previously described.11 RNA from formalin-fixed and paraffin-embedded samples was isolated using the ExpressArt FFPE Clear RNAready kit (AmpTec). RNA (300 ng) was assayed with the nCounter PanCancer Immune Profiling Panel (NanoString Technologies) according to the manufacturer’s protocol.
Statistics
The R software environment for statistical computing (https://www.r-project.org) was used to calculate statistics and generate figures (gplots and ggplots package) and Kaplan-Meier estimates (package survival). Lollipop plots were generated with MutationMapper v1.0.1 (http://www.cbioportal.org/). NanoString data were normalized (geNorm) and analyzed for differential expression with the nSolver analysis software (version 3.0) using the Benjamini-Hochberg method to control the false discovery rate (FDR). Tumor cell abundance was calculated using nSolver nCounter advanced analysis software (version 1.1.4). Cell type abundance was measured as the average log2-scale expression of its characteristic genes (supplemental Table 8). Tumor-infiltrating lymphocyte score is the average of the B-cell, T-cell, CD45, macrophage, and cytotoxic cell scores. Gene set enrichment analysis (http://www.broad.mit.edu/gsea/) was run from the command line using a preranked input gene list (preranked by log2 fold change calculated with the nSolver analysis software). Mutation frequencies between cohorts were compared using 2-sided Fisher’s exact test, and results were FDR corrected.
Results
Clinical characteristics, histopathology, and treatment outcome
The clinical characteristics of the DTFL (n = 38), LSTFL (n = 17), and ASTFL (n = 241) cohorts are summarized in Table 1. The majority of DTFLs were located within the duodenum (22/38, 58%). Histological review of diagnostic DTFL biopsies displayed the typical features of low-grade FL (Figure 1A), with follicular morphology and predominance of centrocytes. The neoplastic cells in all evaluable samples were CD20+ B cells (17/17) that coexpressed CD10 (16/16) and had low proliferation indices by Ki-S5 or Ki67 staining (median 10%, range 1% to 40%). Immunohistochemical stains for BCL2 were positive in 97% of evaluable cases (32/33); BCL2 rearrangements were identified in 91% of cases by FISH (20/23).
Patient and disease characteristics in DTFL, LSTFL, and ASTFL cohorts
| DTFL (n = 38) | LSTFL (n = 17) | ASFL (n = 241) | |||
|---|---|---|---|---|---|
| Age median (range) | 57 (22-84) | 61 (33-85) | 59 (27-83) | ||
| Sex | |||||
| Male | 13 (34%) | 5 (29%) | 127 (53%) | ||
| Localization | Stage | Stage | |||
| Duodenum | 22 | I | 12 | I | 0 |
| Jejunum or ileum | 10 | II | 5 | II | 0 |
| Colon or rectum | 5 | III | 0 | III | 91 |
| Unknown | 1 | IV | 0 | IV | 150 |
| Treatment | Treatment | Treatment | |||
| RT | 12 | RT | 9 | R-CHOP | 137 |
| Rituximab mono | 4 | Rituximab mono | 1 | R-CVP | 104 |
| R-chemo | 3 | R-chemo | 3 | ||
| Surgery | 4 | Surgery | 1 | ||
| No treatment or unknown | 17 | RT + systemic treatment | 2 | ||
| No treatment | 1 | ||||
| DTFL (n = 38) | LSTFL (n = 17) | ASFL (n = 241) | |||
|---|---|---|---|---|---|
| Age median (range) | 57 (22-84) | 61 (33-85) | 59 (27-83) | ||
| Sex | |||||
| Male | 13 (34%) | 5 (29%) | 127 (53%) | ||
| Localization | Stage | Stage | |||
| Duodenum | 22 | I | 12 | I | 0 |
| Jejunum or ileum | 10 | II | 5 | II | 0 |
| Colon or rectum | 5 | III | 0 | III | 91 |
| Unknown | 1 | IV | 0 | IV | 150 |
| Treatment | Treatment | Treatment | |||
| RT | 12 | RT | 9 | R-CHOP | 137 |
| Rituximab mono | 4 | Rituximab mono | 1 | R-CVP | 104 |
| R-chemo | 3 | R-chemo | 3 | ||
| Surgery | 4 | Surgery | 1 | ||
| No treatment or unknown | 17 | RT + systemic treatment | 2 | ||
| No treatment | 1 | ||||
CHOP, cyclophosphamide, doxorubicin, vincristine, and prednisone; CVP, cyclophosphamide, vincristine, and prednisone; R, rituximab.
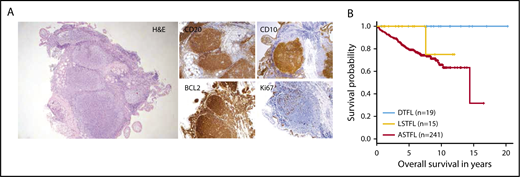
Histopathology and clinical outcome of DTFL. (A) Histopathological features of DTFL. Hematoxylin and eosin staining (H&E) of a representative case (#K17), and immunohistochemical staining for BCL2, CD20, CD10, and Ki67. (B) Kaplan-Meier curves for overall survival of DTFL, LSTFL, and ASTFL cohorts.
Histopathology and clinical outcome of DTFL. (A) Histopathological features of DTFL. Hematoxylin and eosin staining (H&E) of a representative case (#K17), and immunohistochemical staining for BCL2, CD20, CD10, and Ki67. (B) Kaplan-Meier curves for overall survival of DTFL, LSTFL, and ASTFL cohorts.
Patients with DTFL and LSTFL mostly received RT. Other types of therapies included excision only, rituximab monotherapy, immunochemotherapy or no treatment/watchful waiting (Table 1). In contrast, patients with ASTFL uniformly received immunochemotherapy.8 Median follow-up of clinically evaluable patients was 9.1 years for DTFL (n = 19), 4.9 years for LSTFL (n = 15), and 6.1 years for ASTFL (n = 241), respectively. Patients with DTFL had a highly indolent clinical course. Two patients with DTFL developed systemic disease (11%), whereas 4 patients with LSTFL progressed to advanced-stage disease (27%). Histological transformation occurred in 0 DTFL (0%), 1 LSTFL (7%), and 21 ASTFL (9%) cases. Overall survival (OS) was significantly superior for DTFLs as compared with ASTFLs (P = .008; Figure 1B). OS rates for DTFL, LSTFL, and ASTFL were 100%, 75%, and 66% at 10 years, respectively.
The targeted mutational landscape
We first performed a targeted analysis of the mutation status of 82 genes in 31 DTFL and 17 LSTFL cases (supplemental Figure 1, CONSORT diagram). The targeted mutational profiles are shown in Figure 2A. The median number of targeted putative somatic mutations was 8 in DTFL (range 0-17) and 11 in LSTFL (6-29), as compared with 11 in a previously sequenced cohort of 241 ASTFL (range 3-42, P < .001 and P = .78, respectively; supplemental Figure 3).
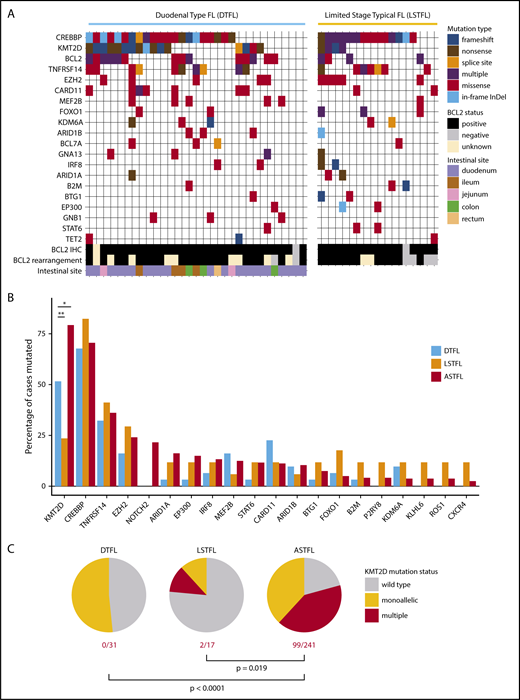
Targeted mutational landscape of DTFL and LSTFL. (A) Mutation plot for DTFL and LSTFL, depicting only genes that were mutated in at least 3 samples and captured by both bait sets. Mutation type and BCL2 status are color coded. BCL2 rearrangement refers to FISH testing. BCL2 IHC refers to immunohistochemistry. (B) Mutation frequencies of recurrently mutated genes in DTFL, LSTFL, and ASTFL cohorts. The bar plot shows mutation frequencies for all genes mutated in >10% of any of the 3 cohorts. *P < .05, **P < .01 for FDR-corrected Fisher’s exact test. (C) Fraction of cases with monoallelic or multiple KMT2D mutations in DTFL, LSTFL, and ASTFL cohorts. P values indicate result of Fisher’s exact test.
Targeted mutational landscape of DTFL and LSTFL. (A) Mutation plot for DTFL and LSTFL, depicting only genes that were mutated in at least 3 samples and captured by both bait sets. Mutation type and BCL2 status are color coded. BCL2 rearrangement refers to FISH testing. BCL2 IHC refers to immunohistochemistry. (B) Mutation frequencies of recurrently mutated genes in DTFL, LSTFL, and ASTFL cohorts. The bar plot shows mutation frequencies for all genes mutated in >10% of any of the 3 cohorts. *P < .05, **P < .01 for FDR-corrected Fisher’s exact test. (C) Fraction of cases with monoallelic or multiple KMT2D mutations in DTFL, LSTFL, and ASTFL cohorts. P values indicate result of Fisher’s exact test.
The mutation patterns for most genes did not differ significantly between DTFL, LSTFL, and ASTFL (Figure 2B and supplemental Table 2). For example, the acetyltransferase CREBBP was mutated in 68% of DTFL, 82% of LSTFL, and 71% of ASTFL cases. Also, mutation types and distribution were not different; that is, the proportions of truncating mutations and missense mutations within the HAT domain of CREBBP were similar in DTFL, LSTFL, and ASTFL (supplemental Figure 2). The cytokine receptor TNFRSF14 (alias HVEM) was mutated in 32% of DTFL, 41% of LSTFL, and 36% of ASTFL cases. Mutations in EZH2, the catalytic subunit of the PRC2 complex, were found in 16% of DTFL, 29% of LSTFL, and 24% of ASTFL cases. EZH2 mutations were all heterozygous and occurred exclusively at previously described gain-of-function hot spots in exons 16 and 18.
However, a significant difference was noticed for the histone methyltransferase KMT2D (alias MLL2). Similar to ASTFL, KMT2D mutations in DTFL and LSTFL were predominately disruptive (Figure 2A). In line with previous reports,12 mutations in KMT2D were highly recurrent and detectable in 79% of ASTFL (191/241) cases. In contrast, only 52% of DTFL cases harbored mutations in KMT2D (16/31; corrected P = .039). KMT2D was also less frequently mutated in LSTFL (24%, 4/17; corrected P < .001). Furthermore, 41% of ASTFL cases harbored multiple KMT2D mutations, likely representing biallelic mutations. In contrast, 0% of DTFL and only 12% of LSTFL cases harbored multiple KMT2D mutations (P < .0001 and P = .019, respectively; Figure 2C).
WES of DTFL
To identify novel coding gene mutations, we performed WES on 11 diagnostic DTFL biopsies and matched germ-line material. We did not detect any recurrently mutated gene (ie, mutated in >2 cases) that had not previously been described in FL. However, 2 genes were mutated more frequently than previously reported. HVCN1 was found to be mutated in 3 cases by WES and in 2 additional cases by targeted resequencing of that gene, for an overall mutation rate of 22% in DTFL (5/23; supplemental Figure 5A). HVCN1 encodes for a voltage-gated protein channel, and mutations have recently been reported to be associated with longer progression-free survival in a heterogeneous cohort of patients with FL.13 Also, the eukaryotic translation elongation factor EEF1A1 was mutated more commonly than previously reported in FL14 for an overall mutation rate of 35% in DTFL (8/23). Mutations were clustered within the N-terminal GTP-binding domain (7/9), and clearly distinct from EEF1A1 mutations in solid tumors, which cluster at a mutational hot spot (T432) within the C terminus (supplemental Figure 5B).
The immune microenvironment
To identify differences in immune response pathways and cellular compositions of the tumor microenvironment, we applied a 730 immune gene expression panel (PanCancer Immune Profiling; NanoString) to 8 DTFLs and 7 LSTFLs. The top differentially expressed genes were markedly enriched for genes from the annotated biological process category “chemokines/cytokines” (Figure 3A), including CCL21, TNFSF15, CCL11, CXCL1, CXCL6, and CCL20 (each with log2 fold change >1 and corrected P < .02). Unsupervised, hierarchical clustering by gene expression levels of all 147 probed chemokines/cytokines separated DTFL and LSTFL into 2 discrete clusters (supplemental Figure 6A). In line with a previous report, CCL20 was among the highest differentially expressed genes in DTFL, supporting the hypothesis that chronic inflammation plays a role in the pathogenesis of DTFL.15 In fact, unsupervised hierarchical clustering of cases by genes associated with chronic inflammation (nCounter PanCancer Immune Profiling Panel annotation) again separated DTFL and LSTFL into 2 discrete clusters (Figure 3B). Furthermore, gene set enrichment analysis for the GO gene set chronic inflammatory response showed a positive, yet nonsignificant enrichment toward upregulated genes in DTFL (supplemental Figure 6B).
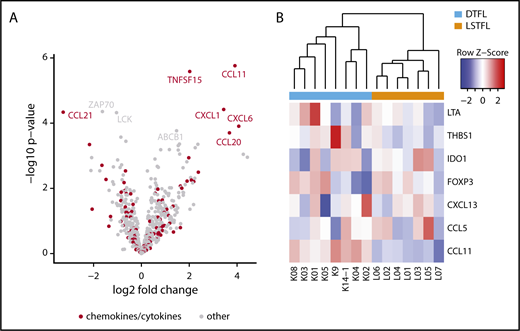
The immune microenvironment of DTFL and LSTFL. (A) Volcano plot indicating differentially expressed genes in DTFL and LSTFL. Chemokine and cytokine genes are highlighted in red. Genes with a log2 fold change >1 and P value <.02 are labeled. (B) Heat map and unsupervised, hierarchical clustering for genes associated with chronic inflammation. Genes associated with chronic inflammation separate DTFL and LSTFL into 2 discrete clusters.
The immune microenvironment of DTFL and LSTFL. (A) Volcano plot indicating differentially expressed genes in DTFL and LSTFL. Chemokine and cytokine genes are highlighted in red. Genes with a log2 fold change >1 and P value <.02 are labeled. (B) Heat map and unsupervised, hierarchical clustering for genes associated with chronic inflammation. Genes associated with chronic inflammation separate DTFL and LSTFL into 2 discrete clusters.
CCL20 is a strong chemotactic factor for lymphocytes and involved in recruitment of proinflammatory interleukin-17-producing helper T cells (Th17) to sites of inflammation.16 We inferred cellular abundance of specific immune cell types from our gene expression data (see “Methods”). There was no significant difference in the abundance of total tumor-infiltrating lymphocytes or B or T cells (supplemental Figure 7A). However, we found a significantly higher abundance of Th17 and activated CD4+ T cells relative to the overall abundance of T cells (P < .001; supplemental Figure 7B) indicating a proinflammatory immune cell environment. Thus, the microenvironment of DTFL is distinct from typical FL and enriched for genes and cell types involved in chronic inflammatory processes.
Progression from DTFL to nodal FL
Two patients progressed from DTFL to ASTFL. The first patient (#K3) had confirmed DTFL of the ileum (Ann Arbor stage IE, grade 1) and developed widespread nodal FL 7 years after surgical resection and consolidating local RT. The second patient (#K14) was a 56-year-old male patient with confirmed DTFL of the colon and no extraintestinal disease (Ann Arbor stage IE, grade 2), who achieved a complete response after local RT but subsequently presented with widespread lymphadenopathy 5 years later. Histopathology of a cervical LN biopsy confirmed grade 1 FL, positive for t(14;18) by FISH. After watchful waiting for 2 years, the patient had progressive disease with infiltration of the urinary bladder. We compared the targeted mutational profile of the initial DTFL and subsequent LN biopsies (Figure 4). Four mutations were shared, confirming that these lymphomas were derived from a CPC. The DTFL harbored 6 additional coding mutations that were not detectable in the LN biopsy; another 12 coding mutations were only detectable in the LN biopsy. This pattern is consistent with divergent evolution as previously described for typical FL.17,18 Gene expression analysis demonstrated that the intestinal biopsy (#K14-1) clustered with the DTFL group, whereas the LN biopsy (#K14-2) clustered with the LSTFL group, both by cytokines/chemokines (supplemental Figure 6C) and by genes associated with chronic inflammation (supplemental Figure 6D), indicating profound differences between the immune microenvironment of the initial DTFL and the subsequent ASTFL.
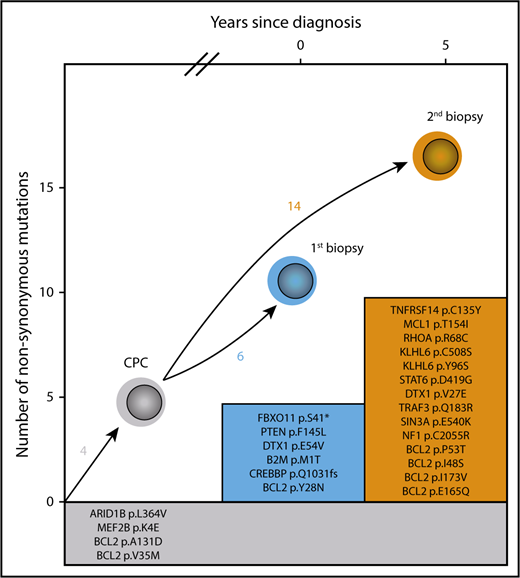
Divergent evolution of DTFL progressing to nodal FL. Mutations listed in the gray box indicate shared mutations defining a common precursor clone (CPC). Mutations in colored boxes indicate mutations found uniquely in each of the biopsies.
Divergent evolution of DTFL progressing to nodal FL. Mutations listed in the gray box indicate shared mutations defining a common precursor clone (CPC). Mutations in colored boxes indicate mutations found uniquely in each of the biopsies.
DTFL vs (subclinical) nodal FL with intestinal manifestation
A 29-year-old male patient (#K12) with grade 1 FL of the terminal ileum was excluded from our DTFL cohort because histopathological review revealed submucosal infiltration. Also, careful histopathological review of an excised, grossly normal mesenteric LN displayed small infiltrates of CD20+/CD10+/BCL2+ lymphocytes and a detectable translocation t(14;18). The patient received local RT but relapsed 2 years later with ASTFL. A cervical LN showed grade 1 FL (CD20+/CD10+/BCL2+, Ki-S5 <5%) without evidence of histological transformation. Sequencing of the intestinal manifestation and the cervical LN biopsies demonstrated shared mutations in CREBBP, KMT2D, TNFRSF14, and ATM, as well as similar mutations in BCL2, confirming identical clonality (supplemental Table 9). Gene expression profiles for chemokines/cytokines and genes associated with chronic inflammation from the intestinal biopsy (collected at initial diagnosis) and the cervical LN biopsy (collected at relapse) both clustered with nodal FL (supplemental Figure 6C-D).
Discussion
Here, we demonstrate that the mutational landscape of DTFL is highly related to typical FL. The majority of genes recurrently mutated in FL, including CREBBP, TNFRSF14, and EZH2, were mutated at similar frequencies in DTFL, LSTFL, and ASTFL. We did not detect novel recurrent gene mutations specific to DTFL by WES. Only 2 genes (HVCN1 and EEF1A1) were mutated at higher rates than previously reported for typical FL,13,14 but this requires validation in larger cohorts as well as further characterization of their biological and clinical relevance. The similarity of DTFL with LSTFL and ASTFL at the mutational level implies highly similar pathogenetic processes and strongly suggests that DTFL indeed belongs to the heterogeneous spectrum of typical FL. In fact, DTFL, when stringently defined, may simply represent very early stages of typical FL. This hypothesis is supported by work from Mamessier and colleagues, who compared array comparative genomic hybridization data of DFTL (n = 4) and in situ follicular neoplasia (ISFN; n = 4) with nodal FL.19 Although the overall frequency of copy number variations was lower in both DTFL and ISFN compared with nodal FL, the patterns of recurrent copy number variations were highly related to typical FL, such as deletions of 1p36 (including TNFRSF14) and gains of 7p (including EZH2). Analogous to our study in DTFL, FL-associated gene mutations have also been identified in ISFN, including EZH2.20 In line with the hypothesis that DTFL represents the ISFN correlate of the intestinal tract, we (1) describe, although uncommon, progression from stringently defined DTFL to nodal FL (ASTFL), with evidence of divergent mutational evolution from a common precursor clone; (2) we further notice a continuous increase in number of gene mutations from DTFL to LSTFL and ASTFL; and (3) most notably, the rate of truncating mutations in KMT2D, particularly multiple, likely biallelic mutations, increases toward ASTFL. KMT2D (alias MLL2) encodes for an H3K4-specific histone methyltransferase and is the single most commonly mutated gene in ASTFL (up to 90% of cases).12 Despite this very high mutation frequency, an earlier study unexpectedly showed that many mutations in KMT2D are not truncal/early events, but rather subclonal/later events.21 Subsequently, elegant studies in mice have shown that loss of Kmt2d in antigen-naïve B cells promotes lymphomagenesis in combination with BCL2, with phenotypic changes increasing from monoallelic to biallelic lesions.22 Within this context, our data provide further evidence of an increasing selection pressure for biallelic/complete KMT2D loss during the development of or progression to ASTFL.
In contrast to the highly similar mutational landscape, we found marked differences in the chemokine/cytokine milieu by digital gene expression profiling of DTFL and nodal FL. Expression levels of chemokines/cytokines clearly distinguished DTFL from LSTFL, indicating highly dissimilar immune microenvironments. Our data confirm a previous report of high CCL20 expression in DTFL.15 CCL20 recruits proinflammatory Th17 cells, which constitute key drivers of inflammation.23 Indeed, we found a higher abundance of Th17 cells in DTFL compared with LSTFL. Furthermore, we noticed that genes associated with chronic inflammation were highly expressed in DTFL as compared with LSTFL, and robustly separated DTFL from LSTFL.
Chronic inflammation of the immune microenvironment may in fact be a more general biological feature of primary intestinal lymphomas, and similarities between DTFL and gastric mucosa-associated lymphoid tissue lymphomas have been reported previously.15 To the best of our knowledge, there is no known association between DFTL and intestinal inflammatory conditions such as inflammatory bowel disease, duodenal ulcers, Helicobacterpylori infection, or celiac disease, but this has not been studied specifically yet. Furthermore, although our study clearly demonstrates that the immune microenvironment of DTFL is distinct from nodal FL, we cannot delineate to what extent these differences are intrinsic to the (normal) intestinal tract, or induced by lymphoma cells. Toward this end, future studies will have to include comprehensive characterization of the immune microenvironment of intestinal biopsies from healthy individuals, of noninfiltrated intestinal biopsies from patients with DTFL, and of lymphoma-infiltrated and noninfiltrated intestinal biopsies from patients with other (gastrointestinal) lymphomas.
Ultimately however, FL cells in DTFL are exposed to a very different immune microenvironment compared with nodal FL cells. It is intriguing to speculate which factors and mechanisms of the chronic inflammatory microenvironment contribute to confine DTFL to the gastrointestinal tract and the remarkably low propensity of DTFL to progress or disseminate. The low levels of the leukocyte chemoattractant CCL21 in DTFL might be involved in this process. Although high expression of CCL21 has been implicated in mediating immune escape, CCL21-deficient tumors induced antigen-specific immunity, which might hinder tumor progression.24
We show that stringently defined DTFL and subsequent ASTFL can arise from a CPC, as defined by the presence of shared gene mutations.14,17,18 Also, we and others have previously shown that circulating BCL2-rearranged cells are detectable many years before clinical manifestation of FL.25,26 Together, this supports a model of divergent evolution: CPCs within the chronic inflammatory microenvironment of the gastrointestinal tract give rise to confined DTFL, whereas CPCs within the nodal immune microenvironment, which is permissive to progression and dissemination, can eventually develop to LSTFL and ASTFL.
Furthermore, our case report of a patient with (subclinical) nodal FL and an intestinal manifestation (#K12), which does not display the chronic inflammatory features seen in DTFL, indicate that the type of FL (DTFL vs nodal FL) rather than the biopsy site (intestinal vs LN) determines, at least in part, the cytokine/chemokine profile of the immune microenvironment. Clearly, functional studies are needed to better understand the specific cross talk between FL subtypes and their specific microenvironments.
In summary, by comparing the targeted mutational landscapes of DTFL, LSTFL, and ASTL, we find that the mutational landscape of DTFL is highly related to typical FL. However, the lower frequency of multiple/biallelic mutations in KMT2D in DTFL and LSTFL indicates an increasing selection pressure for complete KMT2D loss during the development of or the progression to ASTFL. In contrast, the immune microenvironment of DFTL is distinct from typical FL, most notably for highly dissimilar chemokine/cytokine milieus and features of chronic inflammation. Although, it is currently unclear to what extent these differences are FL induced or intestinal intrinsic, they certainly provide important insights into the distinct biology of the disease.
Presented in part at the 59th annual meeting of the American Society of Hematology, Atlanta, GA, 10 December 2017.
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Acknowledgments
This research was supported by the Deutsche Forschungsgemeinschaft (DFG-SFB/CRC-1243, TP-A11) (O.W.). J.C.H. was supported by a Mildred Scheel postdoctoral fellowship of the Deutsche Krebshilfe e.V. G.O. receives funding from the Robert-Bosch-Foundation, and D.M.W. is a Leukemia and Lymphoma Scholar. O.W. is supported by the Max-Eder Junior Research Group Program of the Deutsche Krebshilfe e.V.
Authorship
Contribution: O.W., J.C.H., A.L., W.K., and D.M.W. designed and performed the research; J.C.H. performed statistical and computational analysis; A.L., S.H., S.A., and A.P. contributed to analysis; A.L., M.S., and P.K. collected and analyzed clinical data; A.L., M.S., A.M.S., S.H., R.K., M.-L.H., G.O., A.R., R.D.G., and W.K. provided samples; M.D.D. analyzed next-generation sequencing data; W.H. and M.D. supervised the research; O.W., J.C.H., A.L., and D.M.W. wrote the first version of the manuscript; and all authors read and approved the final manuscript.
Conflict-of-interest disclosure: R.D.G. consulted for Celgene and Seattle Genetics. W.H. consulted for Roche, Janssen, Celgene, and Gilead. M.D. consulted for and received speaker's honoraria from Roche. O.W. consulted for Roche, Incyte, and Cellestia and received research funding from Roche and Novartis. The remaining authors declare no competing financial interests.
Correspondence: Oliver Weigert, University Hospital of the Ludwig-Maximilians-University Munich, Medical Department III, Laboratory for Experimental Leukemia and Lymphoma Research (ELLF), Max-Lebsche Platz 30, 81377 Munich, Germany; e-mail: oliver.weigert@med.uni-muenchen.de.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal