

Key Points
Genome-scale CRISPR knockout screen identifies cullin-RING ligase regulators as top mediators of lenalidomide resistance.
The E2 ubiquitin-conjugating enzymes, UBE2D3 and UBE2G1, play distinct roles in lenalidomide-induced substrate ubiquitination by CRL4CRBN.

Abstract
Lenalidomide mediates the ubiquitination and degradation of Ikaros family zinc finger protein 1 (IKZF1), IKZF3, and casein kinase 1α (CK1α) by facilitating their interaction with cereblon (CRBN), the substrate receptor for the CRL4CRBN E3 ubiquitin ligase. Through this mechanism, lenalidomide is a clinically effective treatment of multiple myeloma and myelodysplastic syndrome (MDS) with deletion of chromosome 5q [del(5q) MDS]. To identify the cellular machinery required for lenalidomide-induced CRL4CRBN activity, we performed a positive selection, genome-scale clustered regularly interspaced short palindromic repeats (CRISPR)–CRISPR-associated protein 9 (Cas9) screen in a lenalidomide-sensitive myeloma cell line. CRBN was the top-ranking gene, with all CRBN-targeting guide RNAs (gRNAs) ranking as the 6 highest-scoring gRNAs. A counterscreen using an IKZF3 degron reporter to assay lenalidomide-induced protein degradation highlighted regulators of cullin-RING ligase neddylation and 2 E2 ubiquitin-conjugating enzymes as necessary for efficient lenalidomide-induced protein degradation. We demonstrated that loss of UBE2M or members of the constitutive photomorphogenesis 9 (COP9) signalosome results in altered neddylation of cullin 4A and impairs lenalidomide-dependent CRL4CRBN activity. Additionally, we established that UBE2D3 and UBE2G1 play distinct roles in substrate ubiquitination by CRL4CRBN, with UBE2D3 acting to prime targets via monoubiquitination and UBE2G1 functioning to extend polyubiquitin chains with lysine 48 linkages. The validation of UBE2D3 and UBE2G1 highlights the functional capacity of CRISPR-Cas9 screening to identify E2 ubiquitin-conjugating enzyme and E3 ubiquitin ligase complex pairings. More broadly, these findings establish key proteins required for lenalidomide-dependent CRL4CRBN function in myeloma and inform potential mechanisms of drug resistance.
Introduction
Lenalidomide is an effective therapy for multiple myeloma and del(5q) myelodysplastic syndrome (MDS). Recent studies have demonstrated that the therapeutic efficacy of lenalidomide is derived from its ability to induce the ubiquitination and proteasomal degradation of disease-relevant proteins by the CRL4CRBN E3 ubiquitin ligase.1-4
E3 ubiquitin ligases are a class of enzymes that catalyze the covalent transfer of the 8-kDa protein ubiquitin from E2 ubiquitin-conjugating enzymes onto target substrates.5 Substrates can be marked with a single ubiquitin or chains of polyubiquitin. The number of ubiquitin moieties added to a target and the lysine residues on ubiquitin used to form the linkages within polyubiquitin chains serve as a “ubiquitin code,” dictating a diverse set of possible outcomes for the ubiquitinated substrate.6 RING-type E3 ubiquitin ligases rely upon E2 ubiquitin-conjugating enzymes to dictate the ubiquitin lysine linkage and chain elongation. The canonical outcome, polyubiquitination with lysine 48 (K48) linkages, leads to rapid degradation of the substrate by the 26S proteasome. Via this mechanism, E3 ubiquitin ligases and their corresponding E2 enzymes play a crucial role in targeted protein degradation and the posttranslational stability of the proteome.
The CRL4CRBN ubiquitin ligase targeted by lenalidomide belongs to the family of cullin-RING ligases, which share a modular, multisubunit architecture based around a cullin backbone.7 Lenalidomide specifically binds cereblon (CRBN), the substrate receptor for the CRL4CRBN E3 ubiquitin ligase,8,9 and increases its affinity for the lymphocyte lineage transcription factors Ikaros family zinc finger protein 1 (IKZF1) and IKZF3,10,11 and the Wnt pathway regulator casein kinase 1α (CK1α).12 As a result of their stabilized interaction with CRL4CRBN, IKZF1, IKZF3, and CK1α are rapidly polyubiquitinated and degraded by the 26S proteasome. Degradation of IKZF1 and IKZF3 underlies the antimyeloma and immunomodulatory properties of lenalidomide,1-3 whereas degradation of CK1α explains the therapeutic benefit in the context of del(5q) MDS.4
Lenalidomide and its immunomodulatory imide drug (IMiD) analogs, thalidomide and pomalidomide, are the first clinically approved small molecules whose therapeutic properties rely on ubiquitin ligase-mediated degradation of disease-relevant targets. The full set of proteins required for lenalidomide’s therapeutic modulation of the CRL4CRBN ubiquitin ligase has not been identified and holds implications for our understanding of drug mechanism, response, and resistance. Positive-selection clustered regularly interspaced short palindromic repeats (CRISPR)–CRISPR-associated protein 9 (Cas9) screens are a robust tool for the unbiased identification of genes involved in a biological pathway of interest.13 Here, we used a genome-scale CRISPR-Cas9 screen to identify genes that, when inactivated, diminish the effects of lenalidomide, and investigated the roles for validated hits in the biology of lenalidomide-induced substrate degradation by CRL4CRBN.
Methods
Genome-scale CRISPR-Cas9 screen
MM1.S cells expressing Cas9 were infected with the Human GeCKOv2 LentiGuide-Puro library (Broad Genetic Perturbations Platform). On day 0, 60 million cells were plated for dimethyl sulfoxide (DMSO) treatment, and 6 replicates of 120 million cells were plated for 1 μM lenalidomide (Selleck Chemicals, Houston, TX) treatment. Cells were dosed every 2 days with DMSO or 1 μM lenalidomide and passaged every 4. On day 12, 60 million cells from the DMSO arm, and all remaining cells in 3 of the lenalidomide arm replicates were pelleted. On day 20, 60 million cells from the DMSO arm and all cells in the remaining 3 lenalidomide arm replicates were pelleted. The guide RNA (gRNA) library was polymerase chain reaction (PCR) amplified from gDNA isolated from the cell pellets and the resulting amplicons sequenced on the Illumina MiSeq. gRNAs were ranked on the basis of their fold-change in read counts (lenalidomide/DMSO) and the STARS algorithm (https://portals.broadinstitute.org/gpp/public/software/stars) was used to calculate false discovery rate (FDR) values for each gene.
IKZF3 reporter counterscreen
MM1.S, NCIH929, and HEK293T cells expressing Cas9 and a fluorescent IKZF3-enhanced green fluorescent protein (eGFP) degradation reporter (Addgene #74459, Cambridge, MA) were infected with a gRNA library targeting the top 30 genes from the genome-scale screen (3 experimental gRNAs per gene and 12 nontargeting control gRNAs). Eleven days after infection the cells were dosed for 20 hours with DMSO or 1 μM lenalidomide. Unsorted controls were harvested from the DMSO and 1 μM lenalidomide treatment groups, and 1 μM lenalidomide-treated cells underwent fluorescence-activated cell sorting (FACS) for eGFP+ cells. gRNA sequences were PCR-amplified from gDNA isolated from the cells and the resulting amplicons were sequenced on the Illumina NextSeq. The fold-change in gRNA read counts (lenalidomide eGFP+/unsorted DMSO) was calculated and normalized to the average fold-change of the control gRNAs. A Wilcoxon rank-sum test statistic was used to calculate significance relative to the controls.
Competition assays
MM1.S cells expressing Cas9 were transduced with vectors expressing either experimental gRNAs and eGFP or control gRNAs without a fluorophore. These cells were then mixed at a 5:95 ratio, respectively, and then grown in DMSO or 1 μM lenalidomide for 20 days. The cells were dosed every 2 days and passaged every 4. With each passage, cells were analyzed by flow cytometry to determine the percentage of eGFP+ cells.
In vitro ubiquitination assays
Recombinant CRL4CRBN complex was generated as previously described.14 To generate an IZKF3 fragment, HEK293T were transfected with IKZF3 aa146-168-hemagglutinin (HA)-eGFP and lysed after 48 hours. HA-sepharose beads were used to pull down IKZF3 aa146-168. The in vitro reaction consisted of HA-eluted IKZF3, 2 μM CRL4CRBN, 1 μg/μL ubiquitin (Boston Biochem, Cambridge, MA), 200 nM UBE1 (Boston Biochem), 1 μM UBE2G1 (Boston Biochem), 1 μM UBE2D3 (Boston Biochem), 1 μM ubiquitin aldehyde (Boston Biochem), 1× Mg-adenosine triphosphate (ATP) (Boston Biochem), 1× E3 ligase reaction buffer (Boston Biochem), and 1 μM lenalidomide or DMSO. Assay was run for 30 minutes at 30°C.
Detailed methods can be found in the supplemental Methods (available on the Blood Web site).
Results
Genome-scale CRISPR-Cas9 screen identifies genes required for lenalidomide’s antimyeloma effects
Degradation of IKZF1 and IKZF3 by CRL4CRBN is responsible for lenalidomide activity in multiple myeloma,2,3 therefore, we hypothesized that inactivation of genes required for lenalidomide-dependent CRL4CRBN activity would result in resistance to lenalidomide. Thus, to gain insight into the molecular machinery required for lenalidomide-mediated degradation of CRL4CRBN targets, we performed a positive selection, genome-scale CRISPR-Cas9 screen in the lenalidomide-sensitive myeloma cell line, MM1.S.
MM1.S cells with stable expression of Cas9 were infected with the human GeCKOv2 lentiguide-puro gRNA library.15 This library targets 19 050 genes, with 6 gRNAs per gene and 1000 control gRNAs. Eight days after infection, the cells were treated with 1 μM lenalidomide or DMSO for 20 days (supplemental Figure 1A). DMSO-treated cells continued to proliferate whereas the 1 μM lenalidomide-treated cells depleted to half their original cell number by day 20 (Figure 1A). At the conclusion of the screen, we harvested genomic DNA from the remaining cells in the lenalidomide and DMSO treatment groups, PCR-amplified the gRNA sequences, performed next-generation sequencing of the amplicons, and compared the frequency of read counts for a given gRNA in the DMSO and lenalidomide experimental conditions. In comparison with the 1000 control gRNAs, the gene-targeting gRNAs at day 20 possessed a greater number of outliers with positive lenalidomide/DMSO fold-changes in read counts, consistent with the acquisition of lenalidomide resistance (Figure 1B).
![Figure 1. Genome-scale CRISPR-Cas9 screen in a lenalidomide-treated myeloma cell line identifies genes required for lenalidomide’s antimyeloma effects. (A) Cell number over the 20-day assay (DMSO; 1 replicate, 1 μM lenalidomide [Len]; average of 3 technical replicates). (B) Log2 fold-enrichment (lenalidomide/DMSO) of read counts for gene-targeting gRNAs and 1000 control gRNAs (average of 3 technical replicates) at day 20. Upper and lower bounds of box correspond to the 75% and 25% quantiles, respectively, central line indicates the median, whiskers extend to 1.5× the interquartile range, datapoints beyond 1.5× the interquartile range are displayed individually. (C) gRNA library ranked according to the lenalidomide/DMSO fold-change in read count (average of 3 technical replicates). Blue lines indicate 3 standard deviations above and below the mean; black line is average of all gRNAs. (D) Results of the STARS algorithm analysis. Genes displayed met a FDR threshold of <0.05. “gRNAs scoring” refers to the number of gRNAs (of 6) whose ranks were considered by the algorithm to have generated the lowest P value and were therefore used to generate the score for that gene.](https://ash.silverchair-cdn.com/ash/content_public/journal/blood/132/12/10.1182_blood-2018-01-821769/4/m_blood821769f1.png?Expires=1770819740&Signature=Ms5yonfYNxG88DgYZgngVYpF-t3siABY8guJPp-XLf0VwIIdf04UW5Q80OLoqu7L7mYI3PEpt6jf-OyT~ZbQ2y0ricm4gkduzTE6BwajD9o1GTUoAeImc6ap01xrWJPGFDgO8SsTMu5LmiG3DM27ja~~quCzpBDZV8b85z2YzDfbt5myCkxpFVc6dCU-yByZlC3PJw9Igxjwdwh8pQN~txMxu9Cc-ltEK3InPfYddIQtYfh7YEh9JRFMmwq8Ccq3xhCxqbWOXsR0xM2-AaRmJD04KzrKCPvyfTNR5SIfnJ~D5OfEVKjRNPbXQbDkRMOZLjlg6GTWrNs-TLPQ8tS3ng__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
Genome-scale CRISPR-Cas9 screen in a lenalidomide-treated myeloma cell line identifies genes required for lenalidomide’s antimyeloma effects. (A) Cell number over the 20-day assay (DMSO; 1 replicate, 1 μM lenalidomide [Len]; average of 3 technical replicates). (B) Log2 fold-enrichment (lenalidomide/DMSO) of read counts for gene-targeting gRNAs and 1000 control gRNAs (average of 3 technical replicates) at day 20. Upper and lower bounds of box correspond to the 75% and 25% quantiles, respectively, central line indicates the median, whiskers extend to 1.5× the interquartile range, datapoints beyond 1.5× the interquartile range are displayed individually. (C) gRNA library ranked according to the lenalidomide/DMSO fold-change in read count (average of 3 technical replicates). Blue lines indicate 3 standard deviations above and below the mean; black line is average of all gRNAs. (D) Results of the STARS algorithm analysis. Genes displayed met a FDR threshold of <0.05. “gRNAs scoring” refers to the number of gRNAs (of 6) whose ranks were considered by the algorithm to have generated the lowest P value and were therefore used to generate the score for that gene.
Genome-scale CRISPR-Cas9 screen in a lenalidomide-treated myeloma cell line identifies genes required for lenalidomide’s antimyeloma effects. (A) Cell number over the 20-day assay (DMSO; 1 replicate, 1 μM lenalidomide [Len]; average of 3 technical replicates). (B) Log2 fold-enrichment (lenalidomide/DMSO) of read counts for gene-targeting gRNAs and 1000 control gRNAs (average of 3 technical replicates) at day 20. Upper and lower bounds of box correspond to the 75% and 25% quantiles, respectively, central line indicates the median, whiskers extend to 1.5× the interquartile range, datapoints beyond 1.5× the interquartile range are displayed individually. (C) gRNA library ranked according to the lenalidomide/DMSO fold-change in read count (average of 3 technical replicates). Blue lines indicate 3 standard deviations above and below the mean; black line is average of all gRNAs. (D) Results of the STARS algorithm analysis. Genes displayed met a FDR threshold of <0.05. “gRNAs scoring” refers to the number of gRNAs (of 6) whose ranks were considered by the algorithm to have generated the lowest P value and were therefore used to generate the score for that gene.
The 6 most-enriched gRNAs in the lenalidomide treatment condition at day 20 all targeted CRBN, the substrate receptor for the CRL4CRBN ubiquitin ligase (Figure 1C). To confirm this finding, we introduced CRBN-targeting gRNAs into MM1.S-Cas9 cells expressing eGFP and then mixed these cells at a 5:95 ratio with control gRNA-infected cells. We then grew the cells in a range of lenalidomide concentrations for 20 days, and used flow cytometry every 4 days to monitor the percentage of eGFP+ cells. The eGFP+CRBN gRNA-infected cells rose from a frequency of 5% at day 0 to 80% to 95% at day 20 across lenalidomide concentrations ranging from 0.16 to 10 μM (supplemental Figure 1B). The strength and consistency of this finding indicates that, on a genome scale, CRBN is the most important, nonessential gene in MM1.S for the response to lenalidomide.
To identify additional genes required for lenalidomide activity, we analyzed the genome-scale screen data with the STARS algorithm,16 which ranks genes on the basis of the fold-enrichment of their respective gRNAs (Figure 1D). At a threshold FDR of <0.05, 30 genes scored, 17 of which are involved in regulation of E3 enzymes, particularly cullin-RING ligases. These genes included an additional subunit of the CRL4CRBN ubiquitin ligase (DDB1), cullin-RING ligase regulators (all 9 subunits of the constitutive photomorphogenesis 9 [COP9] signalosome, CAND1, UBE2M, GLMN), and E2 ubiquitin-conjugating enzymes (UBE2G1, UBE2D3). The remaining 13 genes were involved in the NF-κB pathway (TRAF2), the 5′ messenger RNA (mRNA) decapping complex (XRN1, EDC4), nuclear hormone receptor signaling (NCOR1, RARA), the GATOR complex (DEPDC5), tumor suppressors (PPP6C, SPOP), and genes of unclear function. We did not find evidence of genes whose loss increased sensitivity to lenalidomide.
A subset of genes identified in the genome-scale resistance screen impairs lenalidomide-induced degradation of an IKZF3 degron reporter
We next sought to identify the genes that scored in the genome-scale resistance screen due to impaired CRL4CRBN-mediated degradation of IKZF1 and IKZF3. To monitor IKZF3 degradation, we generated a reporter in which the lenalidomide-responsive IKZF3 degron (aa 130-189)2 was linked to eGFP (Figure 2A; supplemental Figure 2A). We hypothesized that CRISPR-Cas9 inactivation of genes required for optimal CRL4CRBN function would impair degradation of the IKZF3 reporter, whereas those that engendered lenalidomide resistance through alternative pathways would not alter reporter degradation.
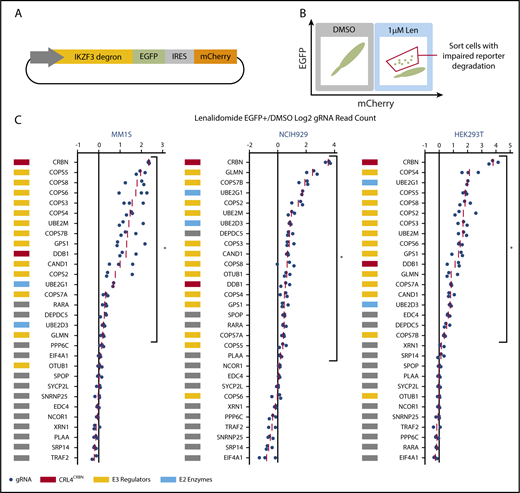
CRISPR-Cas9 inactivation of a subset of genes identified in the genome-scale resistance screen impairs lenalidomide-induced degradation of an IKZF3 degron reporter. (A) Schematic of the IKZF3 degron (aa 130-189) reporter vector. (B) Schematic of flow cytometry–based sorting of cells in which degradation of the IKZF3 degron reporter has been impaired by CRISPR-Cas9 inactivation of target genes. (C) Genes from the reporter screen ranked according to the average fold-change in the log2-transformed gRNA sequencing read counts (lenalidomide-treated eGFP+/DMSO). Fold-change values are normalized to the average fold-change of 12 control gRNAs. Each point represents an individual gRNA, and each point is the average of 3 infection replicates. *FDR < 0.05, 1-sided Wilcoxon rank-sum test.
CRISPR-Cas9 inactivation of a subset of genes identified in the genome-scale resistance screen impairs lenalidomide-induced degradation of an IKZF3 degron reporter. (A) Schematic of the IKZF3 degron (aa 130-189) reporter vector. (B) Schematic of flow cytometry–based sorting of cells in which degradation of the IKZF3 degron reporter has been impaired by CRISPR-Cas9 inactivation of target genes. (C) Genes from the reporter screen ranked according to the average fold-change in the log2-transformed gRNA sequencing read counts (lenalidomide-treated eGFP+/DMSO). Fold-change values are normalized to the average fold-change of 12 control gRNAs. Each point represents an individual gRNA, and each point is the average of 3 infection replicates. *FDR < 0.05, 1-sided Wilcoxon rank-sum test.
To ensure that genes influencing IKZF3 reporter degradation were not cell-type or tissue-type dependent, we performed the screen in lenalidomide-sensitive myeloma cell lines, MM1.S and NCIH929, as well as a lenalidomide-insensitive human embryonic kidney cell line, HEK239T. The Cas9-expressing IKZF3 degron reporter cells were transduced with a pooled library of gRNAs targeting the top 30 genes identified in the genome-scale screen. The library contained 3 gRNAs per gene chosen using an algorithm that ranks gRNAs based on predicted cutting efficiency17 and 12 control gRNAs. Cells transduced with the library were treated with DMSO or 1 μM lenalidomide for 20 hours and then, to identify cells in which degradation of the IKZF3 degron reporter was impaired, we used FACS to isolate cells which remained eGFP+ despite lenalidomide treatment (Figure 2B). We then used next-generation sequencing to quantify the frequency of read counts for a given gRNA in the unsorted DMSO-treated control cells and the sorted eGFP+ cells in the lenalidomide-treated experimental condition (supplemental Figure 2B).
In all 3 cell lines, the gRNAs targeting CRBN exhibited the greatest enrichment in the lenalidomide-treated eGFP+ cells (Figure 2C). Additional genes whose loss impaired degradation of the IZKF3 reporter in the 3 cell lines included CRL4CRBN members (CRBN, DDB1), cullin-RING ligase regulatory factors (GPS1, COPS2, COPS3, COPS4, COPS5, COPS7A, COPS7B, CAND1, UBE2M, GLMN), E2 ubiquitin-conjugating enzymes (UBE2D3 and UBE2G1), and the GATOR complex member DEPDC5 (FDR < 0.05, 1-sided Wilcoxon rank-sum test).
In summary, the genome-scale resistance screen and the IKZF3 degron reporter counterscreen nominated a core set of proteins, notably cullin-RING ligase neddylation cycle enzymes and E2 ubiquitin-conjugating enzymes, for which genetic inactivation promoted both lenalidomide resistance in MM1.S cells and impaired lenalidomide-induced degradation of an IKZF3 degron reporter in MM1.S, NCIH929, and HEK293T cells.
gRNAs targeting COPS5, UBE2M, UBE2D3, and UBE2G1 lead to lenalidomide resistance
We next sought to validate and understand the degree to which inactivation of the cullin-RING ligase regulatory factors and E2 ubiquitin-conjugating enzymes promoted lenalidomide resistance. To do so, we introduced gRNAs targeting COPS5, UBE2M, UBE2D3, and UBE2G1 into MM1.S-Cas9 cells expressing eGFP and mixed these cells at a 5:95 ratio with MM1.S-Cas9 cells transduced with control gRNAs. The cells were then grown for 20 days in DMSO or 1 μM lenalidomide and the proportion of eGFP+ cells was monitored every 4 days using flow cytometry (Figure 3A; supplemental Figure 3A). gRNAs targeting UBE2D3, UBE2G1, and the catalytic subunit of the COP9 signalosome, COPS5, granted a competitive advantage in the context of 1 μM lenalidomide, albeit less so than gRNAs targeting CRBN. Cells transduced with the UBE2M gRNA exhibited minor competitive advantage. From this, we concluded that inactivation of COPS5, UBE2D3, or UBE2G1 promotes lenalidomide resistance in MM1.S cells. The modest competitive advantage of UBE2M gRNA-infected cells may reflect the essentiality of UBE2M for cellular survival.
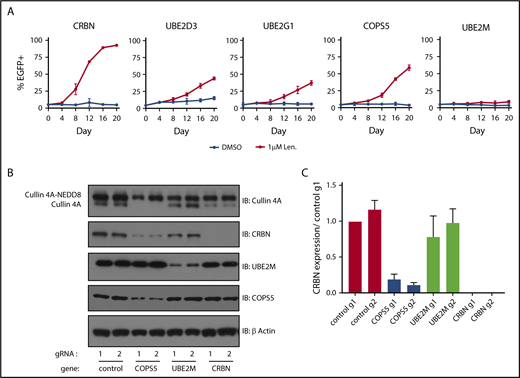
Inactivation of COPS5 and UBE2M promotes lenalidomide resistance and alters cullin 4 neddylation and CRBN levels. (A) MM1.S-Cas9 cells expressing EGFP were infected with gRNAs targeting indicated genes and mixed at a 5:95 ratio with control gRNA-infected cells. The cells were then grown for 20 days in the presence of DMSO or 1 μM lenalidomide and the percentage of EGFP+ cells was quantified every 4 days using flow cytometry. Data points are average of 3 technical replicates and error bars represent a 95% confidence interval. (B) HEK293T single-cell heterozygous for frameshifting mutations in COPS5 and UBE2M were generated via CRISPR-Cas9 editing. Protein lysates were harvested and immunoblotted (IB) for the indicated proteins. Labels indicate neddylated and unneddylated forms of cullin 4A. Data are representative of 3 experimental replicates. (C) Quantification of CRBN levels blots from panel B across 3 experimental replicates. Values are normalized to control guide 1 (g1) and the respective β-actin loading controls. Error bars represent standard error of the mean.
Inactivation of COPS5 and UBE2M promotes lenalidomide resistance and alters cullin 4 neddylation and CRBN levels. (A) MM1.S-Cas9 cells expressing EGFP were infected with gRNAs targeting indicated genes and mixed at a 5:95 ratio with control gRNA-infected cells. The cells were then grown for 20 days in the presence of DMSO or 1 μM lenalidomide and the percentage of EGFP+ cells was quantified every 4 days using flow cytometry. Data points are average of 3 technical replicates and error bars represent a 95% confidence interval. (B) HEK293T single-cell heterozygous for frameshifting mutations in COPS5 and UBE2M were generated via CRISPR-Cas9 editing. Protein lysates were harvested and immunoblotted (IB) for the indicated proteins. Labels indicate neddylated and unneddylated forms of cullin 4A. Data are representative of 3 experimental replicates. (C) Quantification of CRBN levels blots from panel B across 3 experimental replicates. Values are normalized to control guide 1 (g1) and the respective β-actin loading controls. Error bars represent standard error of the mean.
To determine which CRISPR-Cas9-induced DNA edits contributed to competitive advantage in the context of lenalidomide treatment, we collected genomic DNA from the DMSO and 1 μM lenalidomide treated cells at day 0 and day 20, PCR-amplified the gRNA target site, and performed next-generation sequencing of the amplicons. The gRNAs targeting CRBN, COPS5, UBE2D3, and UBE2G1 each generated an outgrowth of frameshifting mutations after 20 days of lenalidomide treatment, with cells infected with CRBN-targeting gRNAs exhibiting the greatest increase in frame-shifting mutations (supplemental Figure 3B). Cells transduced with UBE2M targeting gRNA showed a subtle outgrowth of frameshifting mutations, which may be due to the essentiality of UBE2M.
In sum, the results of the genome-scale screen, counterscreen, and validation assays demonstrate that CRL4CRBN is the fundamental target of lenalidomide, and that a broader network of proteins associated with E3 ubiquitin ligase activity contribute to maximal lenalidomide-mediated function.
Inactivation of the COP9 signalosome and UBE2M alters cullin 4A neddylation and CRBN levels
UBE2M and the COP9 signalosome regulate the activity of cullin-RING ligases via neddylation, a cycle in which the ubiquitin-like NEDD8 protein is covalently added and removed from the cullin backbone of cullin-RING ligases.18 Specifically, UBE2M activates cullin-RING ligases via neddylation of the cullin backbone,19-22 whereas deneddylation is carried out by the COP9 signalosome and returns the ligase to an inactive form.23,24
Having confirmed that gRNAs targeting UBE2M and COPS5 (the catalytic subunit of the COP9 signalosome) increase the survival of MM1.S cells in the presence of lenalidomide and prevent efficient degradation of an IKZF3 reporter in multiple cell lines, we sought to understand how UBE2M and COPS5 loss affects cullin 4 neddylation status and CRBN levels. To do so, we generated HEK293T subclones with heterozygous inactivation of UBE2M (clone 1, 68% frameshifting indels; clone 2, 50% frameshifting indels) or COPS5 (clone 1, 67% frameshifting indels; clone 2, 70% frameshifting indels). We could not isolate cells with homozygous inactivating mutations in UBE2M or COPS5, suggesting that UBE2M and COPS5 may be essential genes in HEK293T cells.
In keeping with the known role of UBE2M as a neddylation enzyme, immunoblots for cullin 4A in UBE2M+/− cells demonstrated an increase in the proportion of cullin 4A in the lower molecular weight band consistent with hyponeddylation, whereas COPS5+/− cells exhibited an increase in the higher molecular weight band of cullin 4A consistent with hyperneddylation (Figure 3B-C; supplemental Figure 3C-D). Hyperneddylation of cullin 4A was in turn associated with a reduction in CRBN levels. The impact on CRBN levels was rescued with neddylation inhibitor, MLN4924, and proteasome inhibitor, MG132 (supplemental Figure 3C).
These results suggest that heterozygous loss of UBE2M influences CRL4CRBN-mediated degradation of IKZF3 by rendering a greater proportion of the available ligases inactive due to hyponeddylation of cullin 4A. Conversely, heterozygous loss of COPS5 was associated with decreased CRBN levels secondary to cullin 4A hyperneddylation, similar to the effects of COP9 signalosome inactivation described for Skip-cullin-Fbox (SCF) E3 ligases and their substrate receptors.25,26
UBE2G1 and UBE2D3 cooperate to polyubiquitinate lenalidomide-induced substrates of CRL4CRBN
E2 ubiquitin-conjugating enzymes dictate the number of ubiquitin moieties added to a substrate as well as which lysine in ubiquitin is used to form the links in polyubiquitin chains. These features in turn determine the outcome of ubiquitination, including changes in subcellular localization, formation of new protein-protein interactions, initiation of NF-κB signaling or DNA repair, or degradation by the 26s proteasome.6 Despite the relevance of E2 ubiquitin-conjugating enzymes to E3 ligase function, the micromolar affinity of the E2-E3 interaction has made it difficult to use binding assays to identify which of the ∼35 E2 ubiquitin-conjugating enzymes are used by a given E3 ligase.
Having used multiple functional genetic assays to establish that loss of UBE2D3 and UBE2G1 impairs lenalidomide function, we next asked whether simultaneous loss of UBE2G1 and UBE2D3 would have an additive or synergistic effect on IKZF3 degradation. To answer this question, we generated UBE2G1−/− and/or UBE2D3−/− HEK293T single-cell clones, transduced them with the IKZF3 degron reporter, and dosed the cells with a range of lenalidomide concentrations (Figure 4A; supplemental Figure 4A). Consistent with our prior data, UBE2G1−/− clones exhibited a greater reduction in IKZF3 reporter degradation in comparison with UBE2D3−/− clones. Moreover, simultaneous loss of UBE2D3 and UBE2G1 resulted in an 50% effective concentration (EC50) shift, which was greater than the additive effects of the single-gene knockouts.
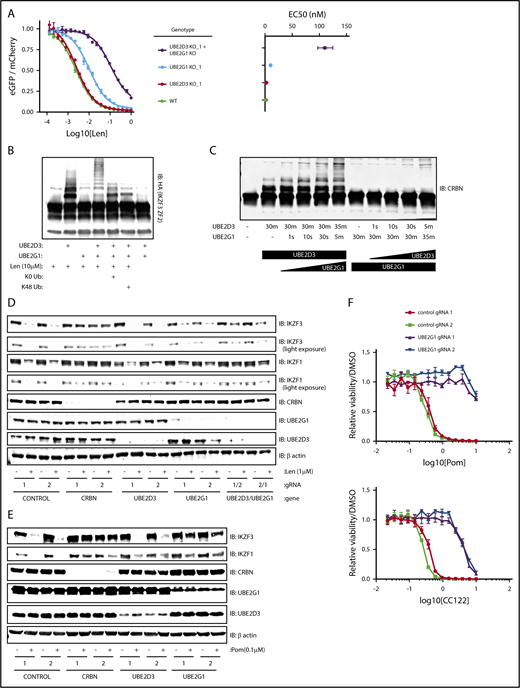
UBE2D3 and UBE2G1 cooperate to polyubiquitinate lenalidomide-induced substrates of CRL4CRBN. (A) HEK293T single-cell clones in which UBE2G1 and/or UBE2D3 were knocked out via CRISPR-Cas9 introduction of frameshifting DNA edits were transduced with an the IKZF3 degron reporter and then treated with a titration of lenalidomide. After 20 hours, the EGFP/mCherry ratio was assayed via flow cytometry. Data points are an average of 3 experimental replicates and error bars represent standard error of the mean. Corresponding EC50 values are shown for each genotype, with error bars representing the 95% confidence interval. (B) An in vitro ubiquitination reaction containing recombinant CRL4CRBN, and HA-IKZF3 (aa146-168) derived from HEK293T cells, and the indicated reaction components was run for 30 minutes and immunoblotted (IB) for HA. Data are representative of 3 experimental replicates. (C) In vitro CRBN autoubiquitination reactions in which UBE2D3 or UBE2G1 were added sequentially, with each enzyme individually incubated with CRBN for 30 minutes followed by addition of the second E2 enzyme for the indicated amount of time. In vitro ubiquitination reactions were immunoblotted as indicated. (D) MM1.S-Cas9 cells were infected with gRNAs targeting the indicated genes, treated with DMSO or 1 μM lenalidomide for 20 hours, then lysates were harvested and immunoblotted as indicated. Data are representative of 3 experimental replicates. (E) NCIH929-Cas9 cells were infected with gRNAs targeting the indicated genes, treated with DMSO or 0.1 μM pomalidomide for 20 hours, then lysates were harvested and immunoblotted as indicated. (F) NCIH929-Cas9 cells infected with control or UBE2G1 gRNAs were treated with DMSO, pomalidomide, or CC-122 for 10 days, with dosing at day 0 and day 5. Dose response curves show viability normalized to DMSO treatment. Data points are an average of 3 experimental replicates and error bars represent standard error of the mean for each drug concentration.
UBE2D3 and UBE2G1 cooperate to polyubiquitinate lenalidomide-induced substrates of CRL4CRBN. (A) HEK293T single-cell clones in which UBE2G1 and/or UBE2D3 were knocked out via CRISPR-Cas9 introduction of frameshifting DNA edits were transduced with an the IKZF3 degron reporter and then treated with a titration of lenalidomide. After 20 hours, the EGFP/mCherry ratio was assayed via flow cytometry. Data points are an average of 3 experimental replicates and error bars represent standard error of the mean. Corresponding EC50 values are shown for each genotype, with error bars representing the 95% confidence interval. (B) An in vitro ubiquitination reaction containing recombinant CRL4CRBN, and HA-IKZF3 (aa146-168) derived from HEK293T cells, and the indicated reaction components was run for 30 minutes and immunoblotted (IB) for HA. Data are representative of 3 experimental replicates. (C) In vitro CRBN autoubiquitination reactions in which UBE2D3 or UBE2G1 were added sequentially, with each enzyme individually incubated with CRBN for 30 minutes followed by addition of the second E2 enzyme for the indicated amount of time. In vitro ubiquitination reactions were immunoblotted as indicated. (D) MM1.S-Cas9 cells were infected with gRNAs targeting the indicated genes, treated with DMSO or 1 μM lenalidomide for 20 hours, then lysates were harvested and immunoblotted as indicated. Data are representative of 3 experimental replicates. (E) NCIH929-Cas9 cells were infected with gRNAs targeting the indicated genes, treated with DMSO or 0.1 μM pomalidomide for 20 hours, then lysates were harvested and immunoblotted as indicated. (F) NCIH929-Cas9 cells infected with control or UBE2G1 gRNAs were treated with DMSO, pomalidomide, or CC-122 for 10 days, with dosing at day 0 and day 5. Dose response curves show viability normalized to DMSO treatment. Data points are an average of 3 experimental replicates and error bars represent standard error of the mean for each drug concentration.
The synergistic effects of UBE2D3 and UBE2G1 inactivation next led us to ask whether UBE2D3 and UBE2G1 have redundant or distinct roles in lenalidomide-induced IKZF3 ubiquitination. To determine their respective roles, we performed an in vitro CRL4CRBN-IKZF3 ubiquitination reaction and immunoblotted for the IKZF3 substrate (Figure 4B). In the presence of UBE2D3 alone, we observed the appearance of 2 higher molecular weight bands, consistent with monoubiquitination at 2 sites of the IKZF3 reporter substrate. However, in the presence of UBE2G1 alone, we did not observe evidence of IKZF3 ubiquitination. When both UBE2D3 and UBE2G1 were added to the reaction we saw a “laddering” pattern of higher molecular weight forms of the IKZF3 substrate, consistent with polyubiquitination. These results indicate that UBE2D3 and UBE2G1 play distinct and complementary roles in lenalidomide-mediated ubiquitination of substrates.
To determine whether the observed ubiquitin chain formation relied on K48 ubiquitin linkages, we added a K0 ubiquitin mutant to the in vitro ubiquitination reaction, which can only participate in monoubiquitination, and a K48R ubiquitin mutant, which retains all lysine residues except K48. In the presence of either the K0 or K48R ubiquitin mutants, the ubiquitin laddering was collapsed to form 2 monoubiquitination bands, suggesting that K48 is the lysine residue required to form polyubiquitin chains on the IKZF3 substrate in the CRL4CRBN-IKZF3 ubiquitination reaction. Additionally, in keeping with the known mechanism of action of the drug, ubiquitination was only observed in the presence of lenalidomide (Figure 4B). These results confirm that UBE2D3 and UBE2G1 cooperate to promote K48-linked polyubiquitination of IKZF3 in vitro.
To determine whether monoubiquitination by UBE2D3 or polyubiquitination by UBE2G1 is rate limiting, we performed an in vitro CRL4CRBN autoubiquitination reaction for 30 minutes in the presence of either UBE2D3 or UBE2G1, then added the second E2 enzyme and assayed ubiquitin chain formation at short intervals (Figure 4C). After 30 minutes of incubation with UBE2D3, we observed a monoubiquitination band, and noted substrate polyubiquitination as early as 10 seconds following the subsequent addition of UBE2G1. Conversely, a 30-minute incubation in the presence of UBE2G1 resulted in no ubiquitination, and the subsequent addition of UBE2D3 did not result in detectable polyubiquitination until 5 minutes. We therefore concluded that lenalidomide-induced monoubiquitination by UBE2D3 is the rate-limiting step in the reaction, after which UBE2G1 is capable of rapid polyubiquitination of the substrate.
Having established that UBE2D3 and UBE2G1 follow a “prime-extend” paradigm to promote CRL4CRBN-induced polyubiquitination of IZKF3 in vitro, we next sought to determine whether UBE2D3 and UBE2G1 are required for lenalidomide-induced degradation of endogenous IKZF1 and IKZF3. To this end, MM1.S-Cas9 and NCIH929-Cas9 cells were infected with gRNAs targeting CRBN, UBE2D3, and UBE2G1, treated with DMSO or 1 μM lenalidomide for 20 hours, and then protein lysates were immunoblotted for IKZF1 and IKZF3 (Figure 4D; supplemental Figure 4B). In both cell lines, introduction of CRBN gRNAs resulted in near-complete stabilization of endogenous IKZF1 and IKZF3 in the presence of lenalidomide. In MM1.S, UBE2G1 and UBE2D3 gRNAs individually had minimal effects, however, cells simultaneously expressing gRNAs targeting UBE2D3 and UBE2G1 exhibited partial stabilization of IKZF3 and IKZF1 in lenalidomide-treated cells. In NCIH929 cells, introduction of gRNAs targeting either UBE2D3 or UBE2G1 resulted in stabilization of IKZF1 and IKZF3 with lenalidomide treatment. These data indicate that UBE2D3 and UBE2G1 are required for efficient lenalidomide-induced degradation of the endogenous IKZF1 and IKZF3 transcription factors in multiple myeloma cell lines.
Lastly, we sought to understand whether additional IMiD analogs of lenalidomide similarly require UBE2D3 and UBE2G1 to mediate efficient ubiquitination and degradation of endogenous IKZF1 and IKZF3. To address this question, NCIH929-Cas9 cells were infected with gRNAs targeting CRBN, UBE2D3, and UBE2G1. Cells were treated for 20 hours with DMSO or 0.1 μM pomalidomide, and protein lysates were immunoblotted for IKZF1 and IKZF3. Although introduction of UBE2D3 gRNAs did not significantly stabilize IKZF1 and IKZF3, UBE2G1 gRNAs demonstrated robust stabilization of IKZF1 and IKZF3 in the setting of pomalidomide treatment. We next investigated the capacity of UBE2G1 to mediate the viability effects of pomalidomide and analog, CC-122. IMiD-sensitive NCIH929 myeloma cells were treated with IMiD for a total of 10 days, with dosing at days 0 and 5. Introduction of UBE2G1 gRNAs impaired efficient killing of NCIH929 cells in both pomalidomide and CC-122 treatment conditions. These results indicate that UBE2G1 is a critical E2 ubiquitin-conjugating enzyme required for maximal pomalidomide and CC-122 mediated CRL4CRBN activity in myeloma cells.
In aggregate, our genetic screens and biochemical studies highlight a network of proteins involved in lenalidomide-dependent function of the CRL4CRBN E3 ubiquitin ligase. The genes identified include enzymes that regulate neddylation and substrate ubiquitination and play a role in a common biochemical pathway (Figure 5).
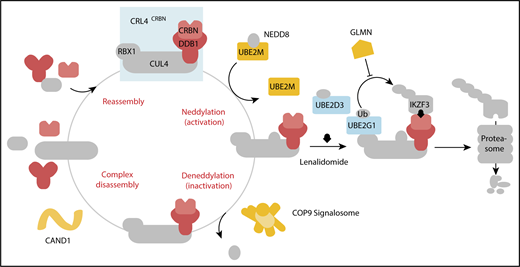
Cullin-RING ligase neddylation enzymes, COP9 signalosome and UBE2M, and the E2 enzymes, UBE2G1 and UBE2D3, regulate lenalidomide-induced CRL4CRBNactivity. Proteins in color scored in the genome-scale CRISPR-Cas9 screen.
Cullin-RING ligase neddylation enzymes, COP9 signalosome and UBE2M, and the E2 enzymes, UBE2G1 and UBE2D3, regulate lenalidomide-induced CRL4CRBNactivity. Proteins in color scored in the genome-scale CRISPR-Cas9 screen.
Discussion
In this study, we used functional genetic screens to identify a set of proteins that are required for the antimyeloma properties of lenalidomide due to their regulation of CRL4CRBN activity and ubiquitination of target substrates. We specifically focused on UBE2M and the COP9 signalosome, enzymes which regulate CRL4CRBN activity via neddylation and deneddylation of CRL4CRBN, and the E2 ubiquitin-conjugating enzymes UBE2D3 and UBE2G1, which operate via a “prime-extend” mechanism to ubiquitinate CRL4CRBN target substrates with K48-linked polyubiquitin chains. The identification of the molecular components that regulate the drug-dependent activity of CRL4CRBN will facilitate a better understanding of the parameters controlling the therapeutic efficacy of lenalidomide, the mechanisms underlying innate and acquired lenalidomide resistance, and the identification of novel disease contexts in which lenalidomide may be beneficial.
The genome-wide CRISPR-Cas9 screen demonstrated that CRBN is the most important nonessential gene required for the antimyeloma effects of lenalidomide. The prevalence of cullin-RING ligase regulatory genes among the strongest hits in the screen suggests, more specifically, that CRBN’s role as a substrate receptor for the CRL4CRBN E3 ubiquitin ligase underlies its requirement for the therapeutic effects of lenalidomide.
Although resistance in del(5q) MDS patients is thought to largely be mediated via inactivation of p53,4,27 a downstream effector of CK1α degradation, the mechanism of lenalidomide resistance in multiple myeloma remains unclear. The necessity of CRBN in our genome-scale screen complements work demonstrating that CRBN is essential for lenalidomide’s antimyeloma effects and ability to degrade IKZF1 and IZKF3,2,3,28,29 and suggests that CRBN is likely to be a central node of acquired resistance in myeloma patients. Loss-of-function mutations in CRBN have not been found in patients who have relapsed on lenalidomide-containing regimens30 ; however, several studies have noted changes in CRBN expression levels and isoform usage at relapse.31-33 Comprehensive assessment of gene or protein expression of the malignant clone in a large number of multiple myeloma patients pre- and postrelapse will clarify the extent to which CRBN is indeed the principal mechanism of lenalidomide resistance in patients and whether the genes identified here may play a role in lenalidomide efficacy in patients.
These results expand our understanding of IMiD mechanism of action and complements previously published lenalidomide-focused RNA interference (RNAi) screens. A multiple myeloma RNAi screen of 6992 “druggable” genes was designed to identify genes whose loss increased the efficacy of lenalidomide.34 In contrast to our approach, this screen identified genes which modulate core multiple myeloma signaling pathways rather than modulators of the ubiquitin proteasome system. Additionally, a genome-scale RNAi screen in a MDS cell line similarly interrogated lenalidomide resistance.35 Our findings diverge in that CRBN did not score in that assay, nor did regulators of CRL4CRBN. This likely reflects different biology in the screened cell types, alternate drug treatment timelines, and the improvement in signal-to-noise ratio observed with genome-scale CRISPR-Cas9 screens. Lastly, our results are concordant with a genome-scale short hairpin RNA screen for factors regulating CRL4CDT2-mediated degradation of the replication origin licensing factor CDT1 in UV-irradiated HeLa cells.36 As in our study, CRL4CDT2 activity requires components of the CRL4CDT2 complex and subunits of the COP9 signalosome. Differences include the identification p97, UFD1, NAE1, and proteasomal subunits, proteins which may not have scored in our assay due to their effects on myeloma cell viability. Additionally, our work adds regulatory factors including the E2 ubiquitin conjugating enzymes, importantly in the context of a drug-inducible system in clinical use.
The micromolar-binding affinity of E2 and E3 enzymes has made it difficult to use binding assays to identify which of the ∼35 E2 enzymes are used by a given E3 ligase.6 Here, we have used functional genetics and in vitro assays to demonstrate that UBE2D3 primes CRL4CRBN target substrates via monoubiquitination, after which UBE2G1 polyubiquitinates them with K48-linked ubiquitin chains. The “prime-extend” E2 paradigm has been observed in the context of other ubiquitin ligases6 and differs from the usage of a single E2, UBE2R1 (Cdc34), by the canonical SCF family of CRLs.37-39 Although UBE2D3 and UBE2G1 are required for maximal CRL4CRBN-mediated degradation of IKZF1 and IKZF3, ubiquitination and degradation still occurs in their absence. It therefore remains to be determined which E2s are redundant with UBE2D3 and UBE2G1. Additional questions remain, including whether there are unique contexts in which CRL4CRBN might rely on a distinct set of E2 enzymes, whether other CRL4-containing E3 ligases use UBE2G1, and what structural motifs on CRL4CRBN or its substrates dictate E2 usage and specificity. More broadly, CRISPR screens may be a generalizable approach for the identification of E2-E3 pairs.
The regulatory biochemical network identified in our assays holds broader implications for what is likely to become a growing class of drugs with the ability to mediate targeted degradation of proteins via cullin-RING ligases. Indeed, the lenalidomide analogs thalidomide, pomalidomide, CC-122,40 and CC-88541 also mediate CRL4CRBN-dependent degradation of therapeutic targets. Even more recently, the compound indisulam was shown to mediate CRL4DCAF15-dependent degradation of the splicing factor RBM39.42,43 Additionally, potential therapeutic agents are being generated in which thalidomide is conjugated to small molecule binders of other proteins, thus creating chimeric compounds that induce CRL4CRBN-mediated degradation of a target protein of interest.44-46 Further work will be required to understand the extent to which the genes identified in our genome-scale screen will similarly be required for these novel compounds. Lastly, the gain-of-function mechanism that lenalidomide and these compounds share will render positive-selection CRISPR-Cas9 screening an effective tool both to highlight potential mechanisms of drug resistance and to deepen our biological understanding of ubiquitin ligase function and regulation.
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Acknowledgments
This work was supported by the National Institutes of Health (National Heart, Lung, and Blood Institute R01HL082945 and National Cancer Institute P30CA006516 [B.L.E.], National Cancer Institute R01CA214608 [E.S.F.]), the Edward P. Evans Foundation (B.L.E.), the Leukemia & Lymphoma Society (B.L.E.), the Starr Cancer Consortium (B.L.E.), and the Howard Hughes Faculty Scholars Program (B.L.E.). The project described was supported by award number T32GM007753 from the National Institutes of Health, National Institute of General Medical Sciences.
The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institute of General Medical Sciences or the National Institutes of Health.
Authorship
Contribution: B.L.E. developed the initial concept for the genome-scale screen; G.S.C. and Q.L.S. designed the parameters for the genome-scale screen and counterscreen; Q.L.S. performed the genome-scale screen, counterscreen, and lenalidomide titration of HEK293T single-cell clones; G.S.C. aided with analysis of the next-generation sequencing results; J.A.G. and Q.L.S. together performed cellular competition assays; J.A.G. performed CRISPR and single-cell cloning of KO cell lines, substrate analysis, neddylation analyses, drug-dependent viability assays and in vitro ubiquitination assays; E.S.F. provided guidance and reagents; and Q.L.S., J.A.G., and B.L.E. analyzed data and drafted the manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
The current affiliation for G.S.C. is Discovery Science, Janssen Research and Development (Johnson & Johnson), Spring House, PA.
Correspondence: Benjamin L. Ebert, Dana-Farber Cancer Institute, 450 Brookline Ave, D1610A, Boston, MA 02115; e-mail: benjamin_ebert@dfci.harvard.edu.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal