

In this issue of Blood, describe key proteins required for the anti–myeloma properties of lenalidomide. By performing CRISPR-Cas9 functional genetic screening, they identify 2 E2 ubiquitin-conjugating enzymes (UBE2D3 and UBE2G1) and the COP9 signalosome as essential for lenalidomide-dependent CRL4CRBN function in myeloma due to their regulation of CRL4CRBN activity and ubiquitination of target substrates.1
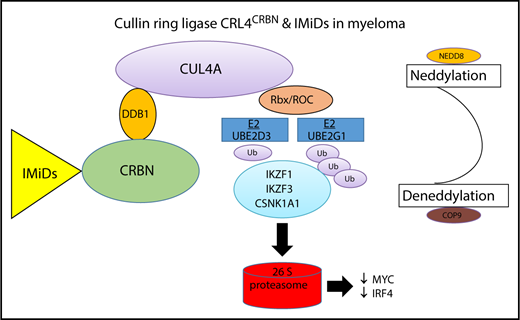
Schematic representation of the CLR4CRBN complex in myeloma cells. IMiDs bind to CRBN, a substrate receptor protein for the CRL4A E3 ubiquitin ligase complex, and cause proteasomal degradation of IKZF1, IKZF3, and CSNK1A1 consecutively downregulating IRF4 and Myc and resulting in MM-associated cytotoxicity. The blue boxes indicate the 2 E2 ubiquitin-conjugating enzymes, UBE2D3 and UBE2G1, reported by Sievers et al as essential for lenalidomide-dependent CRL4CRBN function in MM.
Schematic representation of the CLR4CRBN complex in myeloma cells. IMiDs bind to CRBN, a substrate receptor protein for the CRL4A E3 ubiquitin ligase complex, and cause proteasomal degradation of IKZF1, IKZF3, and CSNK1A1 consecutively downregulating IRF4 and Myc and resulting in MM-associated cytotoxicity. The blue boxes indicate the 2 E2 ubiquitin-conjugating enzymes, UBE2D3 and UBE2G1, reported by Sievers et al as essential for lenalidomide-dependent CRL4CRBN function in MM.
The story of immunomodulatory drugs (IMiDs), such as thalidomide, lenalidomide, and pomalidomide, in multiple myeloma (MM) is quite surreal. In the late 1950s, thalidomide was used to treat morning sickness but was discontinued in the early 1960s after it was found to cause birth defects. Despite its tragic past, thalidomide has reentered clinical practice due to its immunomodulatory and antiangiogenic properties and found to be effective in the treatment of MM in 2006 when was approved by the Food and Drug Administration for the treatment of patients with relapsed MM. Since then, its derivatives lenalidomide and pomalidomide have proven their activity and are now widely used in newly diagnosed and relapsed refractory MM. In 2010, Ito et al2 reported that cereblon (CRBN), a substrate receptor of the CRL4 E3 ubiquitin ligase, was a direct target of IMiDs and was required for the teratogenic activities of thalidomide. We also now recognize that in MM cells IMiDs bind to CRBN and neomorphe the substrates recruitment to the Cul4A Cullin ring E3 ligase, inducing Ikaros (IKZF1), Aiolos (IKZF3), zinc finger protein 91 (ZFP91), and casein kinase 1 α (CSNK1A1) ubiquitylation and proteasomal degradation. These events lead to specific and sequential transcriptional repression of MYC and IRF4, 2 essential factors for myeloma cells’ survival.3,4 However, it is not clear how myeloma cells acquire resistance to IMiDs, “beyond CRBN,” and whether other neo-substrates essential for MM survival are targeted by IMiDs for degradation.
In the last few years, clustered regularly interspaced short palindromic repeats-CRISPR associated nuclease 9 (CRISPR-CAS9) systems have emerged as versatile, reliable, and convenient genome-editing tools and have opened a new era in molecular biology.5 Herein, by performing a genome-scale CRISPR-CAS9 screen in lenalidomide-sensitive MM cell lines, Sievers et al identified genes that, when inactivated, diminish the effects of lenalidomide and investigated their roles in the modulation of the CRL4CRBN ubiquitin ligase.
The ubiquitin proteasome system is an important posttranslational regulatory process for proteins that alters their stability, localization, or interaction properties.6 It is mediated by the successive enzymatic reactions involving activating enzymes, conjugating enzymes (E2), and ligase enzymes (E3), resulting in an isopeptide link between the C-terminus glycine residue of ubiquitin and a specific lysine on a target protein.7 In this process, the E3 ligase determines the substrate specificity and stability. Among the diverse E3 enzymes, the cullin-RING ubiquitin ligases are the largest E3 ligase family in eukaryotes and can be regulated by a process termed neddylation that involves cullin-associated protein Nedd88 and deneddylation, which removes the Nedd8 moiety and requires the isopeptidase activity of the COP9 signalosome.9 Among the 6 human cullins family, the cullin4 (CUL4) subfamily comprises 2 members, CUL4A and CUL4B, which share 83% sequence identity and functional redundancy. It consists of a RING finger domain protein, CUL4 scaffold protein, and DDB1-CUL4 associate substrate receptors. Recent studies have highlighted the role of CUL4A complexes in regulating substrates involved in the cell cycle, signaling, tumor suppression, DNA damage response, and chromatin remodeling and suggested CRL4A as a promising novel target for cancer therapy.10
CRBN is a substrate receptor protein for the CRL4A E3 ubiquitin ligase complex, and drugs like IMiDs have been reported to be able to inhibit or alter the substrate specificity of the E3 ligase activity of CRL4ACRBN. Therefore, the identification of the molecular components that regulate IMiDs-dependent activity of CRL4CRBN will allow a better understanding of the parameters controlling their therapeutic efficacy and help to identify the mechanisms underlying their resistance. As such, through an elegant genome-wide CRISPR-CAS9 screen, the authors identified regulators of cullin-RING ligase neddylation as well as the elusive CRL4ACRBN E2 conjugating enzymes. By using functional genetics and in vitro assays, they demonstrated that UBE2D3 primes CRL4CRBN target substrates via monoubiquitination, after which UBE2G1 polyubiquinates them via K48-linked ubiquitin chains. Furthermore, loss of UBE2M or members of the COP9 signalosome resulted in altered neddylation of CUL4A and impaired lenalidomide-dependent CRL4CRBN activity (see figure).
Overall, the results presented here are novel and pivotal to expand our understanding of IMiDs’ mechanisms of action and likely to elucidate mediators of acquired resistance. Additional questions remain, including whether other CRL4-containing E3 ligases utilize UBE2G1/UBE2D3 conjugating enzymes and what structural motifs on CRL4CRBN or its substrates dictate E2 usage and specificity. Nevertheless, Sievers et al, by CRISPRing the CRL4CRBN ring, established key proteins required for lenalidomide-dependent CRL4CRBN function in MM and provided us with a deeper understanding of this ubiquitin ligase function and regulation. The next step will be to therapeutically exploit these newly identified targets to augment or restore the activities of IMiDs in MM.
Conflict-of-interest disclosure: The author declares no competing financial interests.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal