

Mutations in components of the messenger RNA (mRNA) splicing machinery are the most commonly detected class of acquired genetic variant in blood and marrow cells from patients with myelodysplastic syndromes (MDS). However, how do these spliceosome mutations result in the core pathological features of MDS, such as failed hematopoiesis, cell dysmorphology, and clonal dominance? In this issue of Blood, move us closer to understanding the mechanisms of splicing-induced changes, as they report comprehensive RNA sequencing data from 84 patients with MDS, including 28 with splicing mutations.1
In 2011, Yoshida and colleagues in Japan provided critical insight into MDS pathophysiology when they reported a high frequency of somatic mutations in spliceosome components in MDS and, to a lesser extent, other myeloid neoplasms. The most common spliceosome mutations observed in MDS include U2AF1, with mutation hotspots in the zinc finger domains at codons S34 and R156/Q157; SF3B1, with hotspots such as K700 and K666 in the HEAT domains; and SRSF2, with a hotspot at codon P95, in a hinge region between the RNA recognition domain and a serine-arginine (SR)-rich domain (SR domains are common in spliceosome-associated proteins).2 SF3B1 mutations in MDS are tightly linked to the ring sideroblast phenotype, which is seen in 15% to 20% of patients and is associated with lower-risk disease; almost all patients with ring sideroblasts have SF3B1 mutations.3
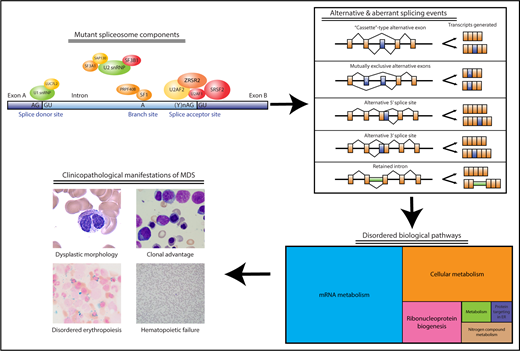
Components of the spliceosome that are recurrently mutated in MDS and their relationship to disease-associated clinicopathological features. Acquired spliceosome mutations, especially in components important for 3′ splice site recognition, are observed in more than one-half of MDS patients, particularly those with ring sideroblasts (SF3B1) or myeloproliferative neoplasm overlap features (SRSF2). The 3 most commonly mutated splicing factors (U2AF1, SRSF2, SF3B1) are depicted in red, whereas other less commonly observed mutations (ZRSR2, SF3A1, U2AF2, PRPF40B, LUC7L2, SF1, SAP130) are depicted in orange-yellow. These mutations result in a wide variety of alternative and aberrant splicing events in CD34+ or more differentiated cells, which can result in reduced, increased, or altered function in encoded proteins important for a broad range of cellular pathways. It is not yet clear how alterations in these pathways as a consequence of aberrant and alternative splicing result in MDS-associated clinical features, including dysplastic blood cell morphology, marrow failure, and clonal outgrowth and instability. ER, endoplasmic reticulum. Photomicrographs in lower left courtesy of ASH Image Bank (imagebank.hematology.org, #2117, 60051, 60183, and 61365) and are copyright the American Society of Hematology. The lower right section of the figure has been modified from Figure 2A in the article by Pellagatti et al that begins on page 1225.
Components of the spliceosome that are recurrently mutated in MDS and their relationship to disease-associated clinicopathological features. Acquired spliceosome mutations, especially in components important for 3′ splice site recognition, are observed in more than one-half of MDS patients, particularly those with ring sideroblasts (SF3B1) or myeloproliferative neoplasm overlap features (SRSF2). The 3 most commonly mutated splicing factors (U2AF1, SRSF2, SF3B1) are depicted in red, whereas other less commonly observed mutations (ZRSR2, SF3A1, U2AF2, PRPF40B, LUC7L2, SF1, SAP130) are depicted in orange-yellow. These mutations result in a wide variety of alternative and aberrant splicing events in CD34+ or more differentiated cells, which can result in reduced, increased, or altered function in encoded proteins important for a broad range of cellular pathways. It is not yet clear how alterations in these pathways as a consequence of aberrant and alternative splicing result in MDS-associated clinical features, including dysplastic blood cell morphology, marrow failure, and clonal outgrowth and instability. ER, endoplasmic reticulum. Photomicrographs in lower left courtesy of ASH Image Bank (imagebank.hematology.org, #2117, 60051, 60183, and 61365) and are copyright the American Society of Hematology. The lower right section of the figure has been modified from Figure 2A in the article by Pellagatti et al that begins on page 1225.
Long before recurrent spliceosome mutations were discovered, investigators had reported individual MDS-associated transcript variants with functional consequences resulting from alternative splicing, as had been observed in other neoplasms.4 For instance, in the 1990s, several groups described an imbalance in antiapoptotic BCL-xL vs proapoptotic BCL-xS in MDS CD34+ cells, an imbalance that contributes to the excessive intramedullary apoptosis and marrow failure characteristic of early MDS.5 Both BCL-xL and BCL-xS are transcriptional variants of the BCL2L1 gene that result from alternative splicing. Similarly, missplicing of cell surface apoptotic regulators such as Fas, tumor necrosis factor, and TRAIL receptors and their ligands results in altered apoptotic sensitivity in myeloid malignancies.6 Aberrant splicing events leading to truncated variants or nonsense mediated decay of ATRX, a chromatin-remodeling factor, are associated with the rare but phenotypically striking syndrome of acquired α−thalassemia in MDS.7
Once recurrent spliceosome mutations were recognized in myeloid neoplasia, the first investigations into the downstream consequences of these mutations focused on iron homeostasis, heme biosynthesis, and erythroid terminal maturation, because of the strong genotype-phenotype link between SF3B1 and ring sideroblasts.8 A pathophysiologic role was described for SF3B1 mutation-induced aberrant splicing in ABCB7, a membrane-associated adenosine triphosphate–binding cassette transporter that is important in shuttling heme from mitochondrion to the cytosol.9 This was an intellectually satisfying observation, because germ line mutations in ABCB7 are associated with a form of X-linked sideroblastic anemia with ataxia, and Boultwood and colleagues had described altered expression of ABCB7 in MDS with ring sideroblasts in 2008.10
The genome-wide work of Pellagatti et al now moves us beyond candidate gene approaches. The investigators studied both CD34+ cells and several subtypes of committed hematopoietic progenitors, which is an important comparison, because alternative splicing patterns can change during cell differentiation. More than 100 genes were noted to be distinctly aberrantly spliced with each of the 3 common spliceosome mutations, and there were also differences in splicing events resulting from distinct alleles of a single splicing factor (eg, U2AF1 S34 vs Q157). The majority of aberrantly spliced genes associated with each mutated splicing factor were different from those associated with other factors, but there was some overlap, and a few variants even correlated with clinical parameters, including survival.
The investigators then performed gene ontogeny and pathway analysis in order to try to gain some insight into likely functional consequences of the panoply of recurrent transcript variants (see figure). Some of the implicated pathways were not surprising, such as RNA processing and translation, but others had not been suspected to be involved in MDS previously, such as sirtuin histone deacetylase signaling. This interesting and comprehensive analysis will serve as a benchmark for future work on the consequences of splicing mutations in MDS.
The next steps must include functional assessment of recurrently observed variants, beginning with confirming predicted protein consequences of mRNA splice variants. Functional studies of this aspect of biology can be challenging, in part because it is difficult to create convincing mouse models of alternative splicing, because splicing differs between organisms. Murine models have not yet allowed us to understand how splicing mutations confer a clonal advantage, for example. Competitive transplantation of cells with SF3B1 loss results in these cells being outcompeted by isogenic wild-type cells, and yet SF3B1 mutations are an early event in human clonal hematopoiesis, convincing evidence of their contribution to clonal dominance.
Pellagatti and colleagues have already begun this necessary functional work by knockdown of the mitosis regulators SEPT2 and AKAP8: factors not previously known to be altered in MDS, but which were recurrent transcriptional variants noted in their RNA sequencing analysis and resulted in impaired erythropoiesis when underexpressed. Genome-wide studies of this type inevitably generate more “leads” than any single group or consortium can follow up on. For those investigators interested in analyzing the consequences of splicing mutations for myeloid biology, these new RNA sequencing findings provide plenty of strange new transcriptional beasts to choose from.
Conflict-of-interest disclosure: D.P.S. is the principal investigator of a multicenter clinical trial of H3B-8800, a splicing modulator developed by H3 Biosciences.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal