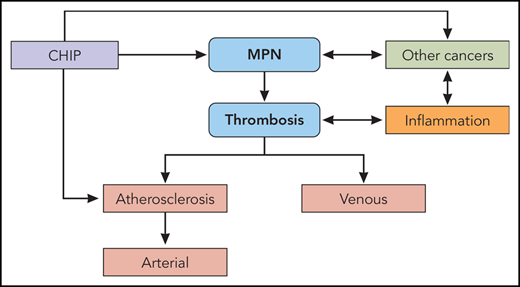
Visual Abstract

Professional illustration by Patrick Lane, ScEYEnce Studios.
Abstract
Thrombotic, vascular, and bleeding complications are the most common causes of morbidity and mortality in the Philadelphia chromosome–negative myeloproliferative neoplasms (MPNs). In these disorders, circulating red cells, leukocytes, and platelets, as well as some vascular endothelial cells, each have abnormalities that are cell-intrinsic to the MPN driver mutations they harbor (eg, JAK2 V617F). When these cells are activated in the MPNs, their interactions with each other create a highly proadhesive and prothrombotic milieu in the circulation that predisposes patients with MPN to venous, arterial, and microvascular thrombosis and occlusive disease. Bleeding problems in the MPNs are caused by the MPN blood cell-initiated development of acquired von Willebrand disease. The inflammatory state created by MPN stem cells in their microenvironment extends systemically to amplify the clinical thrombotic tendency and, at the same time, preferentially promote further MPN stem cell clonal expansion, thereby generating a vicious cycle that favors a prothrombotic state in these diseases.
Pathogenesis of MPNs as it relates to thrombotic, microvascular, and bleeding complications
The Philadelphia chromosome–negative myeloproliferative neoplasms (MPNs) are driven by somatic mutations in JAK2 (V617F, exon 12/13), CALR, or MPL. Approximately 10% of MPNs do not carry any of these driver mutations and are termed “triple-negative” MPNs.1 The principal driver mutations2 and their mutation order3 influence the major phenotypes of the MPNs (primarily polycythemia vera [PV], essential thrombocythemia [ET], and myelofibrosis [MF]). Many patients have additional, nondriver somatic mutations that span a wide range of cancer-associated genes, creating individual variations in their genetic and clonal landscape. These somatic nondriver mutations typically involve epigenetic regulator genes (eg, TET2, DNMT3A), chromatin regulator genes (eg, ASXL1, EZH2), and splicing machinery genes (eg, SF3B1).1 Comprehensive genomic characterization has identified genetic subgroups of MPNs, and integration of genomic information with clinical variables is now beginning to enable personalized predictions of patient outcomes, with potential ramifications for treatment.4 The course of patients with MPNs can be also affected by underlying germline predispositions, modifying mutations, and environmental factors such as aging and inflammation.5
Thrombosis, vascular events, and bleeding are the most common causes of morbidity and mortality in PV and ET.6 In MF, and even in prefibrotic MF, thrombotic complications are as frequent as in ET but less frequent than in PV, whereas bleeding complications occur more commonly than in either ET or PV.7 In a large cohort of PV patients, the cause of death was thrombohemorrhagic in 45%, compared with hematologic transformation in 13%.8 Another large MPN population-based study found a nearly 10-fold increase in the rate of venous thrombosis and a threefold increase for arterial thrombosis within the first 3 months of diagnosis.9 Both genomic alterations and acquired factors (eg, systemic inflammation) are determinants of thrombosis risk in the MPNs.10,11 The JAK2 V617F mutation has been found to carry an increased risk of thrombotic complications, whereas CALR carries a lower risk than the JAK2 mutation.12 Further molecular profiling (eg, by next-generation sequencing), particularly looking for additional mutations in genes involved in DNA methylation and chromatin or spliceosome pathways, promises to provide a more nuanced delineation of thrombosis risk in the MPNs.13
MPNs arise from clonal hematopoiesis of indeterminate potential (CHIP), which in the future (see “Future directions”) might provide insight into the origins of the prothrombotic state of MPNs. CHIP is the presence of a clonal mutation in a driver gene, most often characteristic of hematologic neoplasms, occurring with a variant burden of ≥2% but without any clinical evidence of a hematologic neoplasm. CHIP is now considered to be an inevitable consequence of normal human aging, but those with CHIP have an ∼10-fold increased risk of developing a clinically overt hematologic malignancy, including MPN.14 An unexpected discovery has been the finding of an association between CHIP and an increased risk of coronary artery disease and stroke.15 Initial studies of how CHIP might lead to arterial thrombosis have pointed to an increased number or hyperactivity of CHIP-bearing leukocytes. JAK2 V617–transgenic mice drive clonal hematopoiesis toward the myeloid and granulocytic lineage, the hyperactivation of which promotes inflammation.16,17 A breach in the endothelial integrity of atherosclerotic arteries triggers the recruitment of activated neutrophils from the circulation to those sites. Here, granulocytes elaborate neutrophil extracellular traps (NETs) (see "Leukocytes") that initiate and propagate arterial thrombosis. CHIP with different clonal driver mutations can also lead to distinct proinflammatory profiles, including increased levels of interleukin 1β (IL-1β)18 and IL-6,19 as well as possibly C-reactive protein,20 that likewise promote arterial thrombosis. Experimental evidence of JAK2 V617F–driven atherogenesis is described herein in other sections. There is speculation that arterial thrombotic events, especially those antedating a clinical diagnosis of MPN, may be caused or promoted by an as-yet undefined CHIP-associated prothrombotic state.
Acquired prothrombotic factors in the MPNs
Inflammation
Elevated circulating levels of proinflammatory cytokines generated by JAK2 V617F–mutated MPN cells and stromal cells in their microenvironment create a chronic, systemic, usually subclinical proinflammatory state that contributes to MPN pathogenesis.10,11 In the MPNs, interactions between quantitatively increased blood cells and microparticles, together with qualitative abnormalities of red cells, leukocytes, and platelets (see "Cellular basis of thrombotic, vascular, and bleeding complications of the MPNs") that are cell-intrinsic defects caused by their carriage of MPN driver mutations, promote a prothrombotic phenotype. The prothrombotic state in the MPNs is compounded by activation of the plasma hemostatic system. An acquired form of activated protein C resistance (much as seen in inherited factor V Leiden) has been found to be related to mutation burden in ET and PV.21 Prothrombotic, proinflammatory, and hypoxia-inducible factor–regulated genes (F3, SELP, VEGFA, and SLC2A1 in granulocytes and IL1RAP in platelets) are upregulated in patients with MPN that have thrombosis compared with those without thrombosis.22 The hypercoagulability that results from the functional interplay between abnormal blood cells, the activated coagulation pathway, and dysfunctional endothelium is then heightened and perpetuated by the effects of inflammatory cytokines (a “thromboinflammatory state”). In turn, chronic inflammation promotes the selective clonal evolution of JAK2 V617F and other MPN driver mutation-bearing hematopoietic progenitors, suppressing the wild-type (normal) population.23 Despite the large body of evidence now that inflammation promotes thrombosis and at the same time promotes the selective clonal evolution of MPN stem cells, a direct causal link between inflammation and thrombosis in the MPNs specifically has yet to be conclusively demonstrated.
Aging
Clonal hematopoiesis and systemic inflammation are inextricably linked to aging and thrombosis risk. A state of systemic, chronic inflammation escalates in the course of aging in general. Tumor necrosis factor α (TNF-α) is a key aging-associated proinflammatory cytokine that causes platelet hyperreactivity and increased thrombosis risk in this population. Patients with MPNs likewise exhibit increased systemic levels of TNF-α and mitochondrial changes that mediate prothrombotic platelet hyperreactivity (see “Platelets”).24 Aging is associated with several inflammatory diseases that are leading causes of morbidity and mortality worldwide, such as neurodegenerative diseases, autoinflammatory and autoimmune disorders, inflammatory bowel disease, and rheumatic diseases.25 Aging itself, even in the absence of MPNs, is a progressive risk factor for both venous thrombosis and atherothrombosis.26 Furthermore, each of these types of vascular occlusive events (venous and arterial) has been considered to predispose to the other.27
Cellular basis of thrombotic, vascular, and bleeding complications of the MPNs
Platelets
No significant correlation has been found between the degree of thrombocytosis and initial thrombotic and vascular complications.28-30 In fact, patients with ET with extreme thrombocytosis tend to have bleeding rather than thrombotic complications and, in the absence of leukocytosis (see "Leukocytes"), may even be protected from thrombosis.30
Numerous platelet function abnormalities have been found. Some are distinctive for the MPNs.31 These platelet abnormalities are probably intrinsic to the mutant stem cell–derived platelets. There is a correlation between JAK2 V617F allele burden and platelet activation.32 However, none of the platelet defects has been shown to be causal for clinical thrombosis or bleeding. In fact, in vitro platelet function testing methods in the MPNs can be misleading.33 Direct platelet–red cell interaction has been observed in experimental thrombogenesis, mediated by FasL/FasR,34 possibly contributing to the prothrombotic phenotype of MPNs. One study recently found that platelet activation by stimulation of Toll-like receptors (TLRs; TLR2 and TLR4), which recognize inflammatory signals, was heightened in ET patients, boosting platelet/leukocyte/endothelial interactions and the secretion of inflammatory mediators. These findings highlight the role of platelets as inflammatory sentinels in the prothrombotic scenario of MPNs.35
JAK2 V617F mutation–positive patients with MPN display higher levels of in vivo platelet activation markers. JAK2-mutant allele burden has been related to circulating platelet activation markers, P-selectin and CD40 ligand, in ET.32 Also, among patient with ET, those carrying the JAK2 V617F mutation had higher circulating levels of platelet-expressed tissue factor and platelet/neutrophil aggregates than those who did not carry the mutation.36 More recently, platelets from JAK2 V617F mutation–positive ET patients who had no previous history of thrombosis were found to be significantly more hyperreactive in platelet activation and adhesion assays than those from ET patients with CALR mutations,37 consistent with the clinical finding of higher thrombosis rates with JAK2 than with CALR (see "Pathogenesis of MPNs as it relates to thrombotic, microvascular, and bleeding complications"). In apparent contrast, a JAK2 V617F–transgenic mouse model showed reduced platelet aggregability, increased tendency for bleeding, and smaller injury-induced thrombi than wild-type mice.38
Erythrocytes
Erythrocytosis can cause venous or arterial thrombosis by directly increasing whole-blood viscosity. However, that alone is insufficient to explain the propensity for thrombosis in PV. Individuals with comparable or even higher degrees of secondary erythrocytosis (eg, due to high altitude, chronic obstructive lung disease, etc) are generally not prone to thrombosis. Nonetheless, sustained normalization of the hematocrit (to <45%) in PV patients is associated with a reduction in thrombotic events,39 indicating that erythrocytosis is still an important thrombosis risk in PV.
In patients with PV, increased adhesion of red cells to endothelial cells has been observed, mediated by the laminin α5 chain and the Lutheran/basal cell adhesion molecule (Lu/BCAM).40 A novel erythropoietin receptor-independent Rap1/Akt signaling pathway was identified that is activated by the JAK2V617F mutation, leading to Lu/BCAM phosphorylation and activation in circulating PV red cells.41 Using a proteomic approach, abnormal expression of Lu/BCAM and endoplasmic reticulum proteins has been detected in PV red cell membranes.42 These findings indicate that the JAK2V617F mutation is not only responsible for increased red cell counts but also changes in the repertoire of proadhesive red cell proteins. In atherosclerosis-prone JAK2 V617F–transgenic mice, compared with wild-type, erythrophagocytosis has been found,43 suggesting not only macrophage abnormalities (see next section) but also intrinsic red cell defects and their increased production.
Leukocytes
The relationship between leukocytosis and thrombosis risk in patients with MPN is better, although inconclusively established.11 Leukocyte counts of >15 × 109/L in PV and >11 × 109/L in ET have been independently associated with an increased risk of thrombosis.44,45 A meta-analysis confirmed the role of leukocytosis as an established, and probably independent, risk factor for thrombosis. This relationship was mainly accounted for by arterial rather than venous thrombosis, occurring mainly in patients with ET. In another meta-analysis, leukocytosis was associated with a risk of thrombosis in the MPNs,46 although some conclusions from this study have been questioned.47 Most recent studies support leukocytosis as a risk factor for thrombosis specifically in patients with ET and PV.43-45 However, in a recent study of patients with PV only, leukocyte counts taken at regular intervals found that persistently elevated leukocyte trajectories were not associated with increased thrombosis risk.48
Activated leukocytes are thrombogenic cells that release proinflammatory molecules to influence thrombus formation in concert with activation of platelets and the coagulation system.46,49 Leukocytes and platelets are activated in vivo in MPNs, forming prothrombotic circulating microaggregates.50,51 Activated circulating neutrophils in MPNs also trigger the formation of NETs that are blunted by JAK2-signaling inhibition.52,53 NETs consisting of nuclear material are expelled by activated neutrophils to form an extracellular mesh. This process of “NETosis” plays an important part in in the pathobiology of thrombosis in general. NETs serve as a platform for the processing and activation of IL-1 family cytokines,53 and NETs also induce endothelial cell activation and tissue factor generation.54 A vicious circle is thereby established in which NETosis is sustained by the malignant clone (ie, JAK2 V617F mutation–positive leukocytes and platelets) and also by the chronic inflammatory state that drives clonal evolution.55 NETosis may play an important role in thrombosis development in MPNs56 induced by other thrombogenic factors, including chronic inflammation, leukocytosis, thrombocytosis, in vivo activation of these cells, the JAK2 V617F mutation, and second cancers.54,55
Activated neutrophils in the MPNs release mediators, such as proteolytic enzymes and reactive oxygen species, that damage endothelial cells and thereby activate the coagulation cascade.57 Activated monocytes in the MPNs have heightened surface expression of tissue factor, the main initiator of thrombin generation and fibrin clot formation.58 In atherosclerosis-prone JAK2 V617F–transgenic mice, macrophages exhibit an enhanced inflammatory response, including p38 MAPK activation, compared with wild-type mice.43
In vivo, heightened interactions between leukocytes, platelets, and endothelial cells are central to the prothrombotic state of MPNs. Circulating platelets bind to both neutrophils and monocytes via adhesion molecules like P-selectin (P-selectin glycoprotein ligand-1 counterreceptor) and via platelet surface receptors glycoprotein Ibα and glycoprotein IIb-IIIa (by fibrinogen). Increased expression of P-selectin is involved in the circulating platelet-neutrophil and platelet-monocyte aggregates found in the MPNs. The antioxidant and anti-inflammatory agent, N-acetylcysteine, reduces thrombin-induced platelet-leukocyte aggregate and NET formation.59
Vascular endothelial cells and their interactions
Injury or activation of endothelial cells (ECs) converts their properties from promoting normal blood fluidity to creating a prothrombotic surface. In vivo EC activation in the MPNs is reflected by elevated biomarkers such as von Willebrand factor (VWF), soluble thrombomodulin, and E-selectin.60 JAK2 V617F–expressing ECs in mice were shown to promote thrombosis by increased exposure of P-selectin and VWF, suggesting enhanced adhesive properties.61 Interestingly, leukocytes from patients with MPN adhere more tightly to these ECs, suggesting a proinflammatory prothrombotic phenotype of ECs harboring the JAK2 V617F mutation.61
ECs bearing the JAK2 V617F mutation have been identified in JAK2 V617F mutation–positive patients with MPN, particularly in splanchnic venous vessels. This is not surprising, as mesodermal pluripotent stem cells differentiate into 2 divergent hematopoietic and endothelial lineages.62 Wherever vascular endothelial integrity is breached (eg, by senescence, apoptosis, or injury), reendothelialization can occur from outgrowth of adjacent intact endothelium or potentially by repopulating the intimal surface from circulating EC progenitors.63-67 A somatic MPN driver mutation could be thus carried by ECs from either source.
Normally, there is considerable heterogeneity in morphology, structure, gene expression, function, and responsiveness to signaling molecules of ECs in different regions of the circulation.61,64 ECs that bear MPN driver mutations possibly being restricted to certain vascular regions like the splanchnic venous system is therefore plausible. The JAK2 mutation imparting a thrombogenic phenotype in such ECs could at least partly explain the propensity of patients with MPN to thrombosis of splanchnic veins.
In vitro models of human ECs overexpressing JAK2 V617F and an in vivo model of thrombosis-prone mice with endothelial JAK2 V617F display a proadhesive phenotype with increased expression of EC P-selectin.64 Blockade of P-selectin reduced thrombosis.
Approximately 20% of myocardial infarctions in patients with MPN occur without significant underlying atherosclerotic occlusive disease,68 as opposed to only 3% in patients without MPN. This has led to the recommendation that MPNs should be ruled out in patients who present with myocardial infarction yet do not have obstructive coronary artery disease.69 In such individuals, coronary vasoconstriction has been implicated. JAK2 V617F mice exhibit exaggerated arterial vasoconstriction due to diminished endothelial nitric oxide and heightened oxidative stress. This coronary arterial hyperreactivity, in turn, has been found to be induced by circulating red cell–derived microvesicles.70
Using atherosclerosis-susceptible Ldlr−/− knockout mice transplanted with bone marrow from wild-type or JAK2 V617F mice, JAK2 expression was found to promote early atherosclerotic lesion formation, neutrophil infiltration, and increased complexity in advanced atherosclerosis. The erythrophagocytosis (see "Erythrocytes") in the JAK2 mice promotes atherosclerotic plaque instability.43
Microparticles
Microparticles are shed by the plasma membranes of activated cells. They are important mediators and biomarkers of thrombosis, inflammation, and vascular dysfunction.71 Microparticles can originate from red cells, leukocytes, ECs, and particularly platelets.71 Procoagulant anionic phosphatidylserine (PS) is normally sequestered within the inner plasma membrane leaflets of intact blood cells. This organization is disrupted in shed microparticles, in which PS becomes freely exposed to provide catalytic surfaces for the activation of plasma-clotting factors, thereby efficiently generating downstream thrombin. Circulating levels of microparticles, measured by flow cytometry, are increased in patients with MPN compared with healthy individuals.72,73 Patients with ET have elevated numbers of PS+ microparticles of platelet and EC origin compared with controls.74 Further studies are needed to evaluate the implications of microparticles in procoagulant activity, thrombin generation, and clinical thrombosis risk in patients with MPN.72
Thus, activated blood cell, ECs, and humoral factors, and their interactions with each other, comprise an intravascular prothrombotic and proadhesive milieu in the MPNs (Figure 1).
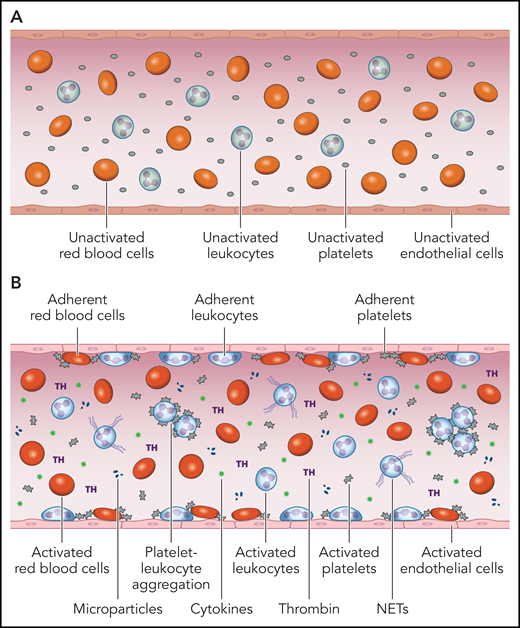
The prothrombotic and proadhesive blood milieu in the MPNs. (A) Under normal conditions, the monolayer of quiescent ECs that line the intimal surface of the circulatory tree displays a thromboresistant, anticoagulant, antiadhesive, anti-inflammatory phenotype that promotes blood fluidity. (B) Activation or dysfunction of ECs in the MPNs changes this milieu to cause activation and adhesion of platelets, leukocytes, and red cells to endothelium, formation of circulating platelet-leukocyte aggregates, NETs and microparticles, generation of thrombin, and release of proinflammatory cytokines. The main triggers for this “thromboinflammatory” state are most likely (i) cell-intrinsic abnormalities of platelets, leukocytes, red cells, and ECs as a function of their origin in a mutant hematopoietic clone and (ii) the establishment of a proinflammatory state, both serving as mutually amplifying factors. Professional illustration by Patrick Lane, ScEYEnce Studios.
The prothrombotic and proadhesive blood milieu in the MPNs. (A) Under normal conditions, the monolayer of quiescent ECs that line the intimal surface of the circulatory tree displays a thromboresistant, anticoagulant, antiadhesive, anti-inflammatory phenotype that promotes blood fluidity. (B) Activation or dysfunction of ECs in the MPNs changes this milieu to cause activation and adhesion of platelets, leukocytes, and red cells to endothelium, formation of circulating platelet-leukocyte aggregates, NETs and microparticles, generation of thrombin, and release of proinflammatory cytokines. The main triggers for this “thromboinflammatory” state are most likely (i) cell-intrinsic abnormalities of platelets, leukocytes, red cells, and ECs as a function of their origin in a mutant hematopoietic clone and (ii) the establishment of a proinflammatory state, both serving as mutually amplifying factors. Professional illustration by Patrick Lane, ScEYEnce Studios.
von Willebrand factor
In many cases of MPN-associated thrombocytosis, the bleeding tendency can be attributed to an acquired form of von Willebrand disease (aVWD). Although aVWD has been associated with extreme thrombocytosis, it is found in >50% of cases with platelets counts of <1000 × 109/L. In aVWD that involves circulatory regions of high shear stress, such as implanted left ventricular assist devices or aortic stenosis, cleavage of high-molecular-weight multimers of VWF appears to be mediated by enhanced activity of ADAMTS-13, the physiologic plasma metalloprotease that modulates circulating VWF size and activity.75 However, ADAMTS-13 has not been noted to be principally involved in the acquired von Willebrand disease of MPNs.76 There is a correlation between JAK2 V617F allele burden and platelet activation.32,77 In that regard, acquired VWD in MPNs can also develop as a result of non–ADAMTS-13-mediated proteolysis of the VWF expressed by activated platelets.
Experimental models of thrombosis in the MPNs
Patient-derived induced pluripotent stem cells and genome editing (CRISPR/Cas9)
MPN-specific induced pluripotent stem cells (iPSCs) have been derived from circulating CD34+ cells of patients with JAK2 V617F mutation–positive PV and MF patients,78 as well as from ET and MF patients with CALR or MPL mutations.5 In contrast to MDS and acute myeloid leukemia cells, MPN cells can be reprogrammed with efficiencies comparable with those of normal cells.5,79 However, long-term engraftment has been found only from acute myeloid leukemia–iPSC-derived hematopoietic stem/progenitor cells at this time.79,80 Genome-editing techniques (eg, clustered regularly interspaced short palindromic repeats [CRISPR]/CRISPR-associated protein 9 [Cas9]) can target the JAK2 V617F allele in PV-iPSCs very effectively.5 Although these approaches have not yet shed any light on the mechanisms of thrombosis in MPNs, using combined CRISPR/Cas9 and iPSC techniques might enable modeling of cooperation between MPN mutations and extrinsic factors (eg, inflammatory microenvironment) in the future.
Animal models
Animal models of both MPNs and various types of thrombosis have been separately established, although each has flaws. The best studied in vivo animal models of MPNs involve mice and zebrafish.5,81 The introduction into zebrafish of jak2a V581F, an ortholog of human JAK2 V617F, has modeled MPNs with features of human PV; and the expression of CALR mutations in zebrafish models MPL-dependent thrombocytosis. These findings suggest that the signaling machinery for MPN phenotype expression is conserved between zebrafish and humans.5 Likewise, the molecular and biochemical machinery for hemostasis and thrombosis has been largely conserved throughout evolution. Vascular injury methods used to induce thrombosis in mammalian models (see next paragraph) have now been adapted to zebrafish. However, thrombosis has not been well characterized in zebrafish.82
Mammalian models of venous or arterial thrombosis unrelated to MPNs have relied on mice, pigs, and nonhuman primates. A nidus for thrombosis can be induced in veins or arteries by various types of injury.83,84 Several experimental methods have been used to provoke thrombosis in mice. Most commonly, these have involved inferior vena cava obstruction, creating distal stasis of blood flow with a ligature, cauterization, or application of ferric chloride (FeCl3) or electrolytic injury to the adventitial surface of veins. Smaller veins (eg, saphenous, femoral) are better suited to intravital microscopy studies. However, none of these venous injury models faithfully reproduces the mechanisms of human deep vein thrombosis because the abrupt injury triggers only rapid clot formation, and the severe damage required is not a realistic scenario in human venous thrombosis.
Murine arterial injury models to induce thrombosis have mainly used the common carotid artery for ease of access and manipulation. Arterial injury is created by FeCl3 or Rose Bengal plus light, both mechanisms being mediated by focally generated oxygen free radicals, mechanical injury, stenosis, or intraluminal collagen.82 All of these techniques depend on creating harsh injury that does not authentically reproduce what occurs pathophysiologically in human arterial thrombosis. Because arterial thrombosis almost invariably occurs on a substrate of underlying atherosclerosis (“atherothrombosis”), mice with experimentally produced atherosclerosis should be used to study arterial thrombosis, with or without subsequent injury. This can be created in transgenically hyperlipidemic mice that are apolipoprotein E–deficient (ApoE−/−) or low-density lipoprotein receptor–deficient (LDLR−/−) that are fed high-fat and high-cholesterol diets.85 Such mice develop accelerated atherosclerotic lesions. The frequency of plaque rupture in these lesions has now been reproducibly demonstrated in ApoE−/− mice in specific regions of the arterial circulation.86
Transgenic MPN mice have been well characterized and are readily available for study. Notwithstanding the limitations noted herein for thrombosis models, some studies have already used experimental models of thrombosis in MPN mice.38 Granulocytes have been found to play a key role in the formation of venous thrombi in JAK2 V617F knock-in mice.38 An acute pulmonary thrombosis model in JAK2 V617F–transgenic mice was used to show that N-acetylcysteine inhibits thrombosis in MPNs. To analyze microcirculatory thrombosis in the MPNs, stalled and transiently occluded brain cortical blood flow was found in transgenic MPN mice in vivo, in real time and without inducing vascular injury, using 2-photon excited fluorescence microscopy. This technology is challenging but it enables imaging of vascular topology and quantification of blood flow in a predetermined region of the circulation by tracking the motion of nonlabeled red cells and rhodamine-labeled leukocytes and platelets, thereby discriminating between these blood cell components in the formation of vascular obstruction.87
Future directions and conclusions
Although much has been learned about the pathobiology of thrombotic, microvascular, and bleeding complications in the MPNs, especially in the past decade, several major issues have to be resolved more clearly in order to translate them into rational clinical applications for prevention and treatment. These issues are included and explored in the following sections.
Comprehensive genomic profiling of thrombosis risk
The JAK2 V617F mutation carries a higher risk for thrombotic complications than CALR mutations in the MPNs. However, a more comprehensive and concurrent genomic profile, mutation order, and allele frequencies, as well as transcriptomic analysis of inflammatory markers for thrombosis risk in individual patients with MPN, are needed. For example, mutations in DNMT3A, ASXL1, and especially TET2 may be independent risk factors for thrombosis in the MPNs.88 Also, patients with MPN who acquire the JAK2 mutation before acquisition of a TET2 mutation (called “JAK2-first patients”) carry a significantly higher risk of both venous and arterial thrombosis than those who are “TET2-first.”3
Better understanding of the role of inflammation in thrombotic problems in the MPNs
Although systemic inflammation has emerged as a major player in thrombosis pathobiology in the MPNs, more should be learned about cause-and-effect relationships in these disorders. As noted, several studies have reported increased circulating levels of cytokines in the MPNs. However, concurrent with more comprehensive genomic profiles in patients with MPN for their association with thrombosis risk, more systematic profiles of established and new proinflammatory (eg, IL-10, IL-6, TNF, C-reactive protein) as well as anti-inflammatory (eg, IL-1rA) cytokines may permit the targeting of specific cytokine signaling pathways for adjunctive treatment89 of patients with MPN at particularly high risk for thrombosis, especially arterial thrombosis. Arterial thrombosis occurs almost invariably on a substrate of preexisting atherosclerotic vessels. It is the rupture of atherosclerotic plaques that triggers thrombus formation. Two important clinical trials in patients without MPN have already shown that specific targeting of IL-1β can significantly reduce cardiovascular event rates independently of lipid or blood pressure control,90 whereas the use of broad-spectrum, nonspecific anti-inflammatory therapy with methotrexate did not do so.91 Ruxolitinib, a relatively nonspecific anti-inflammatory drug, has not been associated with a significant reduction in thrombosis risk in patients with MPN, and in this respect may not be the ideal agent to recapitulate these findings in MPNs. Furthermore, it would not provide much needed proof of principle for inflammation as a major cause of thrombosis in the MPNs. The role of statins as a potential antithrombotic agent in the MPNs remains to be established, considering their anti-inflammatory, antiproliferative, and proapoptotic properties.
Early diagnosis of MPNs and assessment of thrombosis risk
More comprehensive genomic profiling for thrombosis risk should also be explored in individuals with CHIP. In prospective cohorts unselected for coronary arterial events, those with CHIP mutations in DNMT3A, TET2, and ASXL1 had 1.7 to 2.0 times the risk of incident coronary artery disease, whereas the JAK2 V617F CHIP mutation was associated with 12.1 times the risk.15 The prevalence of JAK2 V617F in the general population has been estimated to be 0.1% to 3.1% in different studies. For CALR mutations, prevalence has been reported to be 0.16%.92 The odds ratio for prevalent venous thromboembolism was 2.8 for JAK2 V617F–positive, non-MPN persons with allele burden ≥1% (ie, CHIP), compared with the nonmutated general population. Many of these individuals were found to have elevated blood cell counts without a clinical diagnosis of MPN. These studies argue for the existence of a large population of persons with CHIP, with or without elevated blood cell counts, who are likely to be at increased risk for developing MPNs and are already at risk for at least arterial thrombosis at this point.
Experimental models of thrombosis and MPNs
As emphasized in the previous section, current thrombosis models that can be applied to animals with JAK2 V617F do not faithfully reproduce human thrombosis. Most of them depend on inducing experimental injury to blood vessels to initiate thrombus formation. They also do not consider that heterogeneity of endothelial cells and different shear and other hemodynamic forces exist through different parts of the circulation. Therefore, looking at carotid artery thrombosis, for example, cannot at all necessarily recapitulate mesenteric vascular thrombosis. There is a realistic prospect of simulating human thrombosis in different vascular beds, in vivo and in real time, using methodologies like femtosecond laser-triggered thrombosis and in vivo 2-photon imaging without vascular injury.93 Access to splanchnic vessels may be more challenging, but skin, skeletal muscle, brain, spinal cord, and, even possibly, the heart vasculature can be potentially modeled to visualize blood flow and specific cellular constituents of blood that might be involved in thrombus formation. Merging such sophisticated arterial, venous, and microvascular circulation thrombosis models in different organs in JAK2 V617F–mutated transgenic mice may better recapitulate the human situation and permit the testing of preventive and therapeutic antithrombotic interventions.
In summary, the overarching model that is emerging for the pathogenesis of the hemostatic and vascular complications of MPNs is inevitably related to the pathogenesis of the MPNs themselves. The MPNs and their thrombotic problems are most likely initiated by the mutant (eg, JAK2) clone of hematopoietic stem cells that gains a proliferative advantage over wild-type stem cells in large part because of the inflammatory response it, and its microenvironment, creates. Once this is initiated, the phenomena of mutant clonal expansion and inflammation fuel each other’s actions (Figure 2). This leads to an inexorably vicious cycle in which the primary driver, the mutant MPN clone, and inflammation, its accomplice, become inseparable, leading to the notion that both should be targeted simultaneously for optimal therapy of the MPNs and their thrombotic complications.94-96
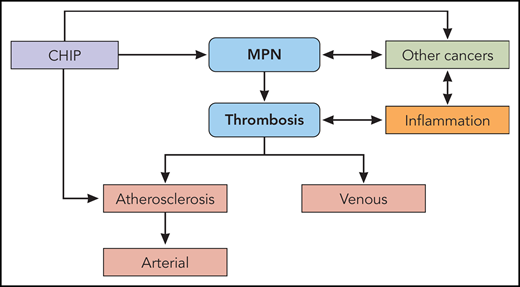
The pathogenesis of thrombosis in the MPNs. MPNs and the CHIP from which they originate are both risk factors for venous thrombosis and atherosclerosis. Atherosclerosis, in turn, is the major substrate for arterial thrombosis. MPNs and systemic inflammation promote each other bidirectionally. Systemic inflammation itself, independent of MPNs, provokes venous thrombosis, atherosclerosis, and arterial thrombosis. Like the MPNs, other cancers and systemic inflammation promote each other bidirectionally. CHIP is also associated with other cancers. Professional illustration by Patrick Lane, ScEYEnce Studios.
The pathogenesis of thrombosis in the MPNs. MPNs and the CHIP from which they originate are both risk factors for venous thrombosis and atherosclerosis. Atherosclerosis, in turn, is the major substrate for arterial thrombosis. MPNs and systemic inflammation promote each other bidirectionally. Systemic inflammation itself, independent of MPNs, provokes venous thrombosis, atherosclerosis, and arterial thrombosis. Like the MPNs, other cancers and systemic inflammation promote each other bidirectionally. CHIP is also associated with other cancers. Professional illustration by Patrick Lane, ScEYEnce Studios.
Authorship
Contribution: H.C.H., M.E., and A.I.S. contributed to the fundamental research and approximately equally wrote and edited the manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Andrew I. Schafer, Weill Cornell Medical College, 1305 York Ave, New York, NY 10021; e-mail: ais2007@med.cornell.edu.