

Abstract
Major causes of morbidity and mortality in myeloproliferative neoplasms are represented by arterial and venous complications, progression to myelofibrosis, and transformation to acute leukemia. The pathogenesis of thrombosis results from a complex interplay of clinical and disease-related factors. Abnormalities of blood cells arising from the clonal proliferation of hematopoietic stem cells involve not only quantitative changes but also qualitative modifications that characterize the switch of these cells from a resting to a procoagulant phenotype. According to age and previous thrombosis, patients are classified in a “high risk” or “low risk”. Novel disease-related determinants such as leukocytosis and JAK2V617F mutational status and/or mutational burden are now under active investigation. In low-risk polycythemia vera patients, only phlebotomy and primary antithrombotic prophylaxis with aspirin is recommended, while in high-risk patients cytotoxic therapy is considered. Whether novel drugs targeting the constitutively active JAK2/STAT pathway will improve the management of thrombosis is a challenge for future studies.
Introduction
Polycythemia vera (PV), essential thrombocythemia (ET), and primary myelofibrosis (PMF) are chronic myeloproliferative neoplasms (MPN) characterized by clonal expansion of an abnormal hematopoietic stem/progenitor cell. Their natural history is marked by thrombohemorrhagic complications and a propensity to transform into myelofibrosis and acute leukemia. The understanding of the MPN pathophysiology dramatically improved following the description of recurrent molecular abnormalities, mainly represented by the V617F mutation in JAK2 exon 14, which involves >95% of PV and ∼60% to 70% of ET and PMF patients.1,2
The aim of this paper is to update the incidence and risk factors, the mechanisms of thrombogenesis, and the management recommendations of vascular complications in MPN.
Incidence and types of thrombosis
In the largest epidemiologic study in PV, the European Collaboration on Low-dose Aspirin (ECLAP), cardiovascular mortality accounted for 41% of all deaths (1.5 deaths per 100 persons per year), mainly due to coronary heart disease (15% of all deaths), congestive heart failure (8%), nonhemorrhagic stroke (8%), and pulmonary embolism (8%). The cumulative rate of nonfatal thrombosis was 3.8 events per 100 persons per year, without a difference between arterial and venous thrombosis.3 In prospective studies in ET, the rate of fatal and nonfatal thrombotic events ranged from 2% to 4% patient-years4,5 and the incidence of arterial events was 2 to 3 times higher than that of venous events.4-6 These rates were observed both in patients with World Health Organization (WHO)-defined ET5,6 and in those defined by the Polycythemia Vera Study Group (PVSG) criteria, which also included cases of early/prefibrotic PMF.4,7 A serious complication typically associated with MPN is splanchnic vein thrombosis. In a large study of 1062 patients with Budd-Chiari syndrome and 855 patients with portal vein thrombosis, the prevalence of MPN was 40.9% and 31.5%, respectively.8 In addition to large vessel thrombosis, ET and PV patients may suffer from microcirculatory symptoms including vascular headaches, dizziness, visual disturbances, distal paresthesia, acrocyanosis, and erythromelalgia.9 In PMF, the prevalence of major thrombosis was assessed in 707 patients followed in 4 European institutions. By excluding thrombosis after splenectomy, the overall cumulative rate of cardiovascular death and nonfatal thrombotic complications was 2.23 events per 100 persons per year. No significant difference between nonfatal venous and arterial thrombosis was registered (0.76% and 0.86% patients per year, respectively). The relatively low thrombotic rate in PMF could depend on other competing events, such as acute leukemia development or other noncardiovascular major complications, including early death.10
Pathogenesis of thrombosis
The pathogenesis of thrombosis in MPN patients is complex. Clinical factors (age, previous history of thrombotic events, obesity, hypertension, and hyperlipemia) as well as the increase in blood cell counts (ie, leukocytosis, erythrocytosis, and thrombocytosis) contribute to the increased risk of thrombosis to a different extent in these patients. Abnormalities of blood cells arising from the clonal proliferation of hematopoietic stem cells involve not only quantitative but also qualitative changes that characterize the switch of these cells from a resting to a procoagulant phenotype (Figure 1). Prothrombotic features include the expression by blood cells of procoagulant and proteolytic properties, the secretion of inflammatory cytokines, and the expression of adhesion molecules. In addition to these mechanisms, prothrombotic changes occur in the normal vascular endothelium in response to the insults of inflammatory cytokines, hyperviscosity, and leukocyte-derived proteases (ie, elastase, cathepsin-G, and myeloperoxidase). Specifically, the upregulation of endothelial adhesion receptors favors the attachment of platelets, erythrocytes, and leukocytes to the vascular wall, with subsequent localization of clotting reactions and fibrin deposition. Therefore, a procoagulant background exists in MPN patients, who present with a hypercoagulable state, a subclinical condition demonstrated by the alterations of plasma thrombotic markers. Among these, the increased levels of circulating procoagulant microparticles (MPs) and the occurrence of an acquired activated protein C (APC) resistance are the most prominent features of hypercoagulability in these subjects.

Pathogenesis of thrombophilia in MPN. The pathogenesis of the acquired thrombophilic state in ET and PV is multifaceted. Mechanisms involved in the pathogenesis of the acquired thrombophilic state associated with these diseases include abnormalities of MPN-clone–derived blood cells (ie, erythrocytes, platelets, and leukocytes), which display prothrombotic features, and abnormalities of normal vascular cells, which become procoagulant in response to inflammatory stimuli. Once activated, neutrophils can also affect the hemostatic system through different pathways. In particular, the release of proteolytic enzymes and of reactive oxygen species can activate or damage platelets and endothelial cells and impair some coagulation proteins. Activated platelets express P-selectin and tissue factor (TF) and release MPs. The increased expression of CD11b on the neutrophil surface allows the adhesion of neutrophils to endothelial cells and platelets and the assembly of coagulation proteases on the neutrophil surface. In addition, abnormalities in red blood cells (RBC), including biochemical changes in the cell membrane and content, may independently impair blood flow also through the formation of RBC aggregates. Furthermore, RBC aggregation facilitates the platelet and leukocyte interaction with the vessel wall.
Pathogenesis of thrombophilia in MPN. The pathogenesis of the acquired thrombophilic state in ET and PV is multifaceted. Mechanisms involved in the pathogenesis of the acquired thrombophilic state associated with these diseases include abnormalities of MPN-clone–derived blood cells (ie, erythrocytes, platelets, and leukocytes), which display prothrombotic features, and abnormalities of normal vascular cells, which become procoagulant in response to inflammatory stimuli. Once activated, neutrophils can also affect the hemostatic system through different pathways. In particular, the release of proteolytic enzymes and of reactive oxygen species can activate or damage platelets and endothelial cells and impair some coagulation proteins. Activated platelets express P-selectin and tissue factor (TF) and release MPs. The increased expression of CD11b on the neutrophil surface allows the adhesion of neutrophils to endothelial cells and platelets and the assembly of coagulation proteases on the neutrophil surface. In addition, abnormalities in red blood cells (RBC), including biochemical changes in the cell membrane and content, may independently impair blood flow also through the formation of RBC aggregates. Furthermore, RBC aggregation facilitates the platelet and leukocyte interaction with the vessel wall.
Principal hemostatic abnormalities of MPN blood and vascular cells
Platelets
In the past, reduced levels of membrane adhesion molecules, acquired storage pool disease, and defective platelet metabolism were reported.11 However, more recent studies show that platelets from MPN patients circulate in an activated status, as assessed by the detection of increased expression of surface P-selectin and tissue factor12-14 and by the increased fraction of platelets phagocytosed by circulating neutrophils and monocytes.15 An enhanced in vivo platelet activation is further suggested by the finding of increased levels of platelet activation products both in the plasma (ie, β-thromboglobulin and platelet factor 4) and urine (ie, thromboxane A2 metabolites 11-dehydro-TxB2 and 2,3-dinor-TxB2).16 The early findings of some platelet function defects in ET are possibly due to the platelet overtiredness and to the dilution technique of platelet-rich plasma needed for some aggregation assays.17 Activated platelets provide a catalytic surface for the generation of thrombin, which further amplifies their own activation. Accordingly, in ET and PV patients, the thrombin generation induced by platelets was recently found to be increased and associated with platelet activation, particularly in carriers of the JAK2V617F mutation.18 Furthermore, immature platelets, the newly formed platelets (reticulated platelets) that show a higher hemostatic activity,19 are more elevated and more reactive than their mature counterparts both in PV and ET patients20 and positively correlate with the presence of the JAK2V617F mutation.20
Red blood cells
The prothrombotic mechanisms of an elevated hematocrit (HCT) have been clearly demonstrated in PV.21 An elevated HCT level can increase the thrombotic risk by multiple pathways (Figure 2). Under the low shear rates, as in the venous bed, a major thrombotic role is played by hyperviscosity, while at high shear rates, the raise of red cell mass displaces platelets toward the vessel wall, thus facilitating shear-induced platelet activation and enhancing platelet–platelet interactions. In addition, in ET and PV, biochemical changes have been reported in the cell membrane and intracellular content of red blood cells leading to the formation of red blood cell aggregates and impaired blood flow.22 Recently, in PV patients, an abnormal adhesion of red blood cells to the subendothelial protein laminin, due to the phosphorylation of Lu/BCAM by a JAK2V617F pathway, has been shown.23

The prothrombotic effect of an elevated HCT in ET and PV patients. Elevated HCT can increase the thrombotic risk by multiple mechanisms: (1) it determines an increase in blood viscosity; (2) at high shear rates, the raise of red cell mass displaces platelets toward the vessel wall, thus facilitating shear-induced platelet activation and enhancing platelet–platelet interactions; (3) under the low shear rates, as in the venous bed, hyperviscosity can increase the thrombotic risk by causing a major disturbance to the blood flow; and (4) biochemical changes in cell membrane and intracellular content of red blood cells.
The prothrombotic effect of an elevated HCT in ET and PV patients. Elevated HCT can increase the thrombotic risk by multiple mechanisms: (1) it determines an increase in blood viscosity; (2) at high shear rates, the raise of red cell mass displaces platelets toward the vessel wall, thus facilitating shear-induced platelet activation and enhancing platelet–platelet interactions; (3) under the low shear rates, as in the venous bed, hyperviscosity can increase the thrombotic risk by causing a major disturbance to the blood flow; and (4) biochemical changes in cell membrane and intracellular content of red blood cells.
Leukocytes
Neutrophils, the most abundant proportion of leukocytes, have a central role in the inflammatory response and in the activation of the blood coagulation system.24 In particular, the release of proteolytic enzymes (ie, elastase and cathepsin G) and reactive oxygen species and the increased expression of CD11b on their surface can activate or damage platelets and endothelial cells and impair some coagulation proteins.25 In patients with ET and PV, the occurrence of neutrophil activation is demonstrated by the detection of specific phenotypic changes (increment in membrane-associated CD11b) and increased plasma concentration of granule-derived proteases (ie, elastase and myeloperoxidase).12,26 The adhesion of platelets to leukocytes and the formation of platelet–leukocyte aggregates mediate the crosstalk among platelets, neutrophils, and monocytes.27 Data suggest that aspirin may inhibit the interaction between neutrophils and platelets.12
Endothelial cells
Several factors may perturb the physiological state of endothelium in MPN patients and turn it into a proadhesive and procoagulant surface. In particular, reactive oxygen species and intracellular proteases released by activated neutrophils can induce detachment or lysis of endothelial cells affecting functions involved in thromboregulation.27 High levels of circulating endothelial cells (resting, activated, apoptotic, and circulating precursor endothelial cells) are measured in MPN.28-30 In addition, high circulating levels of endothelial activation markers, such as thrombomodulin, selectins, and von Willebrand factor, are released and favor the formation of cellular aggregates.14,31,32 Finally, the decrease in endogenous nitric oxide production, a physiologic negative-feedback mechanism for thrombus propagation, vascular hemodynamics, and interactions of leukocytes and platelets with endothelial cells, further contributes to the procoagulant scenario.32
Plasma prothrombotic features
The increased count and/or activation status of blood and vascular cells is likely the basis of the hypercoagulable state of MPN patients, characterized by high concentrations of plasma markers of blood clotting (ie, thrombin-antithrombin complex, prothrombin fragment 1 + 2, and d-dimer) and vascular endothelium activation (ie, thrombomodulin and von Willebrand factor/factor VIII).27 In some instances, these plasma abnormalities correlate well with features of blood cell activation.26 More recently, the findings of incremented circulating MPs, shed by blood cells, and the presence of an acquired resistance to APC, have provided 2 important tools to detect the prothrombotic state in MPN patients, although the role of these and other prothrombotic markers in predicting thrombosis in MPN patients is still undefined.
MPs are membrane fragments released upon activation by all blood cell types (particularly platelets) and endothelial cells and are considered one of the major player in thrombus formation in vivo. They are found elevated in thrombotic diseases and malignancies,33 including patients with MPN.34 In particular, MPs from patients with ET show a high thrombin generation potential. Finally, circulating MPs determine the occurrence of an acquired “thrombomodulin-resistant” phenotype in PV and ET patients.35 The inherited as well as the acquired APC resistance is associated with an increased risk of thrombosis in many conditions (ie, pregnancy, oral contraceptive use, hormone replacement therapy, and cancer) and in MPN can be determined by decreased levels of protein C and protein S.36 By using the thrombin generation assay, an APC resistance phenotype has been demonstrated in ET and PV patients, particularly in JAK2V617F mutation carriers.37 Acquired APC resistance was more frequently found in ET patients with a history of thrombosis.38 A decrease in the free PS level seems to be the major determinant of APC resistance and can be due to the PS cleavage by a protease from platelets.39 Protein S cleavage was indeed significantly increased in patients with ET and an elevated platelet count and returned to normal values in ET subjects receiving hydroxyurea (HU) treatment and with a normal platelet count.40 The results of several studies13,14,41,42 support the evidence that all hemostatic alterations are worse in JAK2V617F mutation carriers than in the wild-type MPN population. However, the role of these biomarkers in identifying MPN patients at higher risk of thrombosis remains to be established. Prospective studies to evaluate this role are warranted to incorporate these markers in the decision-making risk-assessment scores.
Risk factors
Age and previous thrombosis
Increasing age and a history of thrombosis have consistently proven to be independent predictors of future events in PV, ET, and PMF. In the ECLAP study, the incidence of cardiovascular complications was higher in PV patients >65 years (5.0% patient-years; P < .006) or with a history of thrombosis (4.93% patient-years; P = .0017) than in younger subjects with no history of thrombosis (2.5% patient-years).3 In 891 ET patients diagnosed according to the WHO criteria, age >60 years and previous thrombosis were respectively associated with a 1.5 and 1.93 hazard ratio (HR) to develop major thrombosis during a median follow-up of 6.2 years.6
Blood cell counts
A predominant increase of red cell count characterizes the PV hematologic phenotype, and the consequent blood hyperviscosity is a major cause of vascular disturbances that severely impact on morbidity and mortality.43 At variance, no study has demonstrated a significant correlation between platelet number or function and thrombosis in PV and ET. In the ECLAP study, neither the currently proposed therapeutic target of 400 × 109/L nor any of the other platelet-count thresholds evaluated predicted a higher risk of thrombosis.44 In the prospective Primary Thrombocythemia-1 (PT-1) trial, longitudinal blood counts in ET patients treated with aspirin showed a significant association of thrombosis with leucocytosis but not with platelet count. Instead, the risk of major hemorrhage significantly increased (HR = 3.7) during the time periods in which platelet counts were above the normal range (>450 × 109/L) as opposed to those in which platelets were within normal limits. The hemorrhagic risk was approximately 10-fold increased when platelets were >1250 × 109/L.45 These findings suggest that current treatment should not primarily aim at lowering the platelet count for thrombosis prevention. However, platelet-lowering drugs should be considered at platelet counts >1500 × 109/L to reduce the risk of bleeding.46
Leukocytosis was found to be an independent risk factor for arterial thrombosis in MPN.47 PV patients with a white blood cell (WBC) count >15 × 109/L, compared with those with a WBC count <15 × 109/L, had a significant 70% increase of myocardial infarction.48 Three large-cohort studies in ET reported that an increased baseline leukocyte count was an independent risk factor for both thrombosis and inferior survival.49-51 An elevated WBC count developing during follow-up was also correlated with major thrombosis (P < .05) and major hemorrhage (P < .01).45,52 Based on these data, an expert consensus conference indicated that one aim of cytoreduction in ET and PV should be to keep the WBC count within the normal range,46 although this assumption remains to be confirmed in randomized clinical trials. Interestingly, as in PV, the prognostic role of WBC count in ET was mostly observed on the occurrence of arterial thrombosis.6 These findings link WBC and their activation and inflammation in the pathogenesis of thrombosis. This relation was recently demonstrated in PV and ET by the constitutive elevation of the inflammatory biomarkers C-reactive protein and pentraxin-3 and their correlation with JAK2V617F allele burden.53
Other risk factors
Conventional risk factors for atherosclerosis, including hypertension, hyperlipidemia, diabetes, and smoking, have been assessed in multivariable analysis in MPN with variable results.54,55 Some authors suggested that the presence of these conditions can upgrade the low-risk patients with ET to an intermediate-risk or an high-risk category.4,55 In the recently developed “IPSET-thrombosis” score (see below),55 cardiovascular risk factors were among the variables significantly and independently associated with an increased rate of total thrombosis in ET patients.
The influence of the JAK2V617F mutational status and allele burden on the thrombotic risk has been evaluated in several studies. In 173 patients with PV, those harboring greater than 75% JAK2 V617F allele were at higher relative risk (RR) to develop major cardiovascular events during follow-up than were those with <25% mutant allele (RR = 7.1; P = .003).56 In ET, a systematic literature review showed that JAK2 V617F patients have a 2-fold risk of developing thrombosis (odds ratio [OR] = 1.92; 95% confidence interval [CI], 1.45–2.53) both of venous and arterial vessels (OR = 2.49 and 1.77 respectively), but a significant heterogeneity between studies should be pointed out.57 In PMF, the highest incidence of fatal and nonfatal thrombosis was observed when the mutation was present along with leukocytosis (3.9% patient-years; HR = 3.13; 95% CI, 1.26–7.81).10 A high frequency of JAK2V617F was reported in splanchnic vein thrombosis,8 and, interestingly, patients who were JAK2V617F negative were found to harbor JAK2 46/1 haplotype.58 These findings and the identification of JAK2V617F mutation in the liver and spleen endothelial cells of patients with Budd-Chiari syndrome59,60 may suggest a perturbance of endothelium mediated by the mutation with a consequent and possibly local prothrombotic state contributing to the pathogenesis of thrombosis. As to the presence of an MPL mutation, higher rates of arterial thrombosis were found in an Italian study61 but not in the PT-1 trial cohort.62
Instead, in this latter trial, an increased bone marrow reticulin fibrosis was an independent predictor of subsequent thrombotic and hemorrhagic complications.7 This finding was confirmed by other studies showing an increased risk of both bleeding63 and thrombosis64 in patients with early myelofibrosis vs those with ET defined according to the WHO classification.
The frequency of genetic thrombophilic factors, such as factor V Leiden or prothrombin mutations, was higher in MPN with venous thrombosis, suggesting that these tests should be performed in younger patients with a familial or personal history of thrombosis.65-67 On the contrary, in regard to the role of antiphospholipid antibodies68,69 and hyperhomocysteinemia,70,71 data so far produced are too limited to recommend their evaluation in the work-up of patients with MPN.
Risk classification
By incorporating this body of knowledge into a simple, clinically oriented scheme (Table 1), the classification of patients with either PV or ET into a “high-risk” or “low-risk” category according to their age and history of thrombosis is currently recommended.46 More recently, an International Prognostic Score of thrombosis has been developed in WHO-diagnosed ET (IPSET-thrombosis).55 Risk scores were assigned based on multivariable-analysis–derived HRs to age >60 years (HR = 1.5, 1 point), thrombosis history (HR = 1.9, 2 points), cardiovascular risk factors (HR = 1.6, 1 point), and JAK2V617F (HR = 2.0, 2 points). Subsequently, a 3-tiered prognostic model (low-risk, <2 points; intermediate risk, 2 points; and high-risk, >2 points) was devised, allowing a clear identification of the thrombotic risk: 1.03% patient-years vs 2.35% patient-years vs 3.56% patient-years in the 3 groups, respectively. This validated IPSET-thrombosis model may provide objective estimates of the probability of thrombotic events in patients with newly diagnosed ET that can be useful for future prospective clinical studies.
Risk stratification in PV and ET based on thrombotic risk
| Risk category | Age >60 y or history of thrombosis |
|---|---|
| Low | No |
| High | Yes |
| Risk category | Age >60 y or history of thrombosis |
|---|---|
| Low | No |
| High | Yes |
Extreme thrombocytosis (platelet count >1500 × 109/L) is a risk factor for bleeding, not for thrombosis. Both increasing leukocyte count and JAK2 V617F mutation or allele burden have been identified as novel risk factors for thrombosis, but confirmation is required.
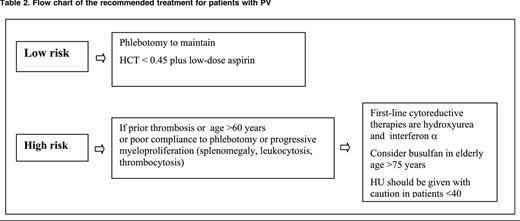

Based on such risk stratification, the potential dangers of cytoreductive therapy with the intent of preventing thrombotic complications may be justified only in high-risk patients.46
How to reduce thrombosis
Interventions on lifestyle
The identification and appropriate management of cardiovascular risk factors and the promotion of a healthy lifestyle in MPN, as in the general population, should be considered a cornerstone of vascular prevention.
Phlebotomy
Phlebotomy (Table 2) is the recommended main tool to control HCT in PV patients. The optimal target of HCT levels for reducing vascular events was a matter of debate.44,72 In a recent large-scale, multicenter, randomized clinical trial (Cyto-PV), 365 JAK2V617F PV patients were assigned to receive either more intensive therapy to maintain an HCT target of <45% or a less intensive treatment with an HCT target of 45% to 50%. After a median follow-up of 31 months, the primary end point of cardiovascular death and major thrombosis was recorded in 5 of 182 patients in the low-HCT group (2.7%) and in 18 of 183 patients in the high-HCT group (9.8%) (HR in the high-HCT group = 3.91; P = .007). This study demonstrates that high HCT is associated with a 4 times higher rate of thrombotic events and supports the importance of the strict control of HCT levels to prevent thrombosis in PV.43
Low-dose aspirin
The efficacy and safety of low-dose aspirin (100 mg daily) in PV has been assessed in the ECLAP double-blind, placebo-controlled, randomized clinical trial.73 In this study, 532 PV patients were randomized to receive 100 mg aspirin or placebo. After a follow-up of about 3 years, data analysis showed a significant reduction of a primary combined end point, including cardiovascular death, nonfatal myocardial infarction, nonfatal stroke, and major venous thromboembolism (RR = 0.4; 95% CI, 0.18-0.91; P = .0277).
In ET (Table 3), the efficacy of aspirin has not been tested in randomized clinical trials. Low-dose aspirin (100 mg daily) controls microvascular symptoms, such as erythromelalgia and transient neurologic and ocular disturbances. A retrospective study74 has suggested that antiplatelet therapy reduces the incidence of venous thrombosis in JAK2-positive patients and the rate of arterial thrombosis in patients with associated cardiovascular risk factors. In regard to bleeding, the annual incidence of major hemorrhages attributed to aspirin in PV and ET ranges between 0.3% and 0.8%, and this rate is higher in ET patients presenting with prefibrotic myelofibrosis (1.39% patient-years), suggesting that major bleeding might be relatively specific to this entity as opposed to WHO-defined ET (0.79% patient-years).63 Interestingly, a recent study has shown that the effect of once-daily low-dose aspirin in ET is shorter-lasting through faster renewal of platelet cyclooxygenase-1 and suggests that impaired platelet inhibition can be rescued by modulating the aspirin dosing interval rather than the dose.75 This new concept deserves to be validated in prospective studies, but prescribing low-dose aspirin twice daily to prevent severe arterial thrombotic recurrence in MPN is now suggested.76
Hydroxyurea
HU is an antimetabolite that prevents DNA synthesis and was introduced in the therapy of PV and ET to reduce thrombosis by the PVSG investigators, who assumed this drug was not leukemogenic.77 In the unique randomized trial comparing HU with another cytoreductive drug in PV, the cumulative incidence of acute myeloid leukemia/myelodysplastic syndrome (AML/MDS) at 10, 15, and 20 years was 6.6%, 16.5%, and 24% in the HU arm and 13%, 34%, and 52% in the pipobroman arm, respectively (P = .004).78 Other studies from registry data and prospective analysis failed to attribute a clear leukemogenic risk to HU.79-81 Overall, the bulk of evidence does not attribute a definite leukemogenic risk of HU, but it should be emphasized that this risk may appear after a long-term exposure to this drug. It is wise to adopt a cautionary principle and to consider carefully the use of this agent in young subjects and in those previously treated with other myelosuppressive agents or carrying cytogenetic abnormalities.
The antithrombotic efficacy of this drug in ET was demonstrated in a seminal randomized clinical trial54 showing that HU reduced the rate of thrombotic events mainly represented by cerebral transient ischemic attacks. Interestingly, HU antithrombotic effect may recognize additional mechanisms of action besides pan myelosuppression, including qualitative changes in leukocytes, decreased expression of endothelial adhesion molecules, and enhanced nitric oxide generation.82
IFN-α
Interferon α (IFN-α) was considered for the treatment of patients with MPN because this agent suppresses the proliferation of hematopoietic progenitors, has a direct inhibiting effect on bone marrow fibroblast progenitor cells, and antagonizes the action of platelet-derived growth factor, transforming growth factor-β, and other cytokines that may be involved in the development of myelofibrosis.83 Two phase 2 studies have shown that pegylated IFN-α 2a therapy led to a high rate of hematologic response and reduced the malignant clone as quantitated by the percentage of the mutated allele JAK2V617F.84,85 In contrast, more limited effects on JAK2 mutational status have been reported after therapy with pegylated IFN-α 2b in a small group of patients with PV and ET.86 The tolerability of pegylated IFN-α 2a at 90 μg weekly was excellent. However, whether this drug is more efficacious than HU in reducing the rate of vascular events remains to be demonstrated in the clinical trials that currently are underway.
Anagrelide
Anagrelide, an imidazoquinazolin compound, has a potent platelet-reducing activity devoid of leukemogenic potential and appears to be an alternative to HU for reducing platelet counts in younger ET patients at high risk of thrombosis and resistant to or intolerant of HU.46 Anagrelide and HU have been compared head to head in 2 randomized clinical trials. The first (PT-1) included 809 ET patients diagnosed according to PVSG criteria and treated with aspirin (100 mg/d).4 Compared with HU, patients in the anagrelide arm showed an increased rate of arterial thrombosis (OR = 2.16; P = .03), major bleeding (OR = 2.61; P = .008), and myelofibrotic transformation (OR = 2.92; P = .01) but a decreased incidence of venous thrombosis (OR = 0.27; P = .006). In the ANAHYDRET (Anagrelide vs Hydroxyurea Efficacy and Tolerability Study in Patients with Essential Thrombocythaemia) noninferiority randomized clinical trial, 259 previously untreated, high-risk WHO-diagnosed ET patients were randomized to HU and anagrelide.5 During the total observation time of 730 patient-years, there was no significant difference between the anagrelide and HU groups regarding incidences of major arterial (7 vs 8) and venous (2 vs 6) thrombosis and severe bleeding events (5 vs 2). Disease transformation into myelofibrosis or secondary leukemia was not reported. According to the ELN, anagrelide is currently recommended as a second-line therapy in high-risk ET patients resistant to or intolerant of HU.46
Secondary prevention of thrombosis
In the largest study that specifically analyzed the incidence of recurrent thrombosis after the first episode,87 thrombosis recurred in 166 of 494 (34%) PV and ET patients, corresponding to 7.6% patient-years. Sex, diagnosis (PV or ET), and presence of vascular risk factors did not predict recurrence, whereas age >60 years did (multivariable HR = 1.67; 95% CI, 1.19-2.32). In this retrospective analysis, the use of a cytoreductive drug together with an antiplatelet agent reduced the risk of recurrence in comparison with both cytoreduction alone (univariate HR = 0.56; 95% CI, 0.24-0.85) and antiplatelet agents alone (univariate HR = 0.67; 95% CI, 0.41-0.99). After the first venous thromboembolic event, long-term oral anticoagulation was associated with a 63% reduction in the risk of recurrence without a significant increase of the incidence of major bleeding (0.9% patient-years) as compared with patients without antithrombotic treatment (1.2% patient-years).
Future perspectives
Current recommendations for the management of MPN should take into consideration the advent of new-generation drugs with JAK2 inhibitory activity. These agents have been found to be effective for the treatment of disease-related splenomegaly or symptoms in adult patients with myelofibrosis. However, no data are available so far on the efficacy of preventing thrombotic complications, which remains the primary goal of therapy in PV and ET.
Acknowledgments
This work was supported by a grant from the Associazione Italiana per la Ricerca sul Cancro (AIRC) (Milano, “Special Program Molecular Clinical Oncology 5x1000” to the AIRC287 Gruppo Italiano Malattie Mieloproliferative, project #1005) (T.B. and G.F.) and by grants from the Italian Association for Cancer Research (AIRC grants IG10558 and “5 per mille” 12237) (A.F.).
Authorship
Contribution: All authors contributed equally to the manuscript, and each author read and approved the final draft.
Conflict-of-interest disclosure: Dr Barbui reports receiving consulting fees from Shire, Novartis, and Italfarmaco. The remaining authors declare no competing financial interests.
Correspondence: Tiziano Barbui, Research Foundation, Ospedale Papa Giovanni XXIII, P.zza OMS, 1, 24127 Bergamo (Bg) Italy; e-mail: tbarbui@hpg23.it.
