

Abstract
Reactive and clonal neutrophil expansion has been associated with thrombosis, suggesting that neutrophils play a role in this process. However, although there is no doubt that activated monocytes trigger coagulation in a tissue factor-dependent manner, it remains uncertain whether stimulated neutrophils can also directly activate coagulation. After more than a decade of debate, it is now largely accepted that normal human neutrophils do not synthetize tissue factor, the initiator of the extrinsic pathway of coagulation. However, neutrophils may passively acquire tissue factor from monocytes. Recently, the contact system, which initiates coagulation via the intrinsic pathway, has been implicated in the pathogenesis of thrombosis. After the recent description of neutrophil extracellular trap (NET) release by activated neutrophils, some animal models of thrombosis have demonstrated that coagulation may be enhanced by direct NET-dependent activation of the contact system. However, there is currently no consensus on how to assess or quantify NETosis in vivo, and other experimental animal models have failed to demonstrate a role for neutrophils in thrombogenesis. Nevertheless, it is likely that NETs can serve to localize other circulating coagulation components and can also promote vessel occlusion independent of fibrin formation. This article provides a critical appraisal of the possible roles of neutrophils in thrombosis and highlights some existing knowledge gaps regarding the procoagulant activities of neutrophil-derived extracellular chromatin and its molecular components. A better understanding of these mechanisms could guide future approaches to prevent and/or treat thrombosis.
Introduction
Clinical disorders characterized by reactive or clonal expansion of neutrophils are commonly associated with an increased risk for thrombosis.1,2 This observation has raised the legitimate question of whether neutrophils, by analogy to activated monocytes, can exhibit a procoagulant phenotype that directly triggers coagulation. Although it is well-established that activated monocytes upregulate expression of tissue factor (TF), the activator of the extrinsic pathway of coagulation,3 the possibility of neutrophil expression of TF has been controversial.4,5 It appears that normal human neutrophils do not synthesize, but rather acquire, TF from activated monocytes in blood, although the mechanism of transfer has not been fully elucidated.6 However, the possibility of neutrophil expression of TF in the case of clonal abnormalities remains unresolved,7,8 which may also be the case in neutrophils from other species.9
Coagulation can also be initiated through the intrinsic pathway, after activation of the contact system, composed of factor XII (FXII), prekallikrein, and high-molecular-weight kininogen. The contribution of this pathway to hemostasis has been largely discounted because deficiency of the individual proteins does not result in a bleeding tendency. Conversely, however, recent evidence from animal models suggests that the contact system plays a major role in thrombosis,10-14 although epidemiological data from human studies are inconsistent.15 Because contact system activation occurs on negatively charged surfaces, several potential in vivo activators have been suggested, among which polyphosphates and DNA are the best characterized.16 Neutrophil-derived DNA could be released after various forms of cell death or activation (reviewed here). Indeed, elevated levels of circulating nuclear components have been detected in patients with diverse disease states associated with an increased risk for thrombosis.17 As the most numerous nucleated cell population in blood, neutrophils are expected to be a major contributor to circulating nuclear material.
In 2004, Brinkmann et al discovered the process of NETosis, whereby neutrophils expel their nuclear material in a meshwork known as neutrophil extracellular traps (NETs).18 Previous reviews have addressed the activation of coagulation by NETs, primarily via contact activation of FXII.19-22 However, subsequent studies have provided additional insights into the ability of NETs to occlude vessels independent of direct activation of coagulation,23 or have addressed the procoagulant activities of extracellular chromatin in its various forms.24 Here, we review current evidence regarding the role of neutrophil-derived extracellular nuclear material in thrombosis.
Neutrophil chromatin
Similar to all eukaryotic cells, genomic DNA (gDNA) found in neutrophils is packaged through the actions of histone proteins. Histones are positively charged and have high affinity for DNA. In addition to a linker histone (H1), 4 major histone subtypes (H2A, H2B, H3, and H4) typically constitute the core nucleosome, with highly conserved amino acid sequences. Each core histone is composed of a histone fold domain of ∼70 amino acids arranged in 3 α helices at the C terminus and a flexible tail at the N terminus. Two H2A-H2B dimers and a single H3-H4 tetramer assemble to form an octameric core.25 Approximately 147 base pairs of DNA is wrapped around the octameric core through the interaction of DNA, with positively charged residues present on the histone fold domain, leading to the formation of the nucleosome, the fundamental repeating unit of chromatin.25 By electron microscopy, the primary structure of chromatin in vitro resembles “beads” (nucleosomes) joined by “strings” (linker DNA).26 Although still a subject of debate, nucleosomes may be assembled into rod-like chromatin fibers of ∼30 nm diameter in vivo.27 These fibers further interact to form higher-order chromatin structures having different particle arrangements, conformations, and densities during interphase.28 The degree of packaging is regulated by a number of mechanisms involving histone and DNA posttranslational modification (PTM), incorporation of histone variants, and the actions of H1.29,30 Indeed, an expanding dictionary of histone PTMs has been described, which occur largely within the tail domains but also in the histone fold domain, and include PTMs such as acetylation, methylation, phosphorylation, and ubiquitination.30 As discussed further here, histone PTMs are believed to play a critical role in NETosis.
In addition to histone PTMs, another major contributor to neutrophil cell fate determination and function is DNA methylation. This epigenetic modification has been well studied and shown to direct cell type specification, gene repression, and imprinting. Mechanistically, DNA methylation can function by modulating the binding of regulatory proteins such as transcription factors to DNA.30 However, whether DNA methylation affects the binding of non-nuclear proteins, such as proteins of the clotting system, to extracellular DNA is unknown.
Mechanisms of extracellular release of neutrophil chromatin
Neutrophil chromatin, which is contained within the nucleus during the quiescent state, has to be released extracellularly to interact with the clotting system. The structure of neutrophil extracellular chromatin may vary according to the mode of release, with apoptosis, necrosis, and NETosis being the most important mechanisms.
Neutrophil apoptosis
Under homeostatic conditions, neutrophils are short-lived cells. On the basis of in vivo labeling studies, their estimated half-life in the circulation and their lifespan in vivo are approximately 3.8 and 5.4 days, respectively.31 Similar to other cells, senescent neutrophils likely undergo constitutive apoptosis. During inflammation, chemotactic factors direct the migration of activated neutrophils to inflamed sites, where they perform phagocytosis and intracellular killing of pathogens. After performing this function, activated neutrophils also die by apoptosis, induced by engulfed pathogens, or by the ligation of certain inflammatory mediators such as tumor necrosis factor-α or Fas-ligand, although controversies exist.32 Morphologically, apoptosis is characterized by nuclear pyknosis (chromatin condensation) and karyorrhexis (chromatin fragmentation). The cell disintegrates into apoptotic bodies, which are small vesicles containing fragments of condensed chromatin and cytoplasmic organelles, surrounded by the cytoplasmic membrane33 (Figure 1). In general, apoptotic cells express a variety of surface molecules, which allow their recognition and clearance by phagocytic cells.34 Overall, disposal of aged or inflamed neutrophils dying by apoptosis is accomplished without the release of intracellular content. However, during severe inflammation, exaggerated neutrophil apoptosis may overwhelm the clearance capacity of scavenger macrophages; nonphagocytosed apoptotic neutrophils may then undergo secondary necrosis (with rupture of the cell membrane),35 leading to extracellular release of fragments of condensed chromatin.
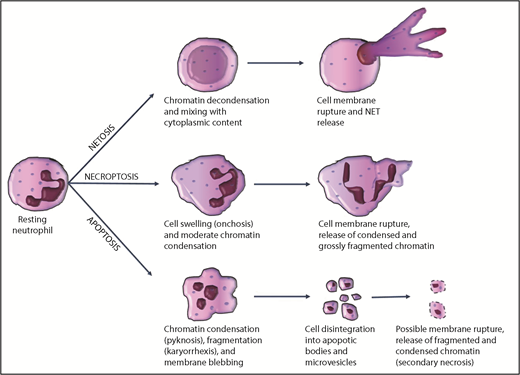
Neutrophil necroptosis
Neutrophil primary necrosis can also occur in a programmed manner (necroptosis), which depends on receptor-interacting protein kinase-1 and receptor-interacting protein kinase-3 pathways.36 Necroptosis is morphologically characterized by moderate chromatin condensation and fragmentation, cell and organelle swelling (oncosis), rupture of the plasma membrane, and extracellular spillage of cell content, including chromatin33 (Figure 1). Neutrophil necroptosis can be triggered in vitro by tumor necrosis factor stimulation, ligation of adhesion receptors, exposure to monosodium urate crystals, or phagocytosis of certain pathogens.37 In contrast to apoptosis, necroptosis leads to extracellular release of condensed chromatin.
NETosis
Extracellular chromatin decorated with cytoplasmic granules is released by neutrophils during NETosis.18 Mast cells, eosinophils, and monocytes/macrophages have also been reported to release NET-like structures in a process known as ETosis.38 A wide variety of artificial and natural stimuli have been shown to induce NETosis in vitro and in vivo,39 including autocrine stimulation by FXII produced by neutrophils.40 The mechanism of NET release, NET structure, and the fate of the NETosing cell seem to depend on the type and duration of the inducing stimulus.41 For example, NETs induced by phorbol myristate acetate are released after at least 3 hours of incubation by a reduced NAD phosphate oxidase- and myeloperoxidase-dependent mechanism. In this scenario, NETs are composed of genomic DNA, as indicated by the association with histones; the cell membrane ruptures during NETosis and cell death accompanies NET release (so called “suicidal NETosis”).41,42 This process appears to be a distinct subroutine of cell death, as it is insensitive to caspase inhibitors and necrostatin-1, inhibitors of apoptosis and necroptosis, respectively.42,43 In contrast, vital NETosis is proposed to be another mechanism whereby neutrophils remain viable and capable of certain functions after NET release.44 In this case, NETosis may be induced by bacteria or fungi within minutes in a reactive oxygen species–independent manner, chromatin moves from the nucleus and across the cytoplasm by vesicular trafficking, NET-containing vesicles fuse with the cytoplasmic membrane, and NETs are released extracellularly without membrane rupture.45,46
However, chromatin decondensation and mixing of cytoplasmic contents with nuclear material before NET release seem to be constant features, regardless of the suicidal or vital pathway of NETosis. These events, in fact, discriminate NETosis from other cell death mechanisms, in which chromatin is instead condensed and/or fragmented, as discussed earlier. The detailed mechanism leading to chromatin decondensation during NETosis is unclear. Neutrophil myeloperoxidase and elastase have been suggested as key players for chromatin decondensation,47 although neutrophil myeloperoxidase and elastase-deficient mice still form NETs.48 Citrullination of arginine on histone tails has been proposed to promote chromatin decondensation and NET formation, likely by affecting histone-DNA interactions. Peptidyl-arginine deiminase 4 (PADI4 or PAD4) has been identified as the enzyme that mediates hypercitrullination of histones during NETosis.49 Interestingly, neutrophils from PAD4 knockout mice do not undergo NETosis, indicating that histone hypercitrullination is indeed a critical step for this process, at least in mice.50 However, it has been reported that some stimuli, such as phorbol myristate acetate, can induce NETosis in human neutrophils without significant histone hypercitrullination.41 Moreover, histone hypercitrullination may not be specific to NETosis, as it also occurs during leukotoxic hypercitrullination and defective mitophagy.51 Overall, it is highly challenging to assess NETosis, especially in vivo, where other nucleated cells are present, but this process can readily be studied in isolated neutrophils. As such, a consensus on how to assess NETosis in vivo is needed to accelerate our understanding of the pathophysiological role of NETs.
Mechanisms of interaction between chromatin components and coagulation pathways
DNA and coagulation activation
The procoagulant activity of DNA has been studied by multiple groups.14,24,52-57 Most of these in vitro studies have used gDNA from intact cells or from the extracellular space after cell death or stimulation (Table 1). DNA is usually rendered protein-free during the purification process, and the procoagulant activity has been assessed by introducing gDNA into whole blood, plasma, or buffer solutions containing purified coagulation proteins. Despite the variety of cellular origins that have included neutrophils, and the variety of DNA purification methods and functional assays used, current reports are consistent in demonstrating that gDNA possesses procoagulant activity. However, some methods of DNA purification may be contaminated with reagent silica particles, which may lead to overestimation of the procoagulant activity through contact activation.58 Indeed, gDNA spiked into plasma shortens clotting times,52,55 triggers thrombin generation independent of TF,14,24,54-57 and amplifies TF-initiated thrombin generation.24,57 Similar to gDNA, bacterial and mitochondrial DNA are also procoagulant.56 Using factor-deficient plasmas and pharmacological approaches, existing reports are consistent with the fact that the procoagulant activity of gDNA is mediated by direct activation of the contact system (Figure 2). Contact activation leads to FXIIa formation, which then activates PK and FXI, ultimately leading to thrombin generation. Indeed, gDNA promotes both FXII and PK auto-activation,14,52,53,55,57 as well as FXIIa- and thrombin-dependent FXI activation24,52,55 in plasma or in purified systems. High-molecular-weight kininogen seems to be necessary for DNA-induced PK auto-activation, while conversely protecting FXI from auto-activation.57 In the activated partial thromboplastin time assay, the contact system is activated on artificial negatively charged surfaces such as kaolin, celite, ellagic acid, or silica. The abundant phosphate moieties in gDNA provide a negatively charged surface for binding of contact system proteins, as well as FXI and thrombin.53,55 In addition to promoting thrombin generation, gDNA impairs fibrinolysis by forming a ternary complex with plasmin and fibrin, thereby preventing plasmin-mediated fibrin degradation.59
Studies examining the procoagulant effects of cell-free DNA on coagulation in vitro
| Reference | Cellular origin | Method of purification | Coagulation Assay | Type of DNA | Conclusions |
|---|---|---|---|---|---|
| Kannemeier et al 200752 | China Hamster ovarian cell line | Silica-based affinity chromatography | Thromboelastography | Genomic | DNA shortens clotting times in whole blood and plasma, activates FXII and amplifies thrombin-dependent FXIa generation in buffer |
| Clotting times in plasma | |||||
| Chromogenic assay in plasma | |||||
| Chromogenic assay in purified system | |||||
| Oemcke et al 200953 | Normal human neutrophils | Guanidinium isothiocyanate detergent + ethanol precipitation | Chromogenic assay for PK hydrolysis in plasma | Genomic | DNA binds FXII and HK and promotes PK hydrolysis in plasma |
| Swystun et al 201154 | Chemotherapy-treated normal whole blood or neutrophils | Silica-based affinity chromatography | Thrombin generation in PPP | Genomic | Extracellular DNA released from chemotherapy-treated blood cells increases thrombin generation in plasma |
| Vu et al 201555 | A549 cells | Silica-based affinity chromatography | Clotting times in plasma | Genomic | DNA shortens clotting times, increases TG in plasma, promotes FXII activation and FXIa generation in buffer |
| Thrombin generation in PPP | |||||
| FXII activation, FXIIa-dependent and thrombin-dependent FXIa generation in purified systems | |||||
| Bhagirath et al 201556 | HEK297 cells | Silica-based affinity chromatography. | Thrombin generation in PPP | Genomic | All types of DNA enhance TG in plasma |
| Bacteria | Bacterial DNA was purchased. | Mitochondrial | |||
| Bacterial | |||||
| Kokoye et al 201614 | Normal human leukocytes | Phenol-chloroform extraction | Western blot for FXII and PK activation in purified systems | Genomic | DNA enhances FXII and PK activation in buffer and enhances thrombin generation in plasma. |
| Thrombin generation in PPP | |||||
| Noubouossie et al 201724 | Normal human neutrophils | Silica-based affinity chromatography | ELISA for FXIa-C1INH complexes in purified contact system | Genomic | DNA triggers contact activation in buffer and thrombin generation in PFP and PRP |
| Chromogenic assays for FXIa generation in buffer | DNA amplifies FXIIa- and thrombin-dependent FXIa generation in buffer | ||||
| Thrombin generation in PFP or PRP | DNA amplifies TF-initiated TG in plasma | ||||
| Ivanov et al 201757 | Normal human leukocytes | Phenol-chloroform extraction | Chromogenic assays for PK and FXI activation | Genomic | DNA promotes PK (in the presence of HK) and FXI activation |
| Thrombin generation in plasma | DNA initiates thrombin generation in plasma independently of TF, and amplifies TF-initiated TG in plasma |
| Reference | Cellular origin | Method of purification | Coagulation Assay | Type of DNA | Conclusions |
|---|---|---|---|---|---|
| Kannemeier et al 200752 | China Hamster ovarian cell line | Silica-based affinity chromatography | Thromboelastography | Genomic | DNA shortens clotting times in whole blood and plasma, activates FXII and amplifies thrombin-dependent FXIa generation in buffer |
| Clotting times in plasma | |||||
| Chromogenic assay in plasma | |||||
| Chromogenic assay in purified system | |||||
| Oemcke et al 200953 | Normal human neutrophils | Guanidinium isothiocyanate detergent + ethanol precipitation | Chromogenic assay for PK hydrolysis in plasma | Genomic | DNA binds FXII and HK and promotes PK hydrolysis in plasma |
| Swystun et al 201154 | Chemotherapy-treated normal whole blood or neutrophils | Silica-based affinity chromatography | Thrombin generation in PPP | Genomic | Extracellular DNA released from chemotherapy-treated blood cells increases thrombin generation in plasma |
| Vu et al 201555 | A549 cells | Silica-based affinity chromatography | Clotting times in plasma | Genomic | DNA shortens clotting times, increases TG in plasma, promotes FXII activation and FXIa generation in buffer |
| Thrombin generation in PPP | |||||
| FXII activation, FXIIa-dependent and thrombin-dependent FXIa generation in purified systems | |||||
| Bhagirath et al 201556 | HEK297 cells | Silica-based affinity chromatography. | Thrombin generation in PPP | Genomic | All types of DNA enhance TG in plasma |
| Bacteria | Bacterial DNA was purchased. | Mitochondrial | |||
| Bacterial | |||||
| Kokoye et al 201614 | Normal human leukocytes | Phenol-chloroform extraction | Western blot for FXII and PK activation in purified systems | Genomic | DNA enhances FXII and PK activation in buffer and enhances thrombin generation in plasma. |
| Thrombin generation in PPP | |||||
| Noubouossie et al 201724 | Normal human neutrophils | Silica-based affinity chromatography | ELISA for FXIa-C1INH complexes in purified contact system | Genomic | DNA triggers contact activation in buffer and thrombin generation in PFP and PRP |
| Chromogenic assays for FXIa generation in buffer | DNA amplifies FXIIa- and thrombin-dependent FXIa generation in buffer | ||||
| Thrombin generation in PFP or PRP | DNA amplifies TF-initiated TG in plasma | ||||
| Ivanov et al 201757 | Normal human leukocytes | Phenol-chloroform extraction | Chromogenic assays for PK and FXI activation | Genomic | DNA promotes PK (in the presence of HK) and FXI activation |
| Thrombin generation in plasma | DNA initiates thrombin generation in plasma independently of TF, and amplifies TF-initiated TG in plasma |
HK, high-molecular-weight kininogen; PK, prekallikrein; PPP, platelet-poor plasma; PRP, platelet-rich plasma; C1INH, C1 esterase inhibitor; TG, thrombin generation.
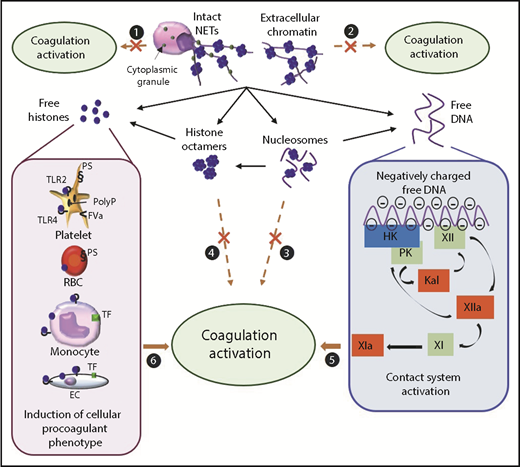
Proposed mechanisms by which NETs/chromatin components activate coagulation in vitro. Intact NETs (1), extracellular chromatin (2), nucleosomes (3), or histone octamers (4) do not directly activate coagulation. (5) Free DNA activates coagulation through the contact pathway. (6) Free histones promote coagulation activation mainly by inducing a procoagulant phenotype on blood and endothelial cells (ECs). FVa, activated factor V; Kal, kallikrein; PolyP, polyphosphate; PS, phosphatidylserine; RBC, red blood cell; TLR, tool-like receptor.
Proposed mechanisms by which NETs/chromatin components activate coagulation in vitro. Intact NETs (1), extracellular chromatin (2), nucleosomes (3), or histone octamers (4) do not directly activate coagulation. (5) Free DNA activates coagulation through the contact pathway. (6) Free histones promote coagulation activation mainly by inducing a procoagulant phenotype on blood and endothelial cells (ECs). FVa, activated factor V; Kal, kallikrein; PolyP, polyphosphate; PS, phosphatidylserine; RBC, red blood cell; TLR, tool-like receptor.
Histones and coagulation activation
Intravenous injection of 75 mg/kg calf thymus histones induces lethality in mice.60 Infusion of sublethal doses induces thrombocytopenia, circulating platelet aggregates, thrombosis in the lungs and kidney, prolongation of the bleeding time, and elevation of plasma thrombin-antithrombin complexes.60-62 The mechanisms by which histones activate coagulation and promote thrombosis appear to be multifactorial (Table 2; Figure 2). In addition to inducing platelet aggregation, histones promote thrombin generation in PRP, but not in platelet-free plasma, in the absence of TF or thrombomodulin.24,63 Indeed, histones induce platelets to release inorganic polyphosphates and to expose membrane-bound phosphatidylserine and activated FV, which support the assembly and enhance the activity of the prothrombinase complex.63 Calf thymus histones shorten TF-initiated clotting times and the time to peak thrombin generation in PPP without the addition of thrombomodulin,64 possibly by promoting prothrombin autoactivation.65 In contrast, it has been reported that purified linker histone H1 prolongs clotting times, delays and reduces thrombin generation in PPP, and promotes fibrinolysis, suggesting paradoxical antithrombotic effects, although the biochemical mechanism remains unresolved.64,66 Histones also impair thrombomodulin-mediated protein C activation, resulting in enhanced thrombin generation in PPP when thrombomodulin is present.64 Furthermore, histones also induce vascular dysfunction60 and promote a procoagulant phenotype in vascular and blood cells (Figure 2). For instance, histones mediate phosphatidylserine exposure on red blood cells that then acquire the ability to assemble the prothrombinase complex and enhance fibrin formation.67 In addition, histones promote red cell aggregation, fragility, and rigidity.68 Incubation of histones with primary endothelial cells or endothelial cell lines results in upregulation of functional TF, whereas thrombomodulin expression is downregulated.69,70 Histones similarly mediate upregulation of TF when incubated with human monocytes and macrophage cell lines, to a level that triggers thrombin generation in plasma.69,71 Using recombinant histone proteins, it appears that the majority of procoagulant and cytotoxic effects attributed to histones are primarily mediated by histones H3 and/or H4, whereas other histone types have no or, at most, a modest effect.60,63,70,71
Studies evaluating the effect of histones on coagulation
| Reference | Histone type | Study design | Main conclusions |
|---|---|---|---|
| Kheiri et al, 199666 | Histone H1 from human acute myelogenous leukemia cells | Incubation in plasma | Prolonged PTT, aPTT and dilute Russell viper venom times, but not thrombin time |
| Xu et al, 200960 | Mixture of calf thymus histones | Intravenous injection into mice | Lethality (75 mg/kg) |
| Sublethal dose induces endothelial damage and microthrombosis in lungs | |||
| Fuchs et al, 201161 | Mixture of calf thymus histones or individual recombinant human histones | Incubation with platelets | Platelet aggregation |
| Intravenous injection of sublethal dose into mice | Thrombocytopenia, prolonged bleeding time, platelet aggregates in lungs | ||
| Semeraro et al, 201163 | Mixture of calf thymus histones or individual recombinant human histones | Incubation with PRP or isolated platelets | Enhanced thrombin generation in PRP, platelet aggregation, expression of phosphatidylserine and FV/Va on platelets, enhanced prothrombinase activity on platelets |
| Ammollo et al, 201164 | Mixture of calf thymus histones or individual recombinant human histones | Incubation with PPP, incubation with thrombin, thrombomodulin and protein C | Shortened clotting times measured by turbidimetry, enhanced thrombin generation in the presence of thrombomodulin; H1 impairs thrombin generation in PPP; histones impair thrombin-thrombomodulin-dependent activation of protein C |
| Abrams et al, 201362 | Mixture of calf thymus or individual recombinant human histones | Tail vein injection of lethal dose into mice | Thrombosis in the lung and kidney, increased plasma TAT complexes |
| Barranco-Medina et al, 201365 | Recombinant human histones H3 or H4 | Incubation with purified coagulation proteins | Histone H4 but not H3 directly promotes prothrombin autoactivation to thrombin |
| Semeraro et al, 201467 | Mixture of calf thymus histones or recombinant human histones H1, H2A, H2B, H3, and H4 | Incubation with isolated red blood cells in suspension | Histone H4 induced phosphatidylserine exposure on red blood cells |
| Yang et al, 201669 | Mixture of calf thymus histones and recombinant human histones H1, H2A, H2B, H3, or H4 | Incubation with primary coronary artery endothelial cells, human umbilical vein endothelial cells, and murine macrophage-like RAW264.7 cells | Histones induced upregulation of tissue factor on endothelial cells and macrophages |
| Kim et al, 201670 | Mixture of calf thymus histones | Incubation with EA.hy926 human endothelial cell line | Histones induced upregulation of tissue factor and protein-disulfide isomerase, and downregulation of thrombomodulin on endothelial cells |
| Gould et al, 201671 | Mixture of bovine histones or recombinant human histones H1, H2A, H2B, H3, and H4 | Incubation with THP-1 cell line and human monocytes | Histones upregulated tissue factor expression and phosphatidylserine exposure by THP-1 cells and human monocytes |
| Noubouossie et al, 201724 | Recombinant human histones H3 or H4 | Incubation in PFP and PRP | Histones triggered thrombin generation in PRP but not in PFP |
| Kordbacheh et al, 201768 | Mixture of calf thymus histones | Incubation in buffer with isolated human erythrocytes or intravenous injection of sublethal dose to mice | Histones induce erythrocyte aggregation, fragility to lysis under shear stress and reduce deformability; in vivo, histones induce thrombocytopenia, anemia and leukopenia |
| Reference | Histone type | Study design | Main conclusions |
|---|---|---|---|
| Kheiri et al, 199666 | Histone H1 from human acute myelogenous leukemia cells | Incubation in plasma | Prolonged PTT, aPTT and dilute Russell viper venom times, but not thrombin time |
| Xu et al, 200960 | Mixture of calf thymus histones | Intravenous injection into mice | Lethality (75 mg/kg) |
| Sublethal dose induces endothelial damage and microthrombosis in lungs | |||
| Fuchs et al, 201161 | Mixture of calf thymus histones or individual recombinant human histones | Incubation with platelets | Platelet aggregation |
| Intravenous injection of sublethal dose into mice | Thrombocytopenia, prolonged bleeding time, platelet aggregates in lungs | ||
| Semeraro et al, 201163 | Mixture of calf thymus histones or individual recombinant human histones | Incubation with PRP or isolated platelets | Enhanced thrombin generation in PRP, platelet aggregation, expression of phosphatidylserine and FV/Va on platelets, enhanced prothrombinase activity on platelets |
| Ammollo et al, 201164 | Mixture of calf thymus histones or individual recombinant human histones | Incubation with PPP, incubation with thrombin, thrombomodulin and protein C | Shortened clotting times measured by turbidimetry, enhanced thrombin generation in the presence of thrombomodulin; H1 impairs thrombin generation in PPP; histones impair thrombin-thrombomodulin-dependent activation of protein C |
| Abrams et al, 201362 | Mixture of calf thymus or individual recombinant human histones | Tail vein injection of lethal dose into mice | Thrombosis in the lung and kidney, increased plasma TAT complexes |
| Barranco-Medina et al, 201365 | Recombinant human histones H3 or H4 | Incubation with purified coagulation proteins | Histone H4 but not H3 directly promotes prothrombin autoactivation to thrombin |
| Semeraro et al, 201467 | Mixture of calf thymus histones or recombinant human histones H1, H2A, H2B, H3, and H4 | Incubation with isolated red blood cells in suspension | Histone H4 induced phosphatidylserine exposure on red blood cells |
| Yang et al, 201669 | Mixture of calf thymus histones and recombinant human histones H1, H2A, H2B, H3, or H4 | Incubation with primary coronary artery endothelial cells, human umbilical vein endothelial cells, and murine macrophage-like RAW264.7 cells | Histones induced upregulation of tissue factor on endothelial cells and macrophages |
| Kim et al, 201670 | Mixture of calf thymus histones | Incubation with EA.hy926 human endothelial cell line | Histones induced upregulation of tissue factor and protein-disulfide isomerase, and downregulation of thrombomodulin on endothelial cells |
| Gould et al, 201671 | Mixture of bovine histones or recombinant human histones H1, H2A, H2B, H3, and H4 | Incubation with THP-1 cell line and human monocytes | Histones upregulated tissue factor expression and phosphatidylserine exposure by THP-1 cells and human monocytes |
| Noubouossie et al, 201724 | Recombinant human histones H3 or H4 | Incubation in PFP and PRP | Histones triggered thrombin generation in PRP but not in PFP |
| Kordbacheh et al, 201768 | Mixture of calf thymus histones | Incubation in buffer with isolated human erythrocytes or intravenous injection of sublethal dose to mice | Histones induce erythrocyte aggregation, fragility to lysis under shear stress and reduce deformability; in vivo, histones induce thrombocytopenia, anemia and leukopenia |
aPTT, activated partial thromboplastin time; PFP, platelet-free plasma; PTT, prothrombin time; TAT, thrombin-antithrombin complexes.
Chromatin/NETs and coagulation
Although apoptosis and necrosis are common mechanisms of (neutrophil) cell death, little is known about the procoagulant activity of chromatin or its primary components derived from these pathways. In contrast, the link between NETosis and the activation of coagulation has been an active area of research. Immunothrombosis has been defined as a process whereby NETs play a beneficial role in host defense by trapping and killing pathogens at the expense of promoting thrombosis.72 Indeed, extracellular chromatin, sometimes containing citrullinated histones, has been detected in arterial, venous, and microvascular thrombi in humans73-77 and in animals.23,78-87 Its presence may not be simply an epiphenomenon, as NETs have been shown to play a critical role in some experimental models of animal thrombosis, hereafter referred to as NET-dependent models.23,78-88 However, the mechanism or mechanisms by which NETs promote thrombosis are unclear. They appear to be multifactorial, with the involvement of blood cells, soluble components of the plasma, the vessel wall, and potential blood flow disturbances (Figure 3).
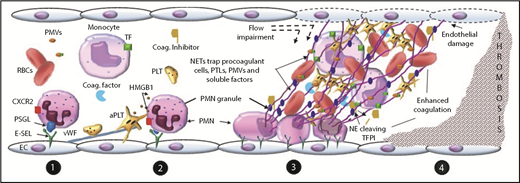
Proposed mechanism of NET-dependent thrombosis in vivo. (1) Neutrophils adhere to the endothelium by interaction between E-selectin and neutrophil P-selectin glycoprotein ligand (PSGL) and CXCR2. Platelets (PLTs) adhere by binding to von Willebrand factor (VWF) attached to the endothelium. (2) PSGL-1 and CXCR2 signaling in neutrophils (PMNs), as well as interactions between adherent neutrophils and platelets mediated by platelet HMGB1 or P-selectin, promote NET release. (3) NETs adhere and damage the vascular wall, impair flow, trap circulating procoagulant actors, impair physiologic anticoagulants, (4) ultimately leading to enhanced coagulation and thrombosis. aPTL, activated platelet; E-SEL, endothelial selectin; NE, neutrophil elastase; PMV, procoagulant microvesicle; TFPI, TF pathway inhibitor.
Proposed mechanism of NET-dependent thrombosis in vivo. (1) Neutrophils adhere to the endothelium by interaction between E-selectin and neutrophil P-selectin glycoprotein ligand (PSGL) and CXCR2. Platelets (PLTs) adhere by binding to von Willebrand factor (VWF) attached to the endothelium. (2) PSGL-1 and CXCR2 signaling in neutrophils (PMNs), as well as interactions between adherent neutrophils and platelets mediated by platelet HMGB1 or P-selectin, promote NET release. (3) NETs adhere and damage the vascular wall, impair flow, trap circulating procoagulant actors, impair physiologic anticoagulants, (4) ultimately leading to enhanced coagulation and thrombosis. aPTL, activated platelet; E-SEL, endothelial selectin; NE, neutrophil elastase; PMV, procoagulant microvesicle; TFPI, TF pathway inhibitor.
In NET-dependent models of venous thrombosis, engagement of PSGL-1 and the chemokine receptor CXCR2 on rolling neutrophils propagates signals that cooperate to induce β2 integrin-dependent neutrophil firm adhesion to endothelium and arrest.89 Neutrophils and platelets adhere to the vessel wall by binding to endothelial selectins and VWF.79,90 PSGL-1 and CXCR2 signaling in neutrophils,89 as well as interactions between adherent neutrophils and platelets (as may be mediated by platelet HMGB1 or P-selectin91,92 ) can lead to NET formation. NETs released into the intravascular space firmly adhere to the vessel wall, resist flow, and promote endothelial damage, likely mediated by the cytotoxic effects of neutrophil enzymes, histones, and/or HMGB1.60,84,93-95 In addition, NETs provide a surface for the binding of a variety of circulating coagulation components including platelets, leukocytes, red blood cells, microvesicles, and soluble clotting factors.53,79,87,96-98 Thus, intravascular NETs impair blood flow and promote accumulation of and interactions between circulating procoagulant cells, microvesicles, and soluble components, which collectively promote thrombin generation and thrombus formation.80,96 In the inferior vena cava stenosis model of thrombosis in mice, thrombosis is critically dependent on NETs, as well as TF expressed by hematopoietic cells, platelets, and FXII,79 all of which NETs serve to colocalize.
NETs also trap tissue factor pathway inhibitor (a major inhibitor of the TF pathway), which colocalizes with neutrophil elastase on NET strands. This proximity promotes neutrophil elastase-mediated degradation of tissue factor pathway inhibitor, leading to enhanced arterial thrombosis in mice.81
Prevention of neutrophil adhesion to endothelium (eg, through blockade of endothelial P-selectin or deletion of PSGL-1), prevention of NET formation, or dismantling of the NET scaffold by various mechanisms protect animals from thrombosis in NET-dependent models.79,80,82,83,85,86,89,92,99 These interventions prevent NET-induced endothelial damage, flow restriction, and/or concentration of circulating procoagulant factors.
NET-independent animal models of thrombosis have also been reported. In the stasis model in which blood flow restriction (usually in the inferior vena cava) is complete, depletion of neutrophils and inhibition of PAD4 or DNAse pretreatment do not protect wild-type mice from thrombosis.100 It is tempting to postulate that NET-induced flow restriction and trapping in the inferior vena cava stenosed at ∼90% were recapitulated by complete flow restriction in this model.
Jiménez-Alcázar et al recently developed a mouse model of in vivo NETosis in the microcirculation, characterized by vascular occlusion, organ damage, and death.23 Although most NET-associated clots harvested from occluded vessels stained for fibrin and VWF, up to 10% did not stain for either component. Strikingly, in contrast to protection conferred by endogenous expression of DNAses, depletion of platelets and pharmacological inhibition of thrombin did not protect DNAse-deficient mice from NET-induced mortality.23 Collectively, these observations suggest that NETs can promote vascular occlusion independent of coagulation activation and fibrin clot formation.
Some studies have reported enhanced thrombin generation in platelet-poor plasma containing NETs generated ex vivo. In these studies, neutrophils were isolated from the peripheral blood of humans57,101 or from mouse bone marrow.102 Thrombin generation was attenuated by pharmacological or genetic blockade of FXII(a) or FXI(a), or by treatment with DNAse, suggesting DNA-dependent activation of the contact pathway. In contrast, our group did not observe any thrombin generation in the presence of intact NETs in platelet-free plasma or in a purified contact system reconstituted in buffer.24 Interestingly, we observed that although gDNA isolated from NETs triggered coagulation, reconstitution of chromatin with gDNA and histones abolished gDNA-induced coagulation activation in plasma and in purified systems.
Pitfalls and knowledge gaps
Although NETs consist of chromatin and not free DNA or free histones, it may be misleading to extrapolate the procoagulant activities demonstrated by studies performed in vitro using gDNA or purified histones to the effect of NETs in promoting thrombosis in vivo. We observed that purified individual histones lose their capacity to trigger thrombin generation in platelet-rich plasma when they assemble into the octameric core histone, whereas gDNA is no longer procoagulant in platelet-free plasma when integrated into a nucleosome or chromatin.24 Likewise, Marsman et al reported that in contrast to purified histones, intact nucleosomes were not cytotoxic to HEK293 in vitro; however, nucleosome cytotoxicity was restored after digestion of the DNA component.103 In addition, Grasso et al observed a significant activation of factor VII activating protease only when histones from NET-derived nucleosomes were made DNA-free.104 These observations strongly support the notion that complexation into octamers or nucleosomes impairs many inflammatory or procoagulant properties of free histones or free DNA. Histones activate cells through TLR-2- and TLR-4-dependent signaling.63,69-71,101 They also intercalate and form pores into the plasma membrane, allowing ion influx-dependent cell activation or death.105 It seems likely that the assembly of individual histone proteins to form octamers, nucleosomes, or chromatin interferes with those activities.
In most studies reporting enhancement of ex vivo thrombin generation by NETs, substantial thrombin generation in plasma is observed at baseline, indicating that initiation of coagulation may be independent of NETs.57,101,102 Most of these studies used the gradient density method for isolating neutrophils from whole blood before stimulation of NETosis (Table 3). In our hands, substantial numbers of platelets remain in neutrophil isolates prepared using this method.24 Moreover, red blood cell lysis performed during the gradient density method may generate red blood cell microvesicles or membrane debris, which are known to trigger coagulation through the contact pathway.106 Overall, careful attention to neutrophil isolation techniques (for purity and absence of activation), to experimental buffer conditions, as well as avoidance of test plasma pre-activation are critical considerations in future experiments addressing the direct procoagulant activity of NETs.
Studies evaluating the procoagulant activity of NETs ex vivo
| Reference | Species | Method of neutrophil isolation | Method/duration of NET induction | Coagulation assay | Main conclusions |
|---|---|---|---|---|---|
| Oemcke et al 200953 | Human | Gradient centrifugation | Glucose oxidase (60 min) or IL8 (60 min) or M1 protein-fibrinogen complexes (60 min) | Chromogenic assay for PK hydrolysis in plasma | NETs bind FXII and HK, and promote PK hydrolysis in plasma |
| Fuchs et al 201098 | Human | Gradient centrifugation | PMA (4 h), glucose oxydase (1U/ml, 4 h) | Electron and fluorescent microscopy after flowing plasma, suspension of platelets or whole blood over NETs seeded in a flow chamber or on glass cover slide | NETs bind platelets and promote platelet activation and aggregation. |
| NETs bind RBCs, VWF, fibrinogen, and fibronectin. | |||||
| NETs promote resistance to tPA-mediated fibrinolysis | |||||
| Gould et al 2014101 | Human | Gradient centrifugation | PMA 150 min | Thrombin generation in PPP or PRP | NETs enhance thrombin generation |
| Noubouossie et al 201724 | Human | Magnetic bead-based negative selection | PMA, calcium ionophore and LPS. | Contact system in buffer | NETS do not trigger contact system activation in buffer, do not trigger thrombin generation in plasma, and do not amplify low TF-initiated TG in PFP. |
| 4 h | Thrombin generation in PFP and PRP | ||||
| Ivanov et al 201757 | Human | Gradient centrifugation | PMA: 90 min | Thrombin generation in plasma | NETs shorten lag time of thrombin generation. |
| Wang et al 2018102 | Mouse | Gradient centrifugation | PMA: 3 h | Thrombin generation in plasma | NETs form complexes with microvesicles to enhance TG in plasma |
| Reference | Species | Method of neutrophil isolation | Method/duration of NET induction | Coagulation assay | Main conclusions |
|---|---|---|---|---|---|
| Oemcke et al 200953 | Human | Gradient centrifugation | Glucose oxidase (60 min) or IL8 (60 min) or M1 protein-fibrinogen complexes (60 min) | Chromogenic assay for PK hydrolysis in plasma | NETs bind FXII and HK, and promote PK hydrolysis in plasma |
| Fuchs et al 201098 | Human | Gradient centrifugation | PMA (4 h), glucose oxydase (1U/ml, 4 h) | Electron and fluorescent microscopy after flowing plasma, suspension of platelets or whole blood over NETs seeded in a flow chamber or on glass cover slide | NETs bind platelets and promote platelet activation and aggregation. |
| NETs bind RBCs, VWF, fibrinogen, and fibronectin. | |||||
| NETs promote resistance to tPA-mediated fibrinolysis | |||||
| Gould et al 2014101 | Human | Gradient centrifugation | PMA 150 min | Thrombin generation in PPP or PRP | NETs enhance thrombin generation |
| Noubouossie et al 201724 | Human | Magnetic bead-based negative selection | PMA, calcium ionophore and LPS. | Contact system in buffer | NETS do not trigger contact system activation in buffer, do not trigger thrombin generation in plasma, and do not amplify low TF-initiated TG in PFP. |
| 4 h | Thrombin generation in PFP and PRP | ||||
| Ivanov et al 201757 | Human | Gradient centrifugation | PMA: 90 min | Thrombin generation in plasma | NETs shorten lag time of thrombin generation. |
| Wang et al 2018102 | Mouse | Gradient centrifugation | PMA: 3 h | Thrombin generation in plasma | NETs form complexes with microvesicles to enhance TG in plasma |
LPS, lipopolysaccharide; PMA, phorbol myristate acetate; tPA, tissue-type plasminogen activator.
Most studies have used gDNA isolated from nonactivated human neutrophils or cell lines to address its procoagulant activities (Table 1). Such gDNA shows a smear with size above 10,000 base pairs on agarose gel electrophoresis.24 It is unlikely that DNA of such a size is found protein-free in vivo. Indeed, extracellular chromatin is likely processed by endogenous nucleases and proteases in vivo.23 DNase1 and DNAse 1-like3 (or DNAse γ) have been identified as 2 major circulating endonucleases. DNAse 1 is secreted by nonhematopoietic cells and preferentially digests protein-free DNA, whereas DNAse1-like3, secreted by immune cells, preferentially digests DNA incorporated into chromatin at internucleosomal sites.107,108 However, it is largely unknown what sizes of DNA circulate in vivo and whether it circulates free or bound to other molecules. Because the commonly used methods to detect circulating DNA use DNA dyes, polymerase chain reaction, or require prior DNA isolation, they may not give information on DNA size or distinguish between protein-free DNA and DNA imbedded into nucleosomes or other molecules. In contrast to polyphosphate, the size-dependency of the contact-dependent procoagulant activity of gDNA has been poorly studied. One study reported that only large fragments of gDNA (≥300 base pairs) promote the formation of thick and densely packed fibrin clot that prolongs tissue-type plasminogen activator-mediated clot lysis time.59
DNA conformation may also determine its procoagulant activity. Some hairpin DNA aptamers are procoagulant, whereas linear ones are not,109 indicating a possible role of DNA secondary structure. As discussed earlier, DNA posttranscriptional modifications such as DNA methylation may alter DNA conformation, flexibility, and protein binding ability, possibly including the binding of proteins of the contact system.110 To our knowledge, no published study has addressed the effect of DNA methylation on procoagulant activity. Likewise, whether histone PTMs that are commonly found in vivo affect the procoagulant activity of histone proteins is unknown. Most studies have used purified calf thymus histones, usually with undetermined PTM patterns, or recombinant human histones that do not have PTMs. However, our group observed similar thrombin generation in PRP triggered by recombinant histone H3 treated or not with PAD4 to induce citrullination.24
NET-dependent thrombosis usually occurs in animals over days,79 whereas ex vivo studies are generally performed for a maximum duration of 4 hours.24 Thus, an ongoing slow procoagulant process in vivo, which is undetectable within the timeframe of ex vivo experimentation, may potentially account for the discrepancy between in vivo and ex vivo studies of the procoagulant role of NETs.
Finally, although the prothrombotic role of NETs has been shown in vivo in animal studies, in vitro studies have mainly used human neutrophils (Tables 1 and 3). It is largely unknown whether neutrophils, DNA, or histones from animals exhibit the same procoagulant profile as their human counterparts. One study used the density gradient method to isolate neutrophils from mouse bone marrow and reported a synergistic effect of NETs in complex with phosphatidylserine-positive, CD41-negative microvesicles in enhancing thrombin generation in plasma.102
Conclusion
In summary, and in contrast to the irrefutable evidence that activated monocytes can directly trigger coagulation in a TF-dependent manner, it remains uncertain whether stimulated neutrophils can directly activate coagulation. NETs are composed of decondensed chromatin and should not be considered interchangeable with gDNA or histones. Convincing evidence indicates that intravascular release of NETs promotes vascular occlusion, with or without fibrin generation. It seems quite likely also that NETs may play a variable role in thrombosis, depending on the anatomic location and size of the vessel. Whether the macromolecular structure of NETs and/or chromatin directly activate coagulation is uncertain. However, the ability of NETs to trap and accumulate circulating cells and soluble components has been consistently demonstrated, and may play an essential role in their thrombogenic potential. For these reasons, intravascular NETs can be considered to be prothrombotic (ie, promote vascular occlusion), but not directly procoagulant (ie, initiate coagulation activation). Accordingly, preventing NET release or enhancing its degradation would represent a novel strategy to prevent thrombosis, without the need for conventional anticoagulants.23 In contrast, purified components of NETs/chromatin, including gDNA and histones, can directly trigger coagulation in vitro. They can therefore be considered to be procoagulant. Because the contact system is clearly dispensable for hemostasis, blocking nuclear material-induced contact activation also represents a potential strategy to prevent thrombosis with minimal to no risk of bleeding. However, further investigations are required to understand whether circulating chromatin components in humans, including those with PTMs, are present in the forms, concentration, and half-life sufficient to trigger coagulation; determine whether the procoagulant activity of nuclear material is similar across animal and human species; and determine whether the underlying disease or clonal abnormalities of neutrophils affect their procoagulant/prothrombotic properties. Finally, to confirm the presence and extent of NETosis in vivo, a consensus on the optimal biomarker profile to detect and quantify NETosis is urgently required.
Authorship
Contribution: D.F.N. and N.S.K. wrote the initial draft; D.F.N. and B.N.R. performed the literature review; B.D.R. provided critical review on histone posttranslational modification text; and all authors contributed to several revisions of the text.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Nigel S. Key, 1079 Genetics Medicine Building, CB #7035, 120 Mason Farm Rd, Chapel Hill, NC 27599; e-mail: nigel_key@med.unc.edu.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal