

Abstract
Primary immunodeficiencies affecting the function of neutrophils and other phagocytic leukocytes are notable for an increased susceptibility to bacterial and fungal infections as a result of impaired leukocyte recruitment, ingestion, and/or killing of microbes. The underlying molecular defects can also impact other innate immune responses to infectious and inflammatory stimuli, leading to inflammatory and autoimmune complications that are not always directly related to infection. This review will provide an update on congenital disorders affecting neutrophil function in which a combination of host defense and inflammatory complications are prominent, including nicotinamide dinucleotide phosphate oxidase defects in chronic granulomatous disease and β2 integrin defects in leukocyte adhesion deficiency.
Introduction
Immune dysregulation is a feature of many primary immunodeficiency (PID) disorders,1-3 including those affecting neutrophil function. Neutrophils and other phagocytes are essential effectors of innate immunity and rapidly respond to the presence of invading bacteria, fungi, and parasites. Inflammatory signals activate adhesion, chemotaxis, phagocytosis, and release of oxidants, proteases, and other molecules aimed at microbial killing. These same processes are important for appropriate responses to wounds or sterile inflammation. Phagocytic leukocytes also synthesize and secrete multiple inflammatory mediators, including leukotrienes, chemokines, and other cytokines to amplify and regulate the inflammatory response and initiate cross talk with adaptive immune cells. Hence, PID with functional phagocyte defects can display recurrent severe bacterial and fungal infections and aberrant inflammation that is not always related to infection.
This review will cover the current understanding of the clinical features and underlying mechanisms driving dysregulated inflammation in 2 inherited defects affecting neutrophil function. A major focus will be on chronic granulomatous disease (CGD), which results from genetic defects in the leukocyte nicotinamide dinucleotide phosphate (NADPH) oxidase. Clinical manifestations in CGD include recurrent bacterial and fungal infections, as well as acute and chronic inflammatory conditions, which reflect the broad impact of reactive oxygen species (ROS) generated from the NADPH oxidase on immune responses. In addition, new insights into the severe periodontal disease associated with leukocyte adhesion deficiency type 1 (LAD-1) will be summarized. This manifestation illuminates another facet of neutrophil biology, in which clearance of senescent neutrophils in tissues is linked to an interleukin-23 (IL-23)–IL-17–granulocyte colony-stimulating factor (G-CSF) cytokine axis that is dysregulated in LAD-1.
CGD
CGD is caused by inactivating X-linked or autosomal-recessive mutations in the leukocyte NADPH oxidase (also referred to as the respiratory burst oxidase) (Figure 1).4,5 Superoxide generated by this enzyme is rapidly converted into other ROS, including H2O2 and myeloperoxidase-catalyzed formation of hypochlorous acid. These oxidants have important microbicidal activity, because their absence leads to susceptibility to a distinctive set of bacterial and fungal pathogens. A second hallmark of CGD is the frequent development of granulomas and other inflammatory disorders. These symptoms reflect the importance of NADPH oxidase–derived ROS for microbial killing, as well as for their pleiotropic impact on other cellular processes.6-10
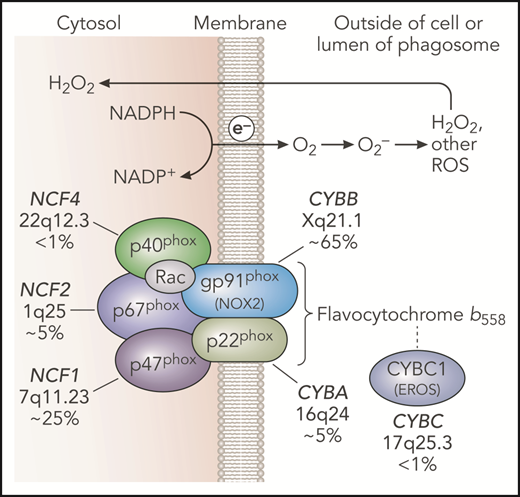
NADPH oxidase and genetic defects in CGD. The leukocyte NADPH oxidase enzyme complex is composed of membrane and cytosolic subunits that are referred to by their molecular mass (kDa) and the designation “phox,” for phagocyte oxidase. CYBB and CYBA refer to cytochrome b-245 β chain and cytochrome b-245 α chain, respectively, the large and small subunits of flavocytochrome b558, whereas NCF refers to neutrophil cytosolic factor, used to designate the cytosolic regulatory subunits of the oxidase. Flavocytochrome b558 is the electron transferase and is located in plasma, specific granules (in neutrophils), and phagosome and some endosome membranes. This heterodimer is composed of gp91phox and p22phox. The gp91phox subunit is sometimes referred to as NOX2. CYBC1 (also known as EROS) is an endoplasmic reticulum protein important for expression of the flavocytochrome b558 heterodimer. The soluble regulatory proteins p47phox, p67phox, and p40phox form a complex in the cytosol; upon leukocyte activation, phosphorylation-induced conformational changes lead to their binding to flavocytochrome b558. The small GTPase Rac is also essential for NADPH oxidase enzymatic activity, which, in its active GTP-bound state, becomes membrane bound and activates p67phox. Together, these regulatory proteins activate the flavocytochrome b558–mediated transfer of electrons from cytosolic NADPH across the membrane via FAD and heme redox centers to molecular oxygen, thereby forming superoxide in the extracellular space or within phagosomes or endosomes. Superoxide is converted into H2O2, which can diffuse across membranes, and other ROS. CGD results from inactivating recessive mutations in any 1 of the 5 phox subunits or CYBC1, as indicated with the approximate incidence, gene, and chromosomal location. Professional illustration by Patrick Lane, ScEYEnce Studios.
NADPH oxidase and genetic defects in CGD. The leukocyte NADPH oxidase enzyme complex is composed of membrane and cytosolic subunits that are referred to by their molecular mass (kDa) and the designation “phox,” for phagocyte oxidase. CYBB and CYBA refer to cytochrome b-245 β chain and cytochrome b-245 α chain, respectively, the large and small subunits of flavocytochrome b558, whereas NCF refers to neutrophil cytosolic factor, used to designate the cytosolic regulatory subunits of the oxidase. Flavocytochrome b558 is the electron transferase and is located in plasma, specific granules (in neutrophils), and phagosome and some endosome membranes. This heterodimer is composed of gp91phox and p22phox. The gp91phox subunit is sometimes referred to as NOX2. CYBC1 (also known as EROS) is an endoplasmic reticulum protein important for expression of the flavocytochrome b558 heterodimer. The soluble regulatory proteins p47phox, p67phox, and p40phox form a complex in the cytosol; upon leukocyte activation, phosphorylation-induced conformational changes lead to their binding to flavocytochrome b558. The small GTPase Rac is also essential for NADPH oxidase enzymatic activity, which, in its active GTP-bound state, becomes membrane bound and activates p67phox. Together, these regulatory proteins activate the flavocytochrome b558–mediated transfer of electrons from cytosolic NADPH across the membrane via FAD and heme redox centers to molecular oxygen, thereby forming superoxide in the extracellular space or within phagosomes or endosomes. Superoxide is converted into H2O2, which can diffuse across membranes, and other ROS. CGD results from inactivating recessive mutations in any 1 of the 5 phox subunits or CYBC1, as indicated with the approximate incidence, gene, and chromosomal location. Professional illustration by Patrick Lane, ScEYEnce Studios.
The concept that the respiratory burst plays a broad role in immune responses was solidified by 2 key experimental observations. In 1 study, administration of sterile Aspergillus hyphae into the lungs of CGD mice induced an excessive acute neutrophilic response and subsequent chronic granulomatous inflammation.11 This finding established the importance of the NADPH oxidase for limiting inflammation, independent of its antimicrobial effects. A second important study made the link between NADPH oxidase deficiency and autoimmunity. Here, positional cloning identified a gene conferring increased severity of experimental arthritis in rats, which unexpectedly proved to be a hypomorphic mutation in an NADPH oxidase subunit, p47phox (NCF1).12
NADPH oxidase and molecular genetics of CGD
The NADPH oxidase is a multi-subunit phagosome and plasma membrane-associated enzyme (see Figure 1 for details) expressed in neutrophils, monocytes and macrophages, dendritic cells, and eosinophils.13 The enzyme is also present in B, and perhaps T, lymphocytes, although here its function is not well understood. The flavocytochrome b558 is composed of 2 integral membrane proteins: gp91phox (also known as NADPH oxidase 2 [NOX2]) and p22phox. The gp91phox subunit contains flavoprotein and heme-binding domains, and p22phox harbors a key docking site for p47phox. Heterodimer formation is mediated, in part, by the endoplasmic reticulum–resident protein essential for ROS (EROS), also known as CYBC1.14 A complex of 3 regulatory subunits, p47phox, p67phox, and p40phox, translocate from the cytoplasm to flavocytochrome b558, along with guanosine triphosphate (GTP)-bound Rac, upon cellular activation with inflammatory or phagocytic stimuli to initiate electron transfer from NADPH to molecular oxygen. Defects in the leukocyte-specific Rac2 isoform lead to only mild impairments in the NADPH oxidase, probably because of redundancy with Rac1, and are not associated with a CGD phenotype.4
CGD occurs in ≈1 in 200 000 live births.15 Approximately two thirds of patients have defects in X-linked CYBB.4 Autosomal-recessive mutations in NCF1 account for ≈25% of CGD. Rarer autosomal-recessive forms of CGD involve mutations in CYBA, NCF2, or NCF4 genes. The incidence of autosomal-recessive CGD is higher in countries with high rates of consanguineous marriage.16,17
Autosomal-recessive null mutations in NCF4 or CYBC1 have only recently been described.18-21 In contrast to the “classic” forms of CGD that lack or have profoundly reduced NADPH oxidase activity, the respiratory burst defects are more nuanced. NCF4 (p40phox) is a specialized subunit that stimulates phagosome oxidase activity via a regulatory domain that binds to phosphatidylinositol 3-phosphate, a membrane lipid present in high concentrations on phagosome and other intracellular membranes.13 As a result, intracellular NADPH oxidase activity is substantially reduced, especially to phagocytic stimuli, and macrophages also display a partial reduction in plasma membrane activity.18,19,22 Thus far, 24 patients with NCF4 defects are described worldwide.18,19 EROS (CYBC1) defects were originally identified in a mouse mutagenesis screen and associated with a profound reduction in macrophage flavocytochrome b558; neutrophils appeared less affected.14 Nine patients are reported, including 8 from Iceland homozygous for the same founder mutation and 1 patient from Saudi Arabia.20,21 Detailed studies are still limited but suggest that defects in human monocyte and macrophage flavocytochrome b558 expression and NADPH oxidase activity are more substantial that those in neutrophils. Of note, phorbol ester–induced dihydrorhodamine-1,2,3 oxidation in neutrophils, a widely used diagnostic test for CGD, may not be abnormal in NCF4 or CYBC1 deficiency.
Clinical features of CGD
Infections
Manifestations of CGD typically begin in infancy or early childhood, although they can be delayed until adolescence or even adulthood.4,15-17,23-27 CGD patients are particularly susceptible to Staphylococcus aureus, Burkholderia cepacia, Nocardia spp., and certain gram-negative enteric bacilli, including Serratia marcescens and Salmonella spp.28 CGD patients have increased risk from Mycobacterium tuberculosis in endemic areas and can develop severe local or systemic disease with bacillus Calmette-Guérin (BCG), an attenuated strain of Mycobacterium bovis, following BCG vaccination.17,24 Invasive fungal infections are a major threat in CGD, most commonly with Aspergillus fumigatus and Aspergillus nidulans.29,30 Frequent sites of infection include the lungs, lymph nodes, skin and soft tissues, the gastrointestinal (GI) tract, including staphylococcal liver abscesses, and osteomyelitis.4,15
Inflammatory complications
Inflammatory manifestations are common in CGD and are often associated with neutrophilic and/or granulomatous inflammation.4-10,31 These symptoms can reflect a dysregulated inflammatory response in the absence of respiratory burst–derived ROS. Excessive acute or persistent inflammation can be seen in the setting of active infection or as a sequelae. However, in other cases, abnormal inflammation develops in the absence of antecedent infection.
Recurrent or chronic inflammatory conditions can involve the skin, GI tract, lung, and liver. A retrospective study of 98 CGD patients from a single institution found that almost 70% had symptoms associated with inflammatory manifestations,32 similar to prior multi-institution registry data.15,25-27 GI symptoms, including abdominal pain, diarrhea, and oral aphthous ulcers, were present in 60 patients. Forty-four patients underwent biopsy, and chronic GI inflammatory lesions were identified in 22 patients. Pulmonary findings, including dyspnea, were reported in 18 patients and were associated with pleural thickening, interstitial lung disease, and/or fibrosis. Twelve patients had episodes of bladder wall granulomas or other inflammation involving the urogenital tract. Chorioretinitis and ocular granulomata were reported in 6 patients, and 7 patients had discoid lupus, leukocytoclastic vasculitis, arthritis, or dermatomyositis. Liver disease with noncirrhotic portal hypertension can also occur and may be an independent risk factor for mortality in CGD.33
The most frequent inflammatory disease of significance is GI disease that is somewhat reminiscent of Crohn’s disease. This can occur in all genetic subgroups of CGD and does not correlate with a higher risk for severe bacterial and fungal infections.28 Although involvement of the anus and rectum is most common, upper GI tract disease is also common.34-36 Microgranulomas pigmented macrophages and tissue eosinophilia are common biopsy findings.35 Granulomatous obstruction of the gastric outlet or urinary tract also occur. X-linked carriers of CGD are also at risk for developing Crohn’s disease, which affected 5 of 93 women in a recent study.37
Macrophage activation syndrome and hemophagocytic lymphohistiocytosis (HLH) can be another complication of CGD. In 63 patients with PIDs that met criteria for HLH but did not have cytotoxicity defects or X-linked lymphoproliferative disorders, 22 had CGD.38 These episodes were triggered by fungi, bacteria (especially B cepacia), or Leishmania in endemic areas. In another new study, only 19 of 101 children with HLH who received genetic testing had biallelic mutations in recognized familial HLH genes. Of the remaining children, 47 underwent whole-exome sequencing, and 14 patients proved to have PID, including 2 with underlying CGD.39
Aberrant inflammation with autoimmune features can also occur in CGD. Up to ≈10% of patients with X-linked CGD develop discoid lupus-like lesions or aphthous ulcers.16,17,23-27 Discoid lupus, photosensitivity, and aphthous ulcers also occur in carrier females, with an incidence of 20% to 50%.37,40,41 Less commonly, CGD patients and X-linked CGD carriers develop systemic autoimmune disease, including lupus, juvenile idiopathic arthritis, antiphospholipid syndrome, idiopathic thrombocytopenic purpura, and immunoglobulin A nephropathy.16,17,23-27,37
Several studies demonstrated skewed immune response parameters in CGD patients studied at times when they were in otherwise good health. Analysis of global gene expression in CGD neutrophils showed constitutively increased expression of inflammation-associated genes, including chemokines and immune receptors.42 Analysis of whole blood in a different study showed increased expression of type 1 interferon–regulated genes in CGD.43 Another group identified increased IL-1β and tumor necrosis factor-α (TNF-α) content in CGD monocytes and a T helper 17 cell bias of CD4+ T cells.44 Human CGD monocytes produced higher levels of IL-1β when stimulated in vitro with particulate and soluble activators of caspase-1 and NLRP3 inflammasome, and 1 study also found increased IL-1α release upon lipopolysaccharide stimulation.45-47 Increased IL-8 release by CGD neutrophils following formyl-methionyl-leucyl phenylalanine stimulation is also reported.48 Finally, many CGD patients, even without colitis, have elevated levels of antimicrobial and other antibodies associated with inflammatory bowel disease (IBD), suggesting an abnormal response to chronic antigen stimulation.49,50
Clinical features in different subgroups of CGD and in X-linked CGD carriers
Patients with X-linked CGD and those with CYBA and NCF2 mutations tend to have a more severe clinical course compared with patients with NCF1 defects.4,15,51 This may reflect, in part, the observation that fungal infections occur slightly less frequently with NCF1 defects and are associated with lower mortality.28 p47phox−/− neutrophils have a small amount (≤2%) of residual enzyme activity, because this subunit functions as an adaptor protein rather than mediating electron transfer. Some X-linked CGD patients with a partially functional gp91phox also have a milder clinical course. These observations suggest that even a small amount of NADPH oxidase activity can partially mitigate the defects in host defense. In contrast, GI disease was not correlated with the degree of residual superoxide production.51
The spectrum of disease with defects in NCF4 or CYBC1, both of which have more superoxide production compared with the 4 original CGD subgroups,18-21 resembles an atypical form of CGD. To date, reported patients have not had invasive bacterial and fungal infections with typical CGD pathogens, including A fumigatus. However, 1 patient with CYBC1 deficiency developed a localized abscess following BCG vaccination20 and a second had miliary tuberculosis,21 perhaps due to a greater impairment in flavocytochrome b biosynthesis in CYBC1-deficient macrophages. More prominent in CYCB1 and NCF4 subgroups are inflammatory manifestations, including granulomatous GI disease; patients with NCF4 defects also frequently exhibited discoid lupus-like skin lesions.19-21
Female carriers of X-linked CGD, as already noted, can develop CGD-associated inflammatory conditions.37,40,41 X-linked inactivation of the chromosome harboring the mutant CYBB allele appears to be random, with a median ≈50% of neutrophils lacking NADPH oxidase activity.4,37 Carriers with ≤10% NADPH oxidase–positive neutrophils can develop infections characteristic of CGD.4,37,40 However, the occurrence of inflammatory and autoimmune complications appears to be independent of the frequency of NADPH oxidase–positive neutrophils. This suggests that even the absence of NADPH oxidase ROS in some leukocytes can promote aberrant inflammation.
Finally, there is a surprising clinical heterogeneity among the >90% of CGD patients who lack or are profoundly deficient in NADPH oxidase activity.4,15 Some develop symptoms beginning during infancy, whereas, at the other end of the spectrum, patients can be well for many years and then develop a serious infection or inflammatory complication. Coexisting genetic variants in other antimicrobial systems and components of innate immunity likely play important roles in modifying disease severity. Variants in the myeloperoxidase, mannose binding lectin, and FcγRIIa genes were associated with a higher risk for granulomatous or autoimmune complications in CGD.52 Each of 2 patients with HLH and X-linked CYBB or NCF1 CGD studied by whole-exome sequencing had a variant in a second gene (DOCK11 or NLRP12) that could impact inflammatory responses. Another study found an increased frequency of single nucleotide polymorphisms associated with IBD risk in 40 unrelated children with CGD who had IBD compared with CGD children without GI inflammation.53 Notably, the number of IBD risk variants in CGD IBD patients was much lower than in other pediatric IBD patients without underlying immunologic defects, supporting the concept that a defective NADPH oxidase is an independent and major risk factor for GI inflammation. Finally, although not yet systematically studied, there is some experimental support for the importance of environmental factors in CGD-associated inflammation. The severity of experimental colitis in NCF1-deficient CGD mice was similar to wild-type littermates, but it became more severe in CGD if the composition of gut microbiota was altered.54
Hypomorphic variants in NADPH oxidase genes are associated with inflammatory and autoimmune diseases
Multiple recent studies identified variant alleles of NADPH oxidase genes as risk factors for autoimmune diseases and inflammatory bowel disease, including lupus (NCF2), rheumatoid arthritis (NCF1, NCF4), Sjögren syndrome (NCF1), Crohn disease (NCF2, NCF4, RAC2), and early-onset IBD (NCF1, NCF2, NCF4, CYBB, RAC2).55-61 These include missense polymorphisms, whereas others are intronic, in promoters, or affect copy number. These variants are not associated with microbial infections. Many of the missense variants reside in important functional domains, but those characterized have only a modest effect on NADPH oxidase activity.55,56,62 These findings illustrate how loci involved as causes of PID overlap with those identified in polygenic disorders of autoimmunity and inflammation.1,63 In PID, rare monogenic mutations with a strong effect cause disease. For more common inflammatory/autoimmune disorders, multiple genes harboring common variants contribute to these conditions. Of note, the most frequent and major genetic associations with lupus are common missense polymorphisms in NCF1 and NCF2,58 and lupus is more severe in NADPH oxidase–deficient mice, including X-linked CGD carriers and mice heterozygous for an NCF2 deletion, in several models of murine lupus.64-66
Mechanisms underlying NADPH oxidase regulation of immune cell functions
ROS are generated by multiple sources and have beneficial and deleterious effects. Mitochondria may be the largest contributor of intracellular ROS, but the NADPH oxidase family,67 which includes the NOX2-containing leukocyte NADPH oxidase, is another important source. ROS can promote harmful oxidative damage to proteins, DNA, and lipids.9,68 On the other hand, H2O2 can oxidize cysteines and methionines to physiologically regulate protein function, including protein kinases and phosphatases. In effect, these modifications use ROS as a signaling mechanism and are reversible by thiol-disulfide and glutathione reductases.9,68 Electron transfer across the membrane by the leukocyte oxidase also has electrogenic effects that can increase neutrophil intracellular calcium levels,69 as well as alter the pH within phagosomes and the activity of digestive proteases.13,70
Thus, NADPH oxidase ROS can have multiple effects (Figure 2), including damaging oxidation that aids microbial killing, but it can be injurious to host tissue if oxidant production is not appropriately regulated or confined. Triggering of NOX2 ROS production has coevolved with activation of other immune responses, and it exerts immunoregulatory effects that balance immune activation (Figure 3). Although specific molecular targets are incompletely understood, a general theme is that deficiency of NOX2-derived ROS increases acute inflammatory responses and can also predispose to autoimmunity. Examples and current insights are summarized below.
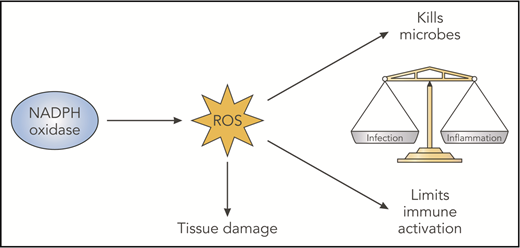
NADPH oxidase–derived ROS have beneficial and deleterious effects. NADPH oxidase–derived ROS have damaging oxidative effects that are important for microbial killing but can damage host tissues. In addition, derivative ROS have immunoregulatory effects, which act to balance immune responses that otherwise promote inflammation and even autoimmunity. Likely factors contributing to abnormal inflammation in the absence of NADPH oxidase ROS include impaired digestion of microbes or debris, increased proinflammatory cytokine production reflecting changes in redox-regulated signals, delayed clearance of inflammatory neutrophils, and altered antigen presentation. Professional illustration by Patrick Lane, ScEYEnce Studios.
NADPH oxidase–derived ROS have beneficial and deleterious effects. NADPH oxidase–derived ROS have damaging oxidative effects that are important for microbial killing but can damage host tissues. In addition, derivative ROS have immunoregulatory effects, which act to balance immune responses that otherwise promote inflammation and even autoimmunity. Likely factors contributing to abnormal inflammation in the absence of NADPH oxidase ROS include impaired digestion of microbes or debris, increased proinflammatory cytokine production reflecting changes in redox-regulated signals, delayed clearance of inflammatory neutrophils, and altered antigen presentation. Professional illustration by Patrick Lane, ScEYEnce Studios.
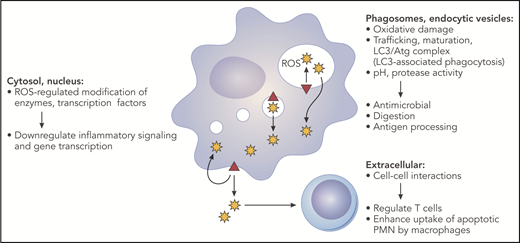
NADPH oxidase–derived oxidants can influence multiple aspects of innate and adaptive immune responses. The phagocyte NADPH oxidase (red triangle) is assembled on the plasma membrane, phagosomes, and endocytic compartments. Superoxide is released inside membrane compartments or the extracellular space and is rapidly converted into derivative ROS. This includes dismutation to H2O2, which is membrane permeant and, thus, can diffuse into the cytosol. Oxidase-generated ROS can have many effects, depending on the site of ROS production, and, thus, can impact multiple pathways important for innate and adaptive immunity. See text for additional details. Professional illustration by Patrick Lane, ScEYEnce Studios.
NADPH oxidase–derived oxidants can influence multiple aspects of innate and adaptive immune responses. The phagocyte NADPH oxidase (red triangle) is assembled on the plasma membrane, phagosomes, and endocytic compartments. Superoxide is released inside membrane compartments or the extracellular space and is rapidly converted into derivative ROS. This includes dismutation to H2O2, which is membrane permeant and, thus, can diffuse into the cytosol. Oxidase-generated ROS can have many effects, depending on the site of ROS production, and, thus, can impact multiple pathways important for innate and adaptive immunity. See text for additional details. Professional illustration by Patrick Lane, ScEYEnce Studios.
Loss of NADPH oxidase ROS can alter redox modifications that otherwise regulate signaling and transcription factors. Proinflammatory NF-κB activity was increased in lipopolysaccharide- or zymosan-stimulated CGD macrophages, reflecting reduced oxidation of nuclear redox factor 1.71,72 CGD mouse neutrophils stimulated with fungal cell walls also exhibited increased phosphorylation of MAPKs and NF-κB activity.73 In both cell types, increased production of proinflammatory chemokines and other cytokines was observed. Activation of the transcription factor nuclear factory erythroid 2–related factor 2, which regulates protective oxidative stress–response genes following oxidation of its inhibitor Keap1, was reduced in zymosan-stimulated CGD mouse macrophages and human monocytes.71 CGD Epstein-Barr virus B-cell lines showed increased p38 mitogen–activated protein kinase activation and cytokine production upon stimulation with Toll-like receptor 7 or 9 agonists.74
NADPH oxidase deficiency is associated with an intrinsically enhanced IL-1 response to various agonists, including necrotic cell lysates, other damage associated molecular patterns (DAMPs), and uric acid crystals. IL-1 induces expression of many proinflammatory genes. Activated human CGD monocytes display increased production of IL-1α and IL-1β,45-47 with similar findings in mouse macrophages and bone marrow–derived dendritic cells.70,75 The underlying mechanism is not understood. One proposal is that enhanced IL-1 production and/or release in CGD reflects reduced activity of autophagy pathways,45 but this is unresolved.
Accumulation of microtubule-associated protein light chain 3 (LC3) on phagosomes, a form of noncanonical autophagy referred to as LC3-associated phagocytosis (LAP), is stimulated by NADPH oxidase ROS.76-78 Triggers of LAP in myeloid cells include microbial cell wall components, immunoglobulin-opsonized particles, and apoptotic cells. The autophagy proteins involved in LAP can accelerate or delay phagosome maturation and impact digestion, depending on the cell type and cargo.70,79 For example, binding and ingestion of apoptotic neutrophils (efferocytosis) activated the respiratory burst in inflammatory murine macrophages, which promoted acquisition of LC3 and rapid acquisition of vacuolar-type H+ ATPases mediating acidification.70 In turn, acidification facilitated efficient proteolysis of efferosome contents. These were impaired in CGD inflammatory macrophages. Defective macrophage LAP following efferocytosis was also associated with increased proinflammatory cytokine production by bone marrow–derived macrophages80 but not by inflammatory macrophages.70
Although LAP is also impaired in CGD neutrophils,76 whether this impacts events within neutrophil phagosomes is uncertain. Proteases and other compounds are delivered to neutrophil phagosomes by fusion with neutrophil granules, rather than the progressive remodeling by sequential fusion of different endocytic compartments as occurs in macrophages. Regulation of luminal pH also differs in neutrophils compared with macrophages.13 Neutrophil phagosomes initially maintain a neutral or even slightly alkaline pH because of their higher levels of NADPH oxidase activity, which consume luminal protons as superoxide dismutates to H2O2; ROS also inhibit V-ATPase accumulation on neutrophil phagosomes.81 Thus, in the absence of the NADPH oxidase, CGD neutrophil phagosomes acidify very rapidly,82,83 although the consequences are not well understood.
The NADPH oxidase can influence antigen presentation, a process that requires controlled degradation to preserve peptides for T-cell recognition. Proteolysis of antigens delivered on small particles was accelerated in mouse and human CGD dendritic cells and macrophages to reduce MHC-1 cross-presentation, which involves enhanced acidification and/or a more reductive environment that maintains cysteine protease activity.84-88 The impact of NADPH oxidase ROS also depends on the cargo. Delayed vacuolar-type H+ ATPase-mediated acidification of CGD macrophage efferosomes slowed digestion and increased cross-presentation of associated antigens.70 The NADPH oxidase also modulates class II processing and antigen repertoire in mouse macrophages and human Epstein-Barr virus B cells.89,90
Neutrophilic inflammation is prominent in CGD. In part, this reflects increased production of proinflammatory mediators by oxidase-deficient myeloid cells, which increases neutrophil recruitment. In addition, spontaneous apoptosis is delayed in oxidase-deficient neutrophils.91,92 Moreover, apoptotic CGD neutrophils generate reduced levels of cell surface lyso-phosphotidylserine,93,94 which otherwise enhances macrophage efferocytosis94 to remove dying cells prior to necrosis and release of DAMPs. An intrinsic impairment in CGD mouse macrophage efferocytosis was also reported in some,95 but not other,70 investigations.
Transcriptional changes driven by hypoxia-inducible factor (HIF) play important roles in regulating the inflammatory response.96 HIF expression triggered by tissue hypoxia at inflamed sites can be impacted by NADPH oxidase–deficient neutrophils as a result of their reduced consumption of oxygen in the absence of a respiratory burst. In an induced colitis model, an excessive infiltration of neutrophil inflammation in CGD mice could be abrogated by pharmacological stabilization of HIF within intestinal epithelial cells.97
The generation of neutrophil extracellular traps (NETs) is a form of neutrophil cell death associated with the release of DNA coated with various proteins into the extracellular environment.98 NETosis may contribute to host defense by trapping microbes, but it also can have deleterious proinflammatory effects. Whether alterations in NETosis contribute to dysregulated inflammation in CGD is unknown. NETosis can be regulated by the NADPH oxidase, particularly in response to microbial stimuli. However, human and mouse CGD neutrophils are capable of generating NETs in response to immune complexes and even spontaneously.64,99 Release of NETS in these settings may be mediated by increased neutrophil mitochondrial ROS, which were detected in lupus patients and some CGD patients.99 This is intriguing, because NETs are postulated to induce autoantibody formation, as well as activate production of type 1 interferon by dendritic cells.100 Hence, there could be a link between aberrant increased NETosis and discoid lupus, systemic lupus, and other autoimmune disease in NADPH oxidase deficiency.
Deficient NADPH oxidase ROS can affect T-cell–dependent inflammation and autoimmunity, although the mechanisms are incompletely understood. Many studies point to a role for NADPH oxidase–derived ROS in limiting T-cell activation in induced arthritis models. This may involve altered oxidation of the T-cell surface, antigen processing, or oxidation of presented autoantigens.101-103 Interestingly, mice with an NCF4 phosphatidylinositol 3-phosphate–binding mutation show enhanced susceptibility to collagen-induced arthritis but not to mannose-induced psoriatic arthritis, whereas NCF1-deficient mice are susceptible to both.104 This suggests that the effects of NADPH oxidase ROS can depend on the level and/or compartment in which they are generated. Other studies in mice support a role for macrophage and dendritic cell NADPH oxidase ROS to induce regulatory T-cell lymphocytes102,105 and suppress the production of interferon-γ, IL-12, and IL-17 by T cells.75,106 NADPH oxidase ROS are also reported to be important for myeloid-derived suppressor cell suppression of T-cell responses in tumor models.107
Given the many processes affected by NADPH oxidase, the etiology of the dysregulated inflammation and autoimmunity associated with NADPH oxidase deficiency is complex, influenced by the inciting agent, and often involves cross talk between different leukocytes. Altered responses by CGD monocytes and macrophages, including increased proinflammatory mediator production, likely play a central role. Restoring oxidase activity to macrophages and monocytes in NCF1-null mice using a CD68 transgene protected against fungal cell wall–induced inflammation and against increased susceptibility to induced arthritis, although the specific pathways responsible are not known.102,108,109 In peritoneal inflammation induced by endogenous DAMPs, increased IL-1α released by CGD sentinel peritoneal macrophages enhanced G-CSF expression, which rapidly mobilized greater numbers of marrow neutrophils for recruitment to the inflamed peritoneal cavity.70 This led to increased acute and prolonged peritoneal inflammation, which could be ameliorated by blocking IL-1α or G-CSF. Interestingly, prolonged peritoneal inflammation in CGD was also associated with impaired activation of invariant natural killer T lymphocytes by efferocytic macrophages, leading to reduced invariant natural killer T production of the proresolution cytokine IL-4.110 NADPH oxidase–deficient neutrophils themselves may also promote excessive inflammation by their increased production of inflammatory mediators, a prolonged lifespan at inflamed sites, and delayed clearance, as discussed earlier.
Management of infections and inflammatory complications in CGD
Management of patients with CGD includes the use of antimicrobials, as well as drugs targeting immune responses.4,5 Prophylactic trimethoprim/sulfamethoxazole (or, in sulfa-allergic patients, dicloxacillin or trimethoprim) and itraconazole, coupled with interferon-γ and aggressive treatment of acute infections and prolonged courses of antimicrobial treatment, have markedly reduced the frequency and severity of infections in CGD.4,111-114 X-linked CGD carriers with a low frequency of NADPH oxidase–positive cells may also benefit from antibiotic prophylaxis.37 Corticosteroids are used to treat clinically significant granulomatous or other inflammatory complications, including CGD-associated GI disease, although with caution because this may increase the risk of fungal infections.4,111 Fulminant pneumonia due to the inhalation of a large number of Aspergillus spores (“mulch pneumonitis”) is also managed with steroids, in addition to antifungal agents, to control the excessive inflammatory response.115 Corticosteroids have also emerged as an important component of treating staphylococcal liver abscesses in CGD, with improved outcomes and oftentimes the avoidance of invasive procedures for abscess drainage.116 TNF-α inhibitors can improve symptoms in CGD IBD but should be avoided because of their predisposing risk of infections.111 Treatment with anakinra, an IL-1 receptor antagonist, relieved symptoms in several CGD patients with colitis,45 but responses were poor or were not sustained in other patients.117 Hydroxychloroquine has been used to treat discoid lupus skin lesions.41 Macrophage activation syndrome and HLH in CGD have been treated with steroids and IV immunoglobulin.38 Because of its immunomodulatory and anti–TNF-α effects, thalidomide has been tried in a few patients with CGD-associated inflammation.118 Intriguingly, pioglitazone, a peroxisome proliferator-activated receptor γ agonist that is approved for type 2 diabetes increased mitochondrial oxidant production and bactericidal function in activated human and mouse CGD phagocytes.119 The increased generation of ROS by an alternative source could potentially benefit both defects in host defense and inflammation in CGD.
Allogeneic hematopoietic stem cell transplantation is curative for CGD, and reduced-intensity conditioning for allogeneic transplantation has emerged as an effective approach.120,121 Gene therapy targeted at autologous hematopoietic stem cells is also under active development.122,123 Currently, selection of patients for these approaches is based on the severity and frequency of infectious or inflammatory complications. Of note, refractory GI disease has been effectively treated with hematopoietic stem cell transplantation, including patients with NCF4 defects and a patient with CYBC1 deficiency.19,20
LAD
Adhesion of neutrophils to the endothelium, tissue matrix, and microbes is essential for their ability to emigrate into sites of infection and eliminate pathogens. Many of these interactions are mediated by integrin and selectin glycoproteins. LAD is a group of autosomal-recessive immunodeficiencies resulting from defects impacting integrin or selectin functions, leading to severe bacterial infections. The most common and first-described LAD subtype is LAD-1, caused by genetic defects in CD18, the common chain of the β2 integrin family, with several hundred patients reported.4,5,124 Patients present with recurrent severe infections, including deep tissue abscesses caused by S aureus or Gram-negative enteric organisms, and also have neutrophilia, but impaired formation of pus, as a result of adhesion and motility defects.
LAD-1 is associated with severe periodontitis, which had been attributed to impaired neutrophil surveillance of periodontal tissue. However, new studies in LAD-1 patients and mice identified a dysregulated IL-23/IL-17 inflammatory axis as the critical mediator of periodontal destruction, rather than bacterial load.125 This appears to reflect disruption of the “neutrostat,” a homeostatic mechanism for linking neutrophil production to disposal of senescent neutrophils in tissues.126,127 In this feedback loop, ingestion of apoptotic neutrophils by phagocytes suppresses their release of IL-23, which controls downstream production of IL-17 by lymphocytes and, in turn, G-CSF by fibroblasts and other cells.127 Because neutrophil emigration is impaired in LAD-1, these cytokines are all markedly increased in LAD-1, leading to the neutrophilia characteristic of LAD-1,127 and, in periodontal tissue, to pathologic IL-17 elevation and inflammatory periodontal bone loss.125 Moreover, compared with healthy individuals and those with aggressive periodontitis not associated with LAD-1, the subgingival microbiome of LAD-1 patients is altered, harboring microbial products that trigger IL-23–related inflammation.128 Taking advantage of these observations, an LAD-1 patient with severe periodontitis and a chronic sacral wound was recently treated with an antibody that blocks the activity of IL-23 and IL-12 and the downstream production of IL-17, which led to resolution of both complications.129
Concluding remarks
Inherited disorders affecting neutrophil function have illuminated cellular pathways that are important for effective control of bacterial and fungal pathogens, as well as for homeostasis, resolution of inflammation, and suppression of autoimmunity. The careful study of affected patients and animal models is leading to new insights into the underlying mechanisms of immune dysregulation and could improve therapeutic approaches for their management.
Acknowledgments
The author thanks Tina McGrath for assistance with manuscript preparation and apologizes to colleagues whose primary work was not cited because of space limitations.
This work was supported by grants from the National Institutes of Health, National Heart, Lung, and Blood Institute (R01HL045635) and the National Institutes of Health, National Institute of Arthritis and Musculoskeletal and Skin Diseases (R01AR072212) and by the Children’s Discovery Institute of Washington University and St. Louis Children’s Hospital.
Authorship
Contribution: M.C.D. reviewed the literature and wrote the manuscript.
Conflict-of-interest disclosure: The author declares no competing financial interests.
Correspondence: Mary C. Dinauer, Department of Pediatrics, Washington University School of Medicine in St. Louis, 660 S. Euclid Ave, Campus Box 8208, St. Louis, MO 63110; e-mail: mdinauer@wustl.edu.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal