

Key Points
NLPHL has an excellent prognosis, irrespective of treatment.
Within the limitations of a retrospective analysis, active surveillance is a viable initial management strategy for selected NLPHL patients.

Abstract
Nodular lymphocyte-predominant Hodgkin lymphoma (NLPHL) is a rare subtype of lymphoma that, like other Hodgkin lymphomas, has historically been treated aggressively. However, in most cases, NLPHL has an indolent course, which raises the question of to what extent these patients require aggressive upfront treatment. We describe the management and outcomes of consecutive NLPHL patients diagnosed at Memorial Sloan Kettering Cancer Center (MSK), with a focus on evaluating active surveillance. All patients aged 16 years or older diagnosed and followed at MSK between 1974 and 2016 were included. Treatment outcomes were compared between management with active surveillance and other strategies. We identified 163 consecutive patients who were treated with radiotherapy alone (46%), active surveillance (23%), chemotherapy (16%), combined modality (12%), or rituximab monotherapy (4%). Median follow-up was 69 months. Five-year progression-free survival (PFS), second PFS (PFS2), and overall survival (OS) estimates were 85% (95% confidence interval [CI], 78-90), 97% (95% CI, 92-99), and 99% (95% CI, 95-100), respectively. Only 1 of 7 deaths was lymphoma related. Patients managed with active surveillance had slightly shorter PFS than those receiving any active treatment, with 5-year PFS of 77% (95% CI, 56-89) vs 87% (95% CI, 79-92; P = .017). This difference did not translate into better PFS2 or OS. Only 10 patients managed with active surveillance (27%) eventually required treatment, after a median of 61 months, and none died. NLPHL has an excellent prognosis. Within the limitations of a retrospective analysis, active surveillance is a viable initial management strategy for selected NLPHL patients.
Introduction
Nodular lymphocyte-predominant Hodgkin lymphoma (NLPHL) is a rare subtype of lymphoma.1 Historically, NLPHL has been considered a subtype of Hodgkin lymphoma (HL) and treated similarly.2,3 However, in most cases, NLPHL will follow a more indolent course than classical HL, with better tumor control and overall survival (OS).3 Histologic and genomic studies have revealed that NLPHL has similarities with classical HL and other more aggressive B-cell lymphomas, particularly T-cell–rich B-cell lymphoma (TCRBCL).4-6 This is in contrast to its indolent clinical course and raises the question of whether, considering its clinical presentation, NLPHL is better regarded as an indolent B-cell lymphoma and managed less aggressively.
Because NLPHL is rare, available evidence regarding its treatment originates from single-arm studies, subgroup analysis of HL trials, and retrospective series. Early-stage patients (Ann Arbor I/II) are usually treated with radiotherapy (RT) only or with a combined modality approach, including RT and chemotherapy (CT).7-11 Advanced-stage patients (Ann Arbor III/IV), who usually cannot be managed with limited RT fields, are often treated with rituximab alone or in combination with CT. Common regimens shown to be highly effective include doxorubicin, bleomycin, vinblastine, and dacarbazine (ABVD), cyclophosphamide, doxorubicin, vincristine, and presnisone (CHOP), and escalated bleomycin, etoposide, doxorubicin, cyclophosphamide, vincristine, procarbazine, and prednisone (BEACOPP), which can all be combined with a CD20 antibody.12,13 The CD20 antibodies rituximab and ofatumumab, which have shown efficacy in relapsed patients, can also be reasonable monotherapy options for newly diagnosed patients who do not want or cannot tolerate more aggressive regimens.14,15 Despite its generally indolent course, NLPHL transformation into an aggressive lymphoma poses a major challenge for treatment.16 Considering the indolent course of the disease, several practitioners advocate an approach similar to that used in indolent B-cell lymphomas: treatment is deferred until considerable burden of disease or symptoms develop. This could spare some patients from treatment-related morbidity without necessarily changing their long-term outcome. Proponents of more aggressive frontline treatment argue that early disease eradication could prevent transformations. Because NLPHL is not clearly associated with decreased survival, rarely causes symptoms, and, like low-grade B-cell non-HL (NHL), is prone to late relapse after initial treatment, initial active surveillance without immediate treatment is an attractive alternative for asymptomatic patients with low tumor burden. Some evidence, especially in the setting of complete surgical excision of localized disease, exists to support this approach,17-20 but data are limited.
At MSK it has been institutional practice to observe certain patients with NLPHL expectantly, as is common with indolent lymphomas. This study evaluated the outcomes of active surveillance compared with upfront treatment at diagnosis.
Patients and methods
Patients and procedures
We included all patients aged 16 years or older with newly diagnosed NLPHL, confirmed by the MSK pathology department, and ≥1 follow-up visit. Diagnosis of NLPHL was based on morphological criteria and the immunophenotype. Common differential diagnoses that frequently resulted in misdiagnosis in the past were considered, such as progressive transformation of germinal centers, T-cell/histiocyte-rich B-cell lymphoma, and lymphocyte-rich classical HL.21 Any ambiguous cases were excluded. Patients presenting at initial diagnosis with transformation, composite lymphoma, prior lymphoma of any type, or prior cancer within 5 years of diagnosis (with the exception of nonmelanoma skin cancer) were excluded.
Data extracted from the electronic medical record were used to calculate the International Prognostic Score (IPS) and Revised International Prognostic Index (R-IPI).22,23 Bulky disease defined as a mass with its long axis ≥10 cm was exceedingly rare in our cohort, present in only 2 patients. Therefore, we defined bulky disease as a mass with its long axis ≥5 cm.
Outcomes
Progression-free survival (PFS) was defined as the time from diagnosis to biopsy-proven disease progression, initiation of further treatment, or death from any cause. Second PFS (PFS2) was defined as the time from disease progression or relapse to second biopsy-proven disease progression, second initiation of further treatment, or death from any cause. Active surveillance was regarded as a line of therapy with its start date at diagnosis. For example, progression after initial management with active surveillance and progression after frontline active treatment (eg, CT) were considered PFS events. Consequently, at the time of a PFS2 event, a patient in the active surveillance group had 1 prior line of active treatment, whereas a patient on any other treatment had 2 prior lines.
Patients were followed as required by the treating physician. Participants who did not have disease progression or had not died were censored at the last known time that the patient was progression-free. We evaluated the rates of transformation, secondary malignancies, and death, which was further classified as associated with lymphoma or not. Patients were censored at the last documented MSK visit.
Statistical analysis
All statistical calculations were performed with R version 3.3.2 (The R Foundation, Vienna, Austria).24 Patient characteristics for the groups on active surveillance and any other treatment were compared using the Fisher’s exact test for categorical variables and the Mann-Whitney U test for continuous variables. Patients with missing variables were excluded from all analyses that were dependent on the respective variable. Median follow-up was calculated using the reverse Kaplan-Meier approach. Survival analysis was done using Kaplan-Meier curves, comparing survival between groups using the log-rank test. Further, univariable survival analyses were performed using the Cox proportional hazard regression, with subsequent multivariable Cox models, including variables with P < .1 in the univariable analyses, treatment type, and treatment interaction terms. The proportional hazard assumption was tested and met for all models by graphical diagnostics and by the absence of a significant relationship between residuals and time. The rates of transformation and secondary cancers per 100 patient-years were calculated by dividing the number of events by the sum of patient-years under observation and then multiplying by 100. Statistical significance was set at α ≤ 0.05, 2-sided.
This study was approved by the MSK Institutional Review Board (16-831).
Results
Patient characteristics
The study included 163 patients diagnosed between 1974 and 2016. Immunohistochemistry was performed in the vast majority of cases (n = 155; 95%). Of the cases with immunophenotype available, 99% stained fully positive for CD20, and 100% and 99% stained fully negative or only weakly/partially positive for CD15 and CD30, respectively. The immunophenotype is summarized in supplemental Table 1, available on the Blood Web site. Median age at presentation was 40 years (range, 16-75 years), with male predominance (n = 103; 63%). Most patients presented with limited-stage disease (n = 121; 74%), with very few presenting with systemic B symptoms (n = 7; 4%) or bulky disease (n = 21; 13%). Few patients presented with extranodal disease (n = 11; 7%), including only 1 in the active surveillance group. Three patients had suspicious bone uptake in positron emission tomography in multiple locations, 3 patients had involvement of the parotid gland, 2 patients had liver involvement, 1 patient had localized involvement of the L4 vertebra, 1 patient had a subcutaneous lesion in the lower upper extremity, and 1 patient had a chest wall infiltration.
Baseline characteristics were similar for patients managed with active surveillance or any other treatment. Notably, there were increased frequencies of advanced-stage disease (38% vs 22%; P = .09) and higher age (median, 47 vs 39 years; P = .03) in the active surveillance group. All patients with splenic involvement (n = 12; 7%) received active treatment (Table 1).
Patient characteristics
| All (N = 163) | Active surveillance (n = 37) | Any other treatment (n = 126) | P | |
|---|---|---|---|---|
| Male | 103 (63) | 22 (60) | 81 (64) | .70 |
| Age at diagnosis, y | 40 (16-75) | 47 (20-75) | 39 (16-74) | .03 |
| Year of diagnosis | 2009 (1974-2016) | 2012 (1998-2016) | 2007 (1974-2016) | <.001 |
| Stage I/II | 121 (74) | 23 (62) | 98 (78) | .09 |
| Involved Ann Arbor regions | 2 (0-12) | 2 (0-7) | 2 (1-12) | .16 |
| B symptoms | 7 (4) | 0 (0) | 7 (6) | .35 |
| Extranodal disease | 11 (7) | 1 (3) | 10 (8) | .46 |
| Mediastinal involvement | 22 (14) | 5 (14) | 17 (14) | >.99 |
| Splenic involvement | 12 (7) | 0 (0) | 12 (10) | .07 |
| Bulky disease ≥5 cm | 21 (13) | 4 (11) | 17 (14) | .79 |
| Bulky disease ≥10 cm | 2 (1) | 0 (0) | 2 (2) | >.99 |
| Disease involvement | ||||
| Above diaphragm | 82 (50) | 14 (38) | 68 (54) | .12 |
| Below diaphragm | 39 (24) | 9 (24) | 30 (24) | |
| Both sides of diaphragm | 42 (26) | 14 (38) | 28 (22) | |
| Hemoglobin, g/dL (n = 137) | 14.4 (10.6-17.2) | 14.0 (11.6-15.8) | 14.5 (10.6-17.2) | .12 |
| WBC, 1000/μL (n = 138) | 6.4 (2.7-12.6) | 6.7 (2.7-11.4) | 6.4 (3.0-12.6) | .55 |
| LDH, U/L (n = 97) | 181 (101-323) | 175 (140-303) | 182 (101-323) | .81 |
| Albumin, g/dL (n = 126) | 4.5 (3.8-5.3) | 4.5 (3.8-5.0) | 4.6 (3.9-5.3) | .27 |
| Percentage of lymphocytes in differential count (n = 134) | 29 (15-57) | 29 (15-44) | 28 (9-57) | .67 |
| IPS (n = 125) | 1 (0-3) | 1 (0-3) | 1 (0-3) | .85 |
| R-IPI (n = 97) | 0 (0-2) | 0 (0-2) | 1 (0-2) | .45 |
| All (N = 163) | Active surveillance (n = 37) | Any other treatment (n = 126) | P | |
|---|---|---|---|---|
| Male | 103 (63) | 22 (60) | 81 (64) | .70 |
| Age at diagnosis, y | 40 (16-75) | 47 (20-75) | 39 (16-74) | .03 |
| Year of diagnosis | 2009 (1974-2016) | 2012 (1998-2016) | 2007 (1974-2016) | <.001 |
| Stage I/II | 121 (74) | 23 (62) | 98 (78) | .09 |
| Involved Ann Arbor regions | 2 (0-12) | 2 (0-7) | 2 (1-12) | .16 |
| B symptoms | 7 (4) | 0 (0) | 7 (6) | .35 |
| Extranodal disease | 11 (7) | 1 (3) | 10 (8) | .46 |
| Mediastinal involvement | 22 (14) | 5 (14) | 17 (14) | >.99 |
| Splenic involvement | 12 (7) | 0 (0) | 12 (10) | .07 |
| Bulky disease ≥5 cm | 21 (13) | 4 (11) | 17 (14) | .79 |
| Bulky disease ≥10 cm | 2 (1) | 0 (0) | 2 (2) | >.99 |
| Disease involvement | ||||
| Above diaphragm | 82 (50) | 14 (38) | 68 (54) | .12 |
| Below diaphragm | 39 (24) | 9 (24) | 30 (24) | |
| Both sides of diaphragm | 42 (26) | 14 (38) | 28 (22) | |
| Hemoglobin, g/dL (n = 137) | 14.4 (10.6-17.2) | 14.0 (11.6-15.8) | 14.5 (10.6-17.2) | .12 |
| WBC, 1000/μL (n = 138) | 6.4 (2.7-12.6) | 6.7 (2.7-11.4) | 6.4 (3.0-12.6) | .55 |
| LDH, U/L (n = 97) | 181 (101-323) | 175 (140-303) | 182 (101-323) | .81 |
| Albumin, g/dL (n = 126) | 4.5 (3.8-5.3) | 4.5 (3.8-5.0) | 4.6 (3.9-5.3) | .27 |
| Percentage of lymphocytes in differential count (n = 134) | 29 (15-57) | 29 (15-44) | 28 (9-57) | .67 |
| IPS (n = 125) | 1 (0-3) | 1 (0-3) | 1 (0-3) | .85 |
| R-IPI (n = 97) | 0 (0-2) | 0 (0-2) | 1 (0-2) | .45 |
Data are presented as median (range) for continuous variables and as n (%) for count variables. If data were not available for all patients (N = 163), the number of patients with available data is shown in parentheses after the variable.
LDH, lactate dehydrogenase; WBC, white blood cell count.
Treatment
Active surveillance was the initial management strategy in 37 patients (23%). Most patients (n = 75; 46%) were treated with RT only. Patients receiving CT alone or as part of combined modality therapy (n = 45; 28%) were treated with an ABVD-based regimen (n = 20; 44%), a CHOP-based regimen (n = 13; 29%), or other regimens (n = 12; 27%) (Table 2; supplemental Table 2).
Treatment regimens
| Treatment | Early stage | Advanced stage |
|---|---|---|
| Active surveillance | 23 (19) | 14 (33) |
| Involved field RT | 54 (45) | 1 (2) |
| Extended field RT | 20 (16) | 0 (0) |
| Combined modality therapy | 13 (11) | 3 (7) |
| Combined modality therapy + rituximab* | 3 (2) | 0 (0) |
| CT | 4 (3) | 9 (22) |
| CT + rituximab* | 2 (2) | 11 (26) |
| Rituximab monotherapy† | 2 (2) | 4 (10) |
| Treatment | Early stage | Advanced stage |
|---|---|---|
| Active surveillance | 23 (19) | 14 (33) |
| Involved field RT | 54 (45) | 1 (2) |
| Extended field RT | 20 (16) | 0 (0) |
| Combined modality therapy | 13 (11) | 3 (7) |
| Combined modality therapy + rituximab* | 3 (2) | 0 (0) |
| CT | 4 (3) | 9 (22) |
| CT + rituximab* | 2 (2) | 11 (26) |
| Rituximab monotherapy† | 2 (2) | 4 (10) |
Data are n (%).
Rituximab was given once per CT cycle.
Rituximab was given 4 times, once weekly, in all but 1 patient, who received 4 weekly doses of rituximab, followed by 2 years of maintenance treatment every 3 months.
Treatment outcomes
Median follow-up was 69 months (range, 4-512), and total follow-up was 1233 patient-years. We observed 40 PFS, 13 PFS2, and 7 OS events overall, with 10, 1, and 0, respectively, in the active surveillance group. Overall, 37 of the 40 PFS events were progressions or relapses. The remaining 3 patients died without experiencing progression or relapse. Of these 37 patients with progression or relapse, 33 events were biopsy proven, and 4 were recorded because of initiation of further treatment without a new biopsy.
One patient each died of progressive disease after transformation to a TCRBCL, breast cancer, respiratory failure, and myocardial infarction. One patient was hospitalized externally during treatment, deteriorated rapidly, and ultimately died; the cause of death could not be established. Two patients died of an unknown cause without evidence of lymphoma.
Overall, the 5-year PFS, PFS2, and OS estimates were 85% (95% confidence interval [CI], 78-90), 97% (95% CI, 92-99), and 99% (95% CI, 95-100), respectively.
Active surveillance vs other treatment approaches
Median PFS was not reached for any subgroup. Of 37 patients managed with active surveillance, 27% progressed during follow-up (8 with NLPHL and 2 with transformed disease). In these patients, median time to initiation of treatment was 61 months (range, 5-278 months), with 4 treated with local RT, 2 with rituximab monotherapy, 3 with chemoimmunotherapy, and 1 never requiring treatment. Only 1 patient experienced a second relapse event (PFS2). As could be expected, active surveillance was associated with a shorter PFS compared with any other treatment, with 5-year PFS of 77% (95% CI, 56-89) vs 87% (95% CI, 79-92; P = .017) (Figure 1A).
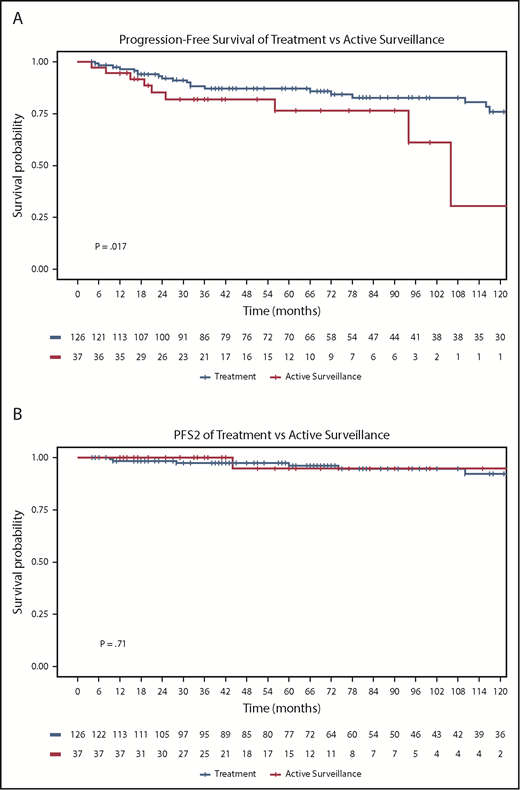
PFS and PFS2 for treatment and active surveillance groups. PFS (A) and PFS2 (B) curves by any other treatment (n = 126) vs active surveillance (n = 37).
PFS and PFS2 for treatment and active surveillance groups. PFS (A) and PFS2 (B) curves by any other treatment (n = 126) vs active surveillance (n = 37).
PFS2 was similar in the 2 groups, with 5-year PFS2 of 95% under active surveillance (95% CI, 68-99) vs 97% with any other treatment (95% CI, 92-99; P = .71) (Figure 1B). No patient in the active surveillance group died, and 5-year OS for the any-other-treatment group was 98% (95% CI, 93-100; P = .38) (Figure 2).
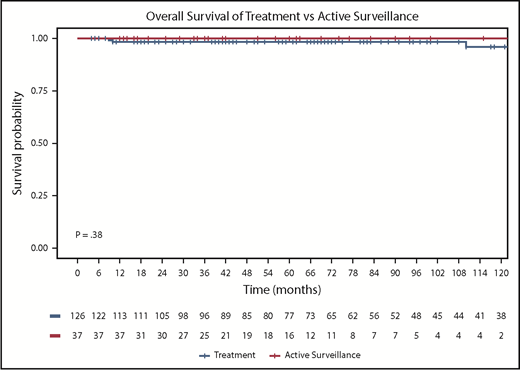
OS for treatment and active surveillance groups. OS curves by any other treatment (n = 126) vs active surveillance (n = 37).
OS for treatment and active surveillance groups. OS curves by any other treatment (n = 126) vs active surveillance (n = 37).
To rule out bias by the inclusion of cases diagnosed decades ago and the very few patients lacking complete immunophenotype, an additional sensitivity analysis including only patients diagnosed after 1999 and with complete immunophenotype (CD20, CD15, CD30; n = 123) was performed. As in the main analysis, active surveillance was associated with a shorter PFS compared with any other treatment, with 5-year PFS of 76% (95% CI, 55-88) vs 89% (95% CI, 81-95; P = .009), respectively. Additionally, PFS2 was again similar in the 2 groups, with 5-year PFS2 of 94% under active surveillance (95% CI, 67-99) vs 97% (95% CI, 89-99) with any other treatment (P = .88). Again, no patient in the active surveillance group died, and 5-year OS for patients receiving any other treatment was 99% (95% CI, 92-100; P = .50).
In early-stage patients specifically, PFS was also inferior with active surveillance, with 5-year PFS of 65% (95% CI, 35-84) vs 94% with any other treatment (95% CI, 87-98; P < .001); however, no PFS2 events or deaths occurred in the active surveillance group (supplemental Figure 1). A subanalysis of advanced-stage patients was not statistically feasible because of the small number of patients and events, but Kaplan-Meier curves are shown.
Table 3 shows the numbers of deaths, transformations, and secondary cancers with active surveillance or any other treatment. A statistical comparison was not performed because of the low event numbers and differences in follow-up time.
Deaths, transformations, and secondary cancers by group
| Deaths | Transformations | Secondary cancers | |
|---|---|---|---|
| Active surveillance (n = 37) | 0 (0.0) | 2 (5.4) | 2 (5.4) |
| Any other treatment (n = 126) | 7 (5.6) | 10 (7.9) | 10 (7.9) |
| Deaths | Transformations | Secondary cancers | |
|---|---|---|---|
| Active surveillance (n = 37) | 0 (0.0) | 2 (5.4) | 2 (5.4) |
| Any other treatment (n = 126) | 7 (5.6) | 10 (7.9) | 10 (7.9) |
Data are n (%).
Of 10 patients under active surveillance who had a PFS event, 1 (10%) had a higher Ann Arbor stage at progression; initially diagnosed with stage I NLPHL, this patient progressed to a stage IV aggressive B-cell lymphoma. Six patients (60%) were diagnosed with early-stage NLPHL and still had a limited-stage disease at progression. Three patients (30%) were diagnosed with advanced-stage NLPHL and had advanced-stage disease at progression.
Transformations
We observed 12 transformations in our cohort, occurring a median of 7.0 years after diagnosis (range, 0.4-15.6 years). The transformation rate was 0.99 per 100 patient-years (Table 3). The diagnosis at transformation was TCRBCL in 8 cases, diffuse large B-cell lymphoma (DLBCL) in 3 cases, and ambiguous between TCRBCL and DLBCL in 1 case.
Secondary cancers
We observed 12 secondary cancers in our cohort, occurring a median of 7.8 years after diagnosis (range, 1.1-24.8). The secondary cancer rate was 1.03 per 100 patient-years, with 4 cases of prostate cancer, 3 cases of nonmelanoma skin cancer, 2 cases of breast cancer, and 1 case each of thyroid cancer, esophageal cancer, and renal cancer (Table 3).
Risk factors for PFS
In univariable analyses, including stage, age, presence of B symptoms, bulky disease, extranodal disease, mediastinal or splenic involvement, IPS,22 and R-IPI,23 the only variables associated (P < .1) with shorter PFS were extranodal disease (hazard ratio [HR], 4.9; 95% CI, 1.8-13.0; P = .002), bulky disease (HR, 3.1; 95% CI, 1.4-6.7; P = .004), splenic involvement (HR, 2.8; 95% CI, 1.0-8.0; P = .06), and advanced-stage disease (HR, 1.9; 95% CI, 1.0-3.7; P = .07).
The final multivariable model, retaining all variables that had a univariable P < .1 and correcting for the type of treatment (RT, CT, rituximab, or absence of active treatment), indicated that bulky disease ≥5 cm (HR, 3.0; 95% CI, 1.3-7.0; P = .01) and extranodal disease (HR, 7.5; 95% CI, 2.1-27.4; P = .002) were risk factors for PFS. Nine of 21 patients with bulky disease ≥5 cm (43%) had a PFS event; notably, 5 of 21 (24%) transformed into a higher-grade lymphoma (2 DLBCL, 3 TCRBCL). Five of 11 patients with extranodal disease (45%) had a PFS event, but none transformed.
PFS by treatment modality
As an exploratory analysis, we compared patients who received RT, CT, or rituximab with those who did not to determine the added benefit of each of these treatment modalities. In the entire cohort, adding RT to patient management was associated with improved PFS (HR, 0.34; 95% CI, 0.17-0.66; P < .001). Separating patients by stage showed that RT was associated with improved PFS in early-stage patients (HR, 0.43; 95% CI, 0.19-0.97; P = .035) (supplemental Figure 2). A separate analysis of advanced-stage patients was not feasible, because only 4 advanced-stage patients received RT. Patients treated with RT had a 5-year PFS of 94% (95% CI, 86-97) vs 73% (95% CI, 60-83) for those not treated with RT. Adding CT was not associated with improved PFS in the entire cohort (HR, 1.01; 95% CI, 0.52-1.97; P = .97) or in early-stage (HR, 0.58; 95% CI, 0.21-1.58; P = .29) or advanced-stage (HR, 1.51; 95% CI, 0.45-5.07; P = .51) patients analyzed separately (supplemental Figure 3). Patients treated with CT had a 5-year PFS of 81% (95% CI, 65-90) vs 87% (95% CI, 79-92) for those not treated with CT. Similarly, adding rituximab to patient management, either alone or in combination with other treatment modalities, such as CT, was not associated with improved PFS in the entire cohort (HR, 1.27; 95% CI, 0.44-3.66; P = .66) or in advanced-stage patients (HR, 0.97; 95% CI, 0.29-3.24; P = .96) analyzed separately (supplemental Figure 4). Patients treated with rituximab had a 5-year PFS of 77% (95% CI, 50-91) vs 86% (95% CI, 78-91) for those not treated with rituximab. Because only 7 early-stage patients received rituximab, these patients were not analyzed separately.
No significant interaction effects were observed. Patients treated with rituximab combined with CT (n = 16) did not have improved PFS compared with those who received CT alone (n = 29) (HR, 1.05; 95% CI, 0.28-4.03; P = .94).
Location of relapse in patients treated with RT
Of 75 patients initially treated with RT, 16 (21%) experienced a relapse. In 12 cases (75%), relapse occurred outside the radiation field, and in 4 cases (25%) relapse occurred within it. All in-field relapses occurred in patients treated with extended-field RT at a dose of 36 to 44 Gy.
Discussion
NLPHL is a rare subtype of lymphoma characterized by an indolent course. There is no consensus regarding the preferred treatment approach, and it has been suggested that certain patients can be managed expectantly. This study aimed to analyze treatment outcomes in a large retrospective cohort of patients with newly diagnosed NLPHL, with a particular emphasis on comparing active surveillance with active treatment. Of 163 patients followed for >5 years, nearly a quarter (n = 37) were managed with active surveillance. Of these, 76% (n = 28) never required treatment, and those who did were mostly managed with local RT or rituximab monotherapy, with a median time to first treatment >5 years. There was no difference in OS between patients treated actively and those managed expectantly. The few deaths that were observed were mostly due to causes other than lymphoma.
As with previous studies, most patients presented with limited-stage disease without bulk and without B symptoms.25-27 Notably, patients with splenic involvement were more likely to be selected for upfront treatment. Patients with advanced-stage disease were more likely to be managed with active surveillance. We speculate that the treating clinicians may have had the impression that splenic involvement could indicate more extensive systemic disease and a higher likelihood of near-term transformation to a more aggressive lymphoma. Indeed, evidence suggests that splenic involvement is a risk factor for transformation.28 This study does not allow analysis of splenic involvement as a risk factor for progression or transformation, because of the more intense treatment that patients with splenic involvement received at the discretion of the treating clinician. Active surveillance was used more often as an upfront management strategy in recent years, possibly because of increasing evidence that NLPHL is rarely the cause of death and usually has an indolent course unless there is transformation.9,11,13,18 This understanding is consistent with the present findings.
The overall outcome was excellent, with only 7 deaths, only 1 due to lymphoma, and ≥1 likely due to treatment-associated complications. Furthermore, consistent with previously published experience, this study showed the tendency of NLPHL to relapse after a long remission, as indicated by the lack of a plateau in PFS.29 An OS similar to that of the general population in the context of a condition defined by very late relapse has been the rationale for advocating deferred therapy in indolent B-cell lymphomas.30,31 In adults with NLPHL, this approach has never been comprehensively studied, but in expectantly managed children with completely resected limited disease, rates of PFS events were similar to ours (ranging from 67%-100%, with varying follow-up). Notably, these pediatric studies reported no deaths,17-20 demonstrating the benign course of NLPHL in an otherwise healthy young population. In keeping with our study, it has been suggested that additional treatment reduces relapse frequency but has no impact on OS.20
This is the first study evaluating PFS2 as an end point in NLPHL. This is an emerging end point that is especially useful for low-grade NHL with common late relapses and a prolonged OS rate.32,33 Notably, comparing PFS in patients initially followed expectantly with those receiving frontline therapy is inherently biased. A more appropriate outcome measure, used in recent studies of indolent lymphomas, is PFS2, which measures the time until subsequent treatment of lymphoma. This measure gauges OS and the long-term requirement for multiple treatments. We demonstrate so few PFS2 events as to suggest no advantage for earlier over deferred treatment. However, the follow-up of this study might be too short to capture late PFS2 events in a disease such as NLPHL, in which these are frequent. Consequently, the total number of PFS2 events in this study is small, and the PFS2 comparisons here should be interpreted with caution.
Most patients received RT, CT, or a combination of both modalities, with or without rituximab. In keeping with the excellent prognosis and indolent course of NLPHL, event numbers were insufficient to conclusively compare specific treatments. Exploratory analyses suggest that addition of RT is associated with improved PFS in early-stage disease. No such association was noted for the incorporation of systemic therapy with rituximab or CT. This is in line with other studies in early-stage NLPHL that found no benefit from CT alone8 or in combination with RT11,34 ; however, at least one study suggested a benefit for the combined-modality approach.9
The rates of transformation and secondary cancers in our cohort were each ∼1% per year per patient, similar to the transformation rate previously reported.25 However, our observation time is likely too short to capture the ultimate incidence of secondary cancers, especially those possibly related to RT, for which multiple decades of follow-up are needed.35 Only 1 patient died due to transformation, although 12 transformations occurred. However, this study does not allow for meaningful conclusions regarding outcome after transformation, because of the small number of transformations and because some of the transformed patients were lost to follow-up shortly after transformation.
Various potential risk factors were analyzed for their ability to predict a PFS event. IPS22 failed to predict PFS. In our final multivariable model, only bulky disease ≥5 cm and extranodal disease were associated with a shorter PFS. Bulky disease was also indicative of a higher likelihood of transformation.
Recently, variant histology has been described to be of prognostic relevance in NLPHL.6 Assessing variant histology might be helpful in identifying patients suitable for active surveillance; however, in this study we did not assess variant histology. Further, collaborative efforts could help to elucidate the outcome of different treatment approaches in variant histology NLPHL.
This study has several limitations that extend beyond its retrospective single-institution design. First, treatment allocation was not random. It is possible that patients managed with active surveillance had less severe disease at presentation; however, this assumption is not supported by the fact that patients managed with active surveillance were nearly 10 years older and had a higher rate of advanced-stage disease. Nevertheless, there may be differences between the active surveillance and other patients in this study that were not captured by our comparison of patient and disease characteristics. Second, patients managed with active surveillance had a shorter follow-up than those receiving any other treatment. However, a median follow-up of 52 months (range, 12-175) for this subgroup, in which >75% of patients did not require treatment, should be sufficient to draw strong conclusions. Third, although this is one of the largest NLPHL cohorts published to date, the numbers of patients and events in this study are still too small to perform adequately powered comparative analyses of relevant subgroups (eg, comparing patients who received CT with or without additional rituximab).
Overall, it can be concluded that NLPHL follows an indolent course, regardless of the treatment approach, with a subset of patients having excellent results with active surveillance. In the absence of randomized trials, it might be advisable to approach NLPHL in selected patients (eg, those without risk factors for progression, such as bulky or extranodal disease) in a similar fashion to indolent NHL, which is treated only upon development of constitutional symptoms, significant burden of disease, or interference with normal organ function. Ultimately, further collaborative efforts, such as meta-analyses using single-patient data, will provide more definitive answers and help to identify subgroups of NLPHL patients who can be safely managed with active surveillance.
Presented in part at the 59th annual meeting of the American Society of Hematology, Atlanta, GA, 11 December 2017.
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Acknowledgments
Editorial support in the preparation of this article was provided by Hannah Rice.
This work was supported in part by National Institutes of Health, National Cancer Institute Cancer Center Support grant P30 CA008748. Additional support was provided by The Lymphoma Foundation, Rob and Karen Schneider and The Louis Schneider and Harry Davis Memorial Trust, The Adam R. Spector Foundation, The David R. and Patricia D. Atkinson Foundation, and Ernest Dicker (D.J.S.).
Authorship
Contribution: D.J.S. conceived and supervised the project, treated patients, analyzed data, and wrote the manuscript; E.J. and S.B. analyzed data and wrote the manuscript; and C.H.M., A.D.Z., A.N., C.S.P., J.F.G., C.L.B., P.D., A.H., P.A.H., A.K., M.J.M., A.J.M., C.N.O., M.L.P., A.Y., P.C.C., and S.M.H. treated patients and approved the manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: David J. Straus, Lymphoma Service, Department of Medicine, Memorial Sloan Kettering Cancer Center, 1275 York Ave, SR‐441B, New York, NY 10065; e-mail: strausd@mskcc.org.
