

Abstract
Neutrophils represent the first line of cellular defense against invading microorganism by rapidly moving across the blood–endothelial cell (EC) barrier and exerting effector cell functions. The neutrophil recruitment cascade to inflamed tissues involves elements of neutrophil rolling, firm adhesion, and crawling onto the EC surface before extravasating by breaching the EC barrier. The interaction between neutrophils and ECs occurs via various adhesive modules and is a critical event determining the mode of neutrophil transmigration, either at the EC junction (paracellular) or directly through the EC body (transcellular). Once thought to be a homogenous entity, new evidence clearly points to the plasticity of neutrophil functions. This review will focus on recent advances in our understanding of the mechanism of the neutrophil transmigration process. It will discuss how neutrophil–EC interactions and the subsequent mode of diapedesis, junctional or nonjunctional, can be context dependent and how this plasticity may be exploited clinically.
Introduction
Neutrophils are the first line of cellular defense against invading microorganisms by crossing the endothelial cell (EC) barrier to reach inflamed tissues.1,2 Neutrophil tissue infiltration is critical for pathogen elimination and tissue repair and must be tightly regulated, as aberrant neutrophil accumulation into tissues causes tissue damage, as seen during acute lung injury, multiple organ dysfunction syndrome, vascular inflammation, and arthritis.3
The recruitment of neutrophils into tissue, also known as the neutrophil extravasation cascade, is a multiple-step process. Floating neutrophils are first captured onto the EC surface in response to inflammatory cytokines and bacteria-derived peptides via upregulation of adhesive molecules on the endothelial luminal surface. These early adhesive interactions are mediated by leukocyte selectins; they are weak and transient, and they promote the “rolling” of neutrophils on ECs. The subsequent stimulation of neutrophils by chemokines that are presented by the endothelial luminal surface triggers the activation of leukocyte integrins, enabling firm adhesion and arrest of neutrophils on the endothelial surface. Once firmly attached, neutrophils flatten and polarize in order to crawl on the endothelial lumen surface and find a permissive site for transmigration across the endothelial barrier. This step is called diapedesis. Finally, the cells must also cross the pericyte layer and breach the venular basal membrane before reaching the inflamed interstitial tissues.1,4,5 Interestingly, neutrophils can cross the EC barrier via 2 distinct routes, either between 2 ECs (paracellular route) or directly through them (transcellular route).6 While neutrophils predominantly migrate at EC junctions, the quantitative contribution of the transcellular route can vary depending on the vascular bed of the tissue as well as the intensity of tissue inflammation.6 Neutrophils can also migrate away from the sites of inflammation, contributing to the resolution7 or dissemination of inflammation,8 depending on the context. This review will summarize old and new key mechanisms of neutrophil transendothelial migration. It will discuss the hypothesis that neutrophil transmigration can be modulated by different factors and that understanding neutrophil transmigration plasticity can be exploited for clinical purposes.
The paradigm of the extravasation cascade: a focus on crawling and diapedesis
The extravasation cascade has been well studied. It is mediated by a series of complex and sequential interactions between leukocytes and the endothelial apical surface via various adhesion receptors. This cascade has been extensively reviewed elsewhere,1,4,9,10 and I will mostly focus on the leukocyte crawling and diapedesis steps (Figure 1).
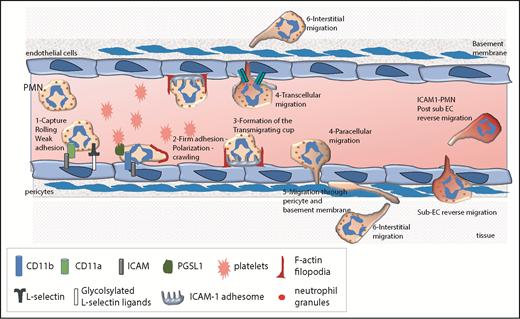
The leukocyte extravasation cascade is controlled by sequential adhesive interactions between leukocytes and ECs. This schema depicts various steps of the extravasation cascade and the adhesive molecules that are involved at each step. The neutrophil extravasation cascade involves a sequence of tethering and rolling along the endothelium, followed by firm adhesion and arrest onto the endothelium. Subsequently, neutrophils undergo lateral migration or crawling on ECs to find a permissive site for transmigration. During this step, the formation of the ICAM-1 adhesome and F-actin–rich filopodia emerging from ECs are critical prior to diapedesis. Diapedesis can occur at EC junctions (paracellular migration) or through the EC body (transcellular migration). Once they have crossed the perivascular basement membrane, neutrophils migrate into the interstitial tissue. They can also return to the blood circulation in a reverse migration process. PMN, polymorphonuclear neutrophil.
The leukocyte extravasation cascade is controlled by sequential adhesive interactions between leukocytes and ECs. This schema depicts various steps of the extravasation cascade and the adhesive molecules that are involved at each step. The neutrophil extravasation cascade involves a sequence of tethering and rolling along the endothelium, followed by firm adhesion and arrest onto the endothelium. Subsequently, neutrophils undergo lateral migration or crawling on ECs to find a permissive site for transmigration. During this step, the formation of the ICAM-1 adhesome and F-actin–rich filopodia emerging from ECs are critical prior to diapedesis. Diapedesis can occur at EC junctions (paracellular migration) or through the EC body (transcellular migration). Once they have crossed the perivascular basement membrane, neutrophils migrate into the interstitial tissue. They can also return to the blood circulation in a reverse migration process. PMN, polymorphonuclear neutrophil.
Leukocyte crawling
Neutrophil firm adhesion to the endothelium is controlled by leukocyte integrins, including lymphocyte function-associated antigen-1 (or αLβ2 integrin or CD11a/CD18), Mac-1 (macrophage-1 antigen [or αMβ2 integrin or CD11b/CD18], and very late antigen-4 (VLA-4 [or α4β1 integrin]), and their endothelial ligands, including intercellular adhesion molecule 1 (ICAM-1) and ICAM-2 and vascular cell adhesion molecule 1, respectively. Once firmly attached, leukocytes flatten and polarize and then crawl onto the endothelial apical surface in search of a permissive site of extravasation.11,12
Leukocyte polarization is necessary for directional migration (Figure 2). During this process, filamentous actin (F-actin) locally polymerizes into lamellipodia on the plasma membrane that faces the chemotactic gradient, the so-called leading edge. Simultaneously, a network of myosin filaments assembles at the opposite end of the cell (ie, the uropod). Myosin filaments associate with bundles of actin and generate cortical tension, which suppresses F-actin protrusions. Cell polarity enables formation of F-actin protrusions at the leading edge only, which is necessary for persistent migration in one direction upon chemokine stimulation.13-15 During infection in vivo, a gradient of chemokines that are sequestered on the endothelial heparin surface allows directional intraluminal crawling.16 Recently, megakaryoblastic leukemia 1 (MKL1) emerged as one important regulator of cytoskeleton in neutrophils.17 MKL1 is a coactivator of the serum response factor, which regulates transcription of actin and actin cytoskeleton–related genes. MKL1 deficiency reduced F-actin content in myeloid cells and caused widespread cytoskeletal dysfunction. These cells had reduced expression of the myosin light chain 9 component of the myosin II complex and overexpression of CD11b integrin. MKL1-deficient neutrophils displayed almost complete abrogation of migration in vitro due to an inability of the cells to properly retract the uropod. Interestingly, the few cells that did migrate exhibited enhanced directional persistence.17 Another important regulator of F-actin dynamics is the nonmuscle myosin class II protein Myh9.18 Using stimulated emission depletion nanoscopy, the authors show that Myh9 is localized in lamellipodia and the uropod. Genetic loss of Myh9 reduced the amount of F-actin polymerization at the front but increased F-actin at the uropod in migrating neutrophils. As a result, Myh9-deficient neutrophils had severely reduced velocity and directional persistence in vitro under both flow conditions and 3-dimensional conditions in the collagen network. It also reduced extravasation into tissue in vivo.18
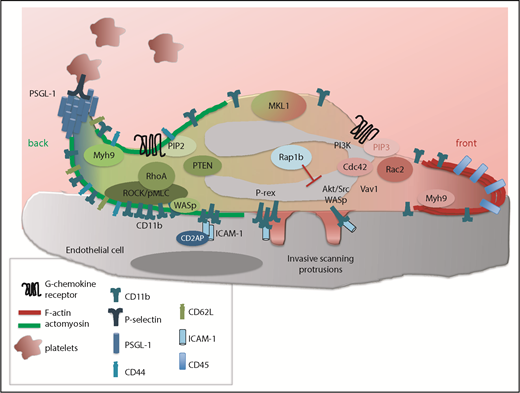
Mechanism of leukocyte polarity and crawling. Upon firm adhesion, neutrophils flatten and adopt a polarized shape with a cell front enriched in polymerized F-actin protrusions (red) and a rear end (or back) enriched in actomyosin contractile filaments (green). This asymmetric shape is controlled by 2 major signaling networks. One is activated at the cell front and involves high levels of PI3K-PIP3 and subsequent Rac1/2-mediated actin polymerization formation. Another is activated at the cell rear and involves phosphatase and tensin homolog (PTEN)–mediated PIP2 and RhoA-driven actomyosin contraction. In addition, Myh9 and MKL1 activity are necessary for proper regulation of F-actin and actomyosin reorganization. Cdc42 signaling, while localized at the cell front, controls the uropod via WASp, which in turn enhances CD11b clustering and RhoA signaling. Lastly, invasive protrusions, which are promoted by Src and Akt signaling but limited by Rap1b, develop at the ventral and lateral part of the leukocyte to scan for a permissive site of transmigration. During polarization, PSGL-1 accumulates at the tip of the uropod and scans for activated platelets in order to maintain neutrophil polarity and crawling properties. In ECs, CD2AP limits the ICAM-1 adhesome. pMLC, phosphorylation of the myosin light chain; ROCK, Rho-associated protein kinase.
Mechanism of leukocyte polarity and crawling. Upon firm adhesion, neutrophils flatten and adopt a polarized shape with a cell front enriched in polymerized F-actin protrusions (red) and a rear end (or back) enriched in actomyosin contractile filaments (green). This asymmetric shape is controlled by 2 major signaling networks. One is activated at the cell front and involves high levels of PI3K-PIP3 and subsequent Rac1/2-mediated actin polymerization formation. Another is activated at the cell rear and involves phosphatase and tensin homolog (PTEN)–mediated PIP2 and RhoA-driven actomyosin contraction. In addition, Myh9 and MKL1 activity are necessary for proper regulation of F-actin and actomyosin reorganization. Cdc42 signaling, while localized at the cell front, controls the uropod via WASp, which in turn enhances CD11b clustering and RhoA signaling. Lastly, invasive protrusions, which are promoted by Src and Akt signaling but limited by Rap1b, develop at the ventral and lateral part of the leukocyte to scan for a permissive site of transmigration. During polarization, PSGL-1 accumulates at the tip of the uropod and scans for activated platelets in order to maintain neutrophil polarity and crawling properties. In ECs, CD2AP limits the ICAM-1 adhesome. pMLC, phosphorylation of the myosin light chain; ROCK, Rho-associated protein kinase.
The reorganization of the cell plasma membrane into 2 separate domains is instrumental for leukocyte polarization (Figure 2). The leading edge is made of lipids that are susceptible to extraction by cold Triton X-100 (detergent-sensitive domains) and contains CD45 and HLA. The uropod is enriched with detergent-resistant lipid domains containing CD44, CD43, l-selectin, heavily glycosylated proteins (eg, PSGL-1), and integrins.19-22 In addition, Mac-1 in neutrophils or lymphocyte function–associated antigen-1 in lymphocytes accumulates along the cell sides and at the uropod during active migration in vitro.22-27 This spatial reorganization is functionally important for leukocyte crawling. For instance, depleting plasma membrane cholesterol levels reversibly inhibits neutrophil polarization and migration.19,20 Blocking Mac-1 or CD18 with monoclonal antibodies significantly blocks monocyte locomotion onto human umbilical vein endothelial cells in vitro12 or neutrophil polarity and persistent migration onto fibrinogen in vitro.27
Members of the small Rho GTPase family, class I phosphoinositide 3 kinase (PI3Kγ and PI3Kδ) and its second messenger, phosphoinositol(3,4,5)tri-phosphate (PIP3), and lipid phosphatases (phosphatase and tensin homolog and signaling inositol polyphosphate phosphatase) are the main regulators of neutrophil polarity.28,29 The Rho GTPase family includes RhoA, Rac, and Cdc42. They cycle between an inactive, guanosine diphosphate–bound form and an active, guanosine triphosphate (GTP)–bound form via guanine nucleotide exchange factors (GEFs) and GTPase-activating proteins (GAPs). Once in a GTP-bound form, Rho GTPases are able to bind and activate specific downstream effectors to carry out their functions.30 In migrating neutrophils, Rac activity is highly polarized at the leading edge and promotes F-actin assembly into lamellipodia by activating the Wiskott–Aldrich Syndrome protein (WASp)/WASP-family verprolin-homologous protein family of proteins.31 In primary neutrophils, which express 2 related Rac proteins, Rac1 and Rac2, Rac2 is the main regulator of actin assembly.32-35 In contrast, RhoA and its effector, Rho-associated protein kinase, stimulate the formation of myosin filaments at the uropod via phosphorylation of the myosin light chain.36,37 Rac activity depends on high levels PI3K activity and PIP3 accumulation,38,39 whereas accumulation of PIP2 at the uropod is important for RhoA activity.40,41 In this model, Cdc42 plays unique and intriguing roles. Cdc42, which localizes at the cell front upon activation, activates Rac GEFs for efficient confinement of Rac activity at the leading edge.42 Cdc42 is also necessary to control the uropod structure by promoting actomyosin contraction and RhoA signaling at the uropod,43,44 notably via activation of its effector, WASp.24 As a consequence, Cdc42-deficient neutrophils exhibit increased F-actin protrusions at the uropod and cannot persistently migrate in one direction in vitro, and this is associated with reduced tissue infiltration in vivo.24,27 Interestingly, Cdc42/WASp stabilizes the uropod by promoting myosin light chain activity via CD11b.24,27
These studies are now being validated and further investigated using new genetic mouse models and advances in intravital imaging. In vivo imaging shows that within seconds after arresting, neutrophils form a lamellipodia-rich domain at the leading edge, whereas CD62L and the glycoprotein ligand for P-selectin, PSGL-1, are enriched in the uropod.45 Although CD11b was found to be distributed throughout neutrophils in this study, CD11b is required for neutrophil intravascular crawling.11 In vivo imaging also confirms the preponderant role of Cdc42 in neutrophil polarity. Cdc42-deficient neutrophils failed to form a durable uropod structure or PSGL-1 clusters and constantly extended protrusions emanating from the cell sides and cell uropod.45 Interestingly, PSGL-1 is functionally important for neutrophil polarity and crawling. The authors show that PSGL-1–deficient neutrophils exhibited an abnormal pattern of CD11b distribution and reduced crawling kinetics in vivo.45 In addition, neutrophils that expressed a mutant form of PSGL-1 that can interact with P-selectin and cluster at the uropod but cannot propagate intracellular signaling showed intravascular migration defects. Interestingly, PSGL-1 can interact with Dock2, a Rac GEF. The Rac GEF Vav1 is required for neutrophil intraluminal crawling in vivo.46 Other studies showed that a Rac-specific GEF, P-Rex1, is required for CD11b-dependent intravascular crawling.47 Hence, in vivo imaging studies confirm and greatly extend our knowledge of the regulation of neutrophil polarization and intraluminal crawling.
The Hidalgo group also revealed an unexpected role of platelets for neutrophil crawling.45 Using intravital imaging, they show that during active crawling, the neutrophil uropod actively protrudes into the vessel lumen such that the luminal space on inflamed vessels is populated of PSGL-1–bearing clusters that undergo continuous collisions with a subset of active platelets through P-selectin. The authors conclude that PSGL-1–bearing microdomains scan for activated platelets present in the bloodstream.45 These dynamic interactions depend on Cdc42 activity and are necessary for efficient neutrophil transmigration in vivo.45 Importantly, during pathogenic inflammation, the uropod becomes the predominant domain for platelet interactions, such as during transfusion-related acute lung injury, suggesting that neutrophil polarization and interactions with accessory cells participate in modulating inflammation.45 This is very intriguing and merits further investigation.
Sensing the vascular bed during neutrophil crawling
During crawling, leukocytes extend highly dynamic membrane protrusions, constantly protruding and retracting onto the EC surface prior to emigration. This phenomenon was first observed in neutrophils by Cinamon et al,23 but it also occurs during lymphocyte crawling.25,48,49 Live-cell imaging combined with immunofluorescence demonstrated that crawling leukocytes generate numerous finger-like protrusions that extend underneath the cells and at the cell periphery. In lymphocytes, these projections are mostly concentrated at the uropod25,48,49 and are stimulated under shear-stress conditions.25 They create deep invaginations onto the EC body and through endothelial junctions. Because of these properties, they were named “invasive protrusions.”25,48,49 Carman et al reported that these structures are rich in actin and surrounded by rings of integrins, closely resembling podosomes seen in myeloid cells.48 The same group then showed that invasive protrusions serve as mechanosensors to “probe” the EC surface in order to find permissive sites for transcellular migration.6,48 Using atomic force microscopy–enabled nanoindentation along with electron and fluorescence microscopy, they show in lymphocytes that the protrusions sense the level of resistance of EC junctions and the stiffness of ECs in order to identify areas of weak endothelial actin density where the cells then transmigrate.50
The transmigration cup
Prior to diapedesis, the neutrophils closely interact with ECs via dense clusters of ICAM-1 that form around the cells.49,51,52 Some studies have reported that actin-rich microvilli containing ICAM-1 clusters arise from the endothelial surface and surround the neutrophils prior to extravasation.51,52 This was recently confirmed in a study by Heemskerk et al in which they used ECs expressing the fluorescent actin reporter Lifeact-GFP.53 They show that during diapedesis, filopodia-like protrusions emerge from the apical site of ECs and wrap around the transmigrating neutrophils, whereas a ring of cortical actin develops at the basolateral site.53 These filopodia-like structures are observed for both the paracellular and transcellular routes. They are important for neutrophil adhesion and crawling, but not for diapedesis per se,53 and depend on Cdc42 activity and RhoA-mediated actomyosin contractility in ECs.53,54 In addition, a new interaction partner of ICAM-1, CD2-protein–associated protein (CD2AP), was recently identified as an endothelial actin-binding protein. Interestingly, CD2AP negatively controls ICAM-1 clustering on the endothelial surface. Loss of CD2AP stimulates the dynamics of ICAM-1 clustering, which facilitates the formation of ICAM-1 complexes on the endothelial surface. Consequently neutrophil adhesion is increased, but crawling is decreased.55
Paracellular diapedesis
Paracellular diapedesis is itself a multistep process, which involves engagement of several adhesion molecules, including ICAM-1/2, vascular cell adhesion molecule 1, junctional adhesion molecule A (JAM-A) and JAM-C, PECAM-1, CD99, and endothelial cell–selective adhesion molecule.5,56 This was demonstrated by studies using genetic mouse models and intravital microscopy, which identified the exact location where leukocyte transmigration is blocked. Following firm adhesion, interactions between neutrophils and ECs occur via ICAM-1 and β2-integrin, which in turn trigger the loosening of the adherent EC junctions by phosphorylating vascular-endothelial cadherin.57 Subsequently, the leukocytes migrate across the EC junction via sequential interactions with JAM-A58,59 and then PECAM.60 The exact role of PECAM during diapedesis has remained contentious. For instance, mouse PECAM-deficient neutrophils were mostly arrested at the level of the perivascular basement membrane in vivo.58,59 However, studies with human neutrophils showed that PECAM neutralizing antibodies arrested the neutrophils between ECs in vitro.61,62 Other important experiments using sequential addition and removal of anti-PECAM and anti-CD99 blocking antibodies or vice versa further suggested that CD99 is required at a later stage of the transmigration process than PECAM.61,62 Blocking CD99 or CD99L2 trapped neutrophils in vivo between ECs and the underlying basement membrane in the inflamed cremaster tissue.63 Recently, the Muller group shed light on these seemingly discrepancies. They used 4-dimensional intravital microscopy to show that the site and order of function of PECAM and CD99 in vivo depends on the strain of mice. In FVB/n mice, PECAM functions upstream of CD99, as in human cells in vitro, and blocking antibodies against either molecule arrest neutrophils before they traverse the endothelium.64 However, in C57BL/6 mice, the same antibodies arrest leukocyte migration through the endothelial basement membrane.64 Interestingly, PECAM-1 interactions stimulate the recruitment of unligated adhesion molecules (eg, PECAM, JAM-A, and CD99) that leukocytes can interact with within the endothelial junction, likely to guide leukocytes moving across the junction. Unligated molecules are recruited to the EC border via certain types of vesicles called the endothelial lateral border recycling compartment (LBRC).65 Finally, during this process, neutrophils generate nuclear lobes that insert into the lamellipodia at the front of the cells, which bend the endothelial actin network and squeeze through the EC barrier.66 Contractile stresses exerted by neutrophils and ECs are also necessary to perturb endothelial junctional tensions in order to open gaps for transmigration and for neutrophils to push themselves across the gap.67 Hence, diapedesis can be mechanically regulated by transmigrating leukocytes and proinflammatory signals that increase EC contractility.
A recent study is shedding important light into the ill-defined role of chemokine receptors. The Nourshargh group just reported that CXCL1, which is produced mainly by tumor necrosis factor–stimulated ECs and pericytes, supports luminal and sub-EC neutrophil crawling. Conversely, neutrophils are the main producers of CXCL2, and this chemokine is critical for correct breaching of EC junctions.68
Transcellular diapedesis
Despite a number of studies providing convincing evidence that neutrophils migrate transcellularly in vivo, the physiological purpose of this process has remained controversial, perhaps because of the difficulty to investigate this phenomenon.69 Studies using transmission electron microscopy of tissue sections clearly showed that in skin tissues, neutrophils take the transcellular route in response to the bacterial chemoattractant formyl-Met-Leu-Phe (fMLP) in vivo.70 The relative contribution of each route of migration depends on the tissue and the vascular bed in vivo, as well as the EC model used for in vitro studies. In venules of the cremaster muscle, only 10% to 15% of neutrophils use the transcellular route in vivo.8,11 In contrast, the frequency of nonjunctional migration increases when neutrophils cross ECs in the skin vasculature.70 Some studies reported that neutrophils or monocytes mostly use transcellular migration in vitro when the EC junctions are too tight, such as across brain ECs,71,72 whereas others showed that neutrophils significantly used junctional migration as well.73
Transcellular migration enables leukocytes to cross the EC barrier away from the EC junctions by forming a transcellular channel between the apical and basal membranes of ECs and leaving the EC junctions intact (Figure 3).48 Like for paracellular migration, transcellular migration is always preceded by intravascular crawling onto the endothelial surface, during which neutrophils or lymphocytes use “scanning/invasive” protrusions25,48,50,73 to identify a permissive site of transmigration. ICAM-1 clusters form as well, and PECAM-1, CD99, and JAM-A are all necessary for transcellular diapedesis.6,49,52,74 Once the site of transcellular migration has been identified, podosomes/protrusions extend into long “invasive-like” protrusions to facilitate the transcellular channel, likely via recruitment of specialized EC vesicles that provide the cellular membrane, cytoskeleton components, and adhesive molecules.48,49 As seen in lymphocytes, transcellular migration involves membrane fusion between leukocytes and ECs, a process that depends on SNARE-containing membrane fusion complexes and lipid raft-rich membranes.49 This latter point and the nature of vesicles recruited to the site of migration perhaps represent unique aspects of the transcellular process, as caveola and vesicular vacuolar organelles were not observed during paracellular migration, and require further investigation.
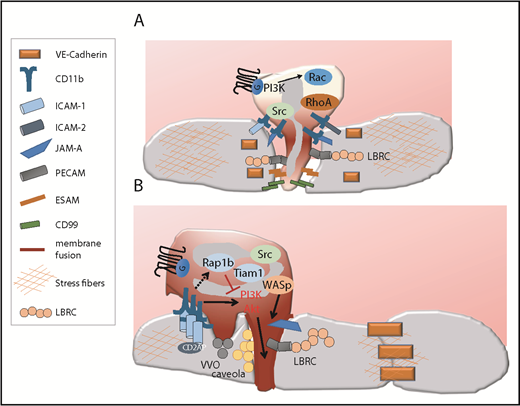
Route of diapedesis. (A) The paracellular route. Paracellular migration is associated with the disruption of the EC junction to form a gap through which the cells migrate, and it involves the sequential engagement of several adhesive receptors, some of which are recruited to the EC plasma membrane via the LBRC. Paracellular migration is favored when the EC content in stress fibers is high, which helps open EC junctions. (B) The transcellular route. During transcellular migration, EC junctions remain intact. Instead, neutrophil-EC contacts fuse (represented in dark red) and remodel into a transcellular channel forming a path for leukocytes. This necessitates the recruitment of an actin-rich membrane, ICAM-enriched caveola, and vesicle vesicular vacuolar organelles (VVO) as well as the recruitment of various adhesive molecules via the LBRC. Several signaling mechanisms important for invasive protrusions and transcellular have been identified. High ICAM density, high integrin signaling, low Rap1b/Tiam1, and subsequent high PI3K/Akt signaling trigger neutrophil invasive protrusions and transcellular migration. CD2AP in ECs destabilizes ICAM clusters and limits transcellular migration. Transcellular migration is favored when the EC content in stress fibers is low but the EC junctions are tight. ESAM, endothelial cell–selective adhesion molecule.
Route of diapedesis. (A) The paracellular route. Paracellular migration is associated with the disruption of the EC junction to form a gap through which the cells migrate, and it involves the sequential engagement of several adhesive receptors, some of which are recruited to the EC plasma membrane via the LBRC. Paracellular migration is favored when the EC content in stress fibers is high, which helps open EC junctions. (B) The transcellular route. During transcellular migration, EC junctions remain intact. Instead, neutrophil-EC contacts fuse (represented in dark red) and remodel into a transcellular channel forming a path for leukocytes. This necessitates the recruitment of an actin-rich membrane, ICAM-enriched caveola, and vesicle vesicular vacuolar organelles (VVO) as well as the recruitment of various adhesive molecules via the LBRC. Several signaling mechanisms important for invasive protrusions and transcellular have been identified. High ICAM density, high integrin signaling, low Rap1b/Tiam1, and subsequent high PI3K/Akt signaling trigger neutrophil invasive protrusions and transcellular migration. CD2AP in ECs destabilizes ICAM clusters and limits transcellular migration. Transcellular migration is favored when the EC content in stress fibers is low but the EC junctions are tight. ESAM, endothelial cell–selective adhesion molecule.
Studies from my laboratory have identified a pathway regulated by the Ras-related small GTPase Rap1b that allows the exploitation of nonjunctional migration. Rap1b deficiency increases neutrophil transmigration across ECs in response to chemokines in vitro and in vivo and promotes a hyperinflammatory reaction.75 When plated onto lipopolysaccharide (LPS)–activated ECs, Rap1b-deficient neutrophils remained away from the EC junctions, contrary to wild-type (WT) neutrophils, yet they formed a transcellular pore and transmigrated through ECs more efficiently than WT cells. Electron microscopy and immunofluorescence images revealed that Rap1b−/− neutrophils extended long protrusions that penetrated deeper into endothelial surfaces than those formed by WT cells. Hence, Rap1b limits neutrophil migration by suppressing the transcellular migration process.75 Interestingly, enhanced Akt activity specifically mediates Rap1b−/− neutrophil transcellular migration but has no effect on paracellular migration. The findings have now been validated in vivo, showing that Rap1b−/− neutrophils use the transcellular migration at higher frequency in response to intradermal inflammatory challenge (Chanchal SurChowdhury and M.-D.F., manuscript submitted March 2019). Also, treatment of neutrophils with a pharmacological activator of Akt activity is sufficient to enhance the frequency of neutrophil transcellular migration in vitro (Chanchal SurChowdhury and M.-D.F., manuscript submitted March 2019). Consistent with our studies, the Rap1b effector Tiam1, which is also a Rac activator, has been involved in transcellular migration.76 Tiam1 deficiency in lymphocytes increased the frequency of transcellular migration across ECs in vitro and increased transcellular pore formation away from endothelial junctions.76 Together, these findings reveal the existence of a neutrophil signaling pathway that specifically controls the transcellular migration process. It will be important to investigate how Rap1b is regulated in vivo under normal or pathologic inflammatory conditions. Equally important will be to investigate the protein composition and regulation of neutrophil invadopodia-like protrusions in order to provide critical insights on how transcellular migration is regulated.
The last steps of the extravasation cascade: crossing pericytes and the perivascular basement membrane
Finally, once the neutrophils have passed the EC barrier, they must cross a layer of pericytes77 lining the blood vessel and then breach the perivascular basement membrane. This is perhaps the least understood step of the transmigration process. Neutrophils tend to migrate at the site of the basement membrane containing low expression of lamin 511 and collagen IV.78-81 Expression of VLA-3 (integrin α3β1, also known as CD49c/CD29)82 and VLA-6 (integrin α6β1, also known as CD49f/CD29),83 2 laminin-binding integrins on neutrophils, as well as neutrophil elastase83 are all required for migration. A recent study showed that serine/threonine protein kinase 4, which encodes mammalian sterile 20–like kinase 1,84 is critical for the translocation of vesicles containing VLA-3, VLA-6, and neutrophil elastase to the neutrophil surface by cooperating with the Rab27 effector protein JFC1. This translocation enables neutrophils to penetrate the basement membrane and extravasate into the interstitial space.84
Factors that influence neutrophil transmigration behavior
Neutrophils have long been thought to be a homogenous entity with “predicted” effector functions. However, it is now recognized that neutrophil functions are plastic and can adapt to specific inflammatory environments. Depending on the tissue and intensity of inflammation, subsets of neutrophils can exhibit distinct life spans, synthesize various amount of cytokines,85 or undergo metabolic changes.86 Further, the neutrophil recruitment process is organ and insult specific.87,88 Neutrophil recruitment to the lung is selectin dependent in response to LPS from Escherichia coli, but not in response to LPS from Streptococcus pneumoniae or Salmonella enteritidis.89,90 Likewise, the dependency on integrins of neutrophil migration to the lungs varies with the inflammatory insult.91-94 Neutrophils also use noncanonical extravasation cascades in the lung, liver, and kidney. I refer the readers to excellent reviews on this topic.87,88 The exact molecular mechanisms of these events are still unclear.
There is also documented evidence suggesting that environmental factors can modulate the route of migration, junctional or not. As suggested earlier, the nature of the vascular bed is a critical factor determining the path of migration. Studies have pointed to the importance of EC stiffness and the tightness of the EC junctions.50,95 It is known that the tightness of EC junctions is higher in brain ECs than in ECs from other organs. Increasing endothelial junction tightness in heart or lung ECs is sufficient to increase the frequency of transcellular migration of T lymphocytes.50 This is consistent with the notion that leukocytes use the transcellular pathway at higher frequency when crossing the tight blood–brain barrier.71,72 EC stiffness also greatly impacts leukocyte diapedesis. In this case, increasing EC stiffness can lead to stabilization of ICAM-1 adhesome and promote intravascular crawling. It also increases EC stress fibers, which in turn can destabilize EC junctions, thus favoring paracellular migration.95 Conversely, soft substrate promotes significantly more transcellular diapedesis by reducing stress fibers in ECs and thus their stiffness. Other components, such as the level of ICAM-1 expression on the EC surface, influence the choice of route of migration.95 High ICAM-1 expression on ECs favors transcellular migration, at least in vitro.96,97 Consistently, loss of CD2AP, which facilitates the formation of ICAM-1 complexes on the endothelial surface, enhances neutrophil adhesion and promotes transcellular migration.55 Hence, increased neutrophil adhesion seems to be positively correlated with transcellular migration. Finally, components in ECs are also important. Caveolin-1 is more enriched around lymphocytes that take the transcellular route than around those migrating at the junction. Knockdown of caveolin-1 in ECs specifically reduced transcellular migration.49 Consistently, another group reported that high levels of caveolin-1 in ECs favored the transcellular path, whereas its downregulation promoted the paracellular route.96
Some of the factors discussed above are important for neutrophil intravascular crawling, raising the interesting question of the relationship between crawling efficiency and the route of transmigration. In some cases, there seems to be a positive association between crawling efficiency and junctional migration. EC stiffness, which promotes crawling efficiency, also favors paracellular migration.95 Conversely, high ICAM-1 expression and adhesion of neutrophils onto ECs tend to limit crawling but favor transcellular migration.96,97 Finally, CD11b-null neutrophils have defective intravascular crawling and largely fail to transmigrate, but the few cells that transmigrate do so transcellularly.11 Other models, such as Tiam1-deficient or Rap1b-deficient neutrophils, also exhibit defective crawling associated with enhanced transcellular migration.75,76 It is thus possible that transcellular migration is a rescue mechanism to enable cells that fail to reach the endothelial junction to cross the blood vessel. However, contrary to CD11b-null neutrophils,11 Rap1b-deficient neutrophils show increased transmigration and adhesion.75 In fact, the level of junctional migration of Rap1b-deficient neutrophils is similar to that of WT, but their ability to use the transcellular pathway is increased, which accounts for their global enhancement of transmigration. In other models, increased adhesion to ECs was also associated with increased transcellular migration.96,97 Hence, transcellular migration could be a compensatory mechanism and a highly regulated process that is favored by heightened adhesion and reduced crawling, as well as other yet-to-be-discovered factors.
In this regard, one such factor could be the level of leukocyte activation. For instance, neutrophil activation with fMLP increases transcellular migration events both in vitro74 and in vivo in a model of intradermal injection of fMLP into ear skin, where EC junctions are not tight.70 Enhanced Akt signaling is sufficient to drive neutrophil transcellular migration.75 Hence, the environment and signaling intensity in neutrophils can influence the decision of the route of migration. It is noteworthy that heightened Akt signaling in patient neutrophils has been tightly correlated with the severity of acute lung injury.98 It will be important to determine whether the relative contribution of transcellular migration of neutrophils varies with the tissue and type of inflammation and whether it represents a pathological process.
The inflammatory milieu can also promote a “reverse migration” behavior. Neutrophils can migrate away from inflammatory foci within tissues.7 In addition, transmigrating neutrophils can undergo abluminal to luminal migration across the endothelium, which is called reverse transendothelial migration (rTEM).8 rTEM is predominantly paracellular and specifically depends on JAM-C. This type of migration is most prevalent during sterile inflammation caused by ischemia-reperfusion injury (I-R), suggesting that rTEM is a pathological process. I-R induced a reduction in expression of JAM-C at EC junctions, which increased the frequency of rTEM events.8 Interestingly, leukotriene B4 released during I-R caused neutrophil-dependent elastase activity to cleave EC JAM-C at sites of neutrophil transmigration and promote rTEM.99 Interestingly, neutrophils that had undergone rTEM exhibit enhanced ICAM-1 expression and can return to the blood circulation and infiltrate distant organs, favoring secondary organ inflammation.8 Hence, the local inflammatory milieu contributes to neutrophil migration plasticity and modulates inflammatory responses.
Perspectives and conclusions: can we exploit neutrophil transmigration plasticity for clinical purposes?
It is now clear that neutrophil transmigration depends on the organ and the inflammatory insult. It is also established that the process of transmigration, during which neutrophils encounter a number of interactions with ECs, causes functional changes in neutrophil effector cell functions. Transmigration is thought to have “priming” effects and increase expression of adhesion molecules, chemokine receptors, and proteases as well as alter neutrophil functions, including increasing their life span and cytotoxicity.85 During acute intestinal inflammation, transmigrating neutrophils cause a local depletion of oxygen, which is sufficient to change the transcriptional profile of tissue epithelial cells and influence resolution of inflammation.100 Neutrophils that have undergone abluminal to luminal rTEM express higher levels of ICAM-1, which seems to drive the dissemination of inflammation.8 The induction of neutrophil ICAM-1 is associated with enhanced phagocytosis and reactive oxygen species generation.101 These findings raise the interesting hypothesis that transcellular migration could induce distinct alterations in neutrophil functions, and this needs to be explored in detail. We need to understand whether transcellular migration occurs during physiological inflammation or pathological conditions and determine if and how it may modulate neutrophil effector functions. Equally important will be to investigate the relationship between the route of neutrophil transmigration and the integrity of the vascular environment.
Understanding neutrophil migration plasticity will offer opportunities for specificity in targeting inflammation. Neutrophils are double-edged swords. Finding a therapeutic window for treating patients with inflammatory diseases without compromising their anti-infection defense mechanisms has been an unreachable challenge. My lab and others have shown that transcellular and paracellular migration of neutrophils can have specific regulatory mechanisms; exploring further these mechanisms may provide a rationale to identify specificity in targeting one route of migration only in order to reduce hyperinflammation while hoping to leave intact some neutrophil effector cell functions.
Hence, diapedesis is a critical step of inflammation that needs further investigation. We need to understand how leukocyte and EC interactions modulate leukocyte effector cell functions and investigate the mechanism of neutrophil migration in a tissue-specific manner in order to design targeted therapy for inflammatory conditions, both normal and pathologic.
Acknowledgments
The work was supported by the National Institutes of Health, National Institute of General Medical Sciences (grant GM112792) (M.-D.F.).
Authorship
Contribution: M.-D.F. wrote and edited the manuscript.
Conflict-of-interest disclosure: The author declares no competing financial interests.
Correspondence: Marie-Dominique Filippi, Division of Experimental Hematology and Cancer Biology, S7.605, Cincinnati Children’s Research Foundation, 3333 Burnet Ave, Cincinnati, OH 45229; e-mail: marie-dominique.filippi@cchmc.org.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal