

Key Points
CDX2 deregulation and UBTF fusion define a B-ALL subtype with distinct immunophenotype, expression profile, and high-risk feature.
Somatic 13q12.2 deletions spanning FLT3 promoter lead to upregulation of CDX2 through a mechanism of enhancer retargeting.

Abstract
Transcriptome sequencing has identified multiple subtypes of B-progenitor acute lymphoblastic leukemia (B-ALL) of prognostic significance, but a minority of cases lack a known genetic driver. Here, we used integrated whole-genome (WGS) and -transcriptome sequencing (RNA-seq), enhancer mapping, and chromatin topology analysis to identify previously unrecognized genomic drivers in B-ALL. Newly diagnosed (n = 3221) and relapsed (n = 177) B-ALL cases with tumor RNA-seq were studied. WGS was performed to detect mutations, structural variants, and copy number alterations. Integrated analysis of histone 3 lysine 27 acetylation and chromatin looping was performed using HiChIP. We identified a subset of 17 newly diagnosed and 5 relapsed B-ALL cases with a distinct gene expression profile and 2 universal and unique genomic alterations resulting from aberrant recombination-activating gene activation: a focal deletion downstream of PAN3 at 13q12.2 resulting in CDX2 deregulation by the PAN3 enhancer and a focal deletion of exons 18-21 of UBTF at 17q21.31 resulting in a chimeric fusion, UBTF::ATXN7L3. A subset of cases also had rearrangement and increased expression of the PAX5 gene, which is otherwise uncommon in B-ALL. Patients were more commonly female and young adult with median age 35 (range,12-70 years). The immunophenotype was characterized by CD10 negativity and immunoglobulin M positivity. Among 16 patients with known clinical response, 9 (56.3%) had high-risk features including relapse (n = 4) or minimal residual disease >1% at the end of remission induction (n = 5). CDX2-deregulated, UBTF::ATXN7L3 rearranged (CDX2/UBTF) B-ALL is a high-risk subtype of leukemia in young adults for which novel therapeutic approaches are required.
Introduction
B-cell acute lymphoblastic leukemia (B-ALL) is the most common childhood tumor and an important cause of morbidity in adults. Despite advances in understanding the biology of B-ALL and new treatment approaches, prognosis is substantially worse in adolescents and adults than in younger children, with cure rates of <40% in patients 40 years old and older.1,2 Conventional genetic analyses including karyotyping, fluorescence in situ hybridization, and targeted molecular analyses detect subtypes defined by aneuploidy, chromosomal rearrangements, and/or known gene fusions,3 but a residual minority of cases lacking a genomic driver remain, commonly termed “B-other.” Sequencing, particularly whole-transcriptome sequencing (RNA-seq), has identified multiple previously unrecognized subtypes,4 including those with rearrangements of DUX4,5-8MEF2D,9NUTM1,10 and ZNF384,11-14 cases with gene expression profiles that phenocopy established subtypes (eg, Ph-like,15ETV6::RUNX1–like,5,16KMT2A-like16), and cases driven by single point mutations PAX5 P80R and IKZF1 N159Y.16,17 These subtypes are associated with varying outcomes in children and adults,18,19 and comprehensive genomic analysis has become an increasingly widely adopted clinical diagnostic approach.20,21 However, prior studies have shown that a subset of cases lack a driver despite careful analysis of RNA-seq data, including cases with distinct gene expression profile, suggesting an unidentified, subtype-defining driver lesion(s). Integrated analysis of RNA-seq with whole-genome sequencing (WGS) has been informative in ALL of T- or ambiguous lineage in identifying genomic alterations and rearrangements leading to enhancer hijacking, for example cases with deregulation of BCL11B and GATA2.22,23 Using a similar integrated approach in B-ALL, we report the genomic basis of a subtype of B-ALL presenting with a distinct immunophenotype and gene expression profile, driven by genetic alterations resulting in deregulation of CDX2 (caudal type homeobox 2) and deletion of the 3′ region of UBTF (upstream binding transcription factor) resulting in an in-frame UBTF::ATXN7L3 fusion.
Methods
Patients and sample collection
We studied patients with B-ALL at diagnosis or relapse and available diagnostic material from the following studies and institutions: St. Jude Children’s Research Hospital (clinicaltrials.gov identifier #NCT00549848),16,19 the Eastern Cooperative Oncology Group (ECOG) and the American College of Radiology Imaging Network (ACRIN) Cancer Research Group (ECOG-ACRIN),18 the Children's Oncology Group,16 the INotuzumab Ozogamicin trial to inVestigAte Tolerability and Efficacy (INO-VATE),24 City of Hope,25 Peter MacCallum Cancer Centre/Royal Melbourne Hospital, South Australian Health and Medical Research Institute, MD Anderson Cancer Center (#NCT00623870 [R05045337; MDM2 inhibitor] and #NCT01134575 [inotuzumab ozogamicin]), and the Munich Leukemia Laboratory (MLL)26 (supplemental Table 1). Patients and/or their guardians provided written informed consent in accordance with the Declaration of Helsinki. The study was approved by the Institutional Review Board of St. Jude Children’s Research Hospital.
Transcriptome sequencing
We used publicly available RNA-seq data from our previous study.16 Additional RNA-seq was performed as previously described.16 Sequencing reads were mapped to the GRCh37 human genome reference by STAR (version 2.4.2a),27 using the suggested 2-pass mapping pipeline. FusionCatcher (version 1.10; https://github.com/ndaniel/fusioncatcher) and STAR-Fusion (version 1.5.0)28 were used to detect fusions. To quantitate gene expression, we used HTSeq (version 0.13.5)29 to calculate read counts for each transcript and the ComBat function in the sva30 R package to correct for batch effects as we previously described.16 The DESeq2 Bioconductor R package31 was used for gene expression normalization and differential expression analysis. For a 2-dimensional t-distributed stochastic neighbor embedding (tSNE) plot and hierarchical clustering, the Rtsne and pheatmap R packages were used with the top 1000 most variable genes based on median absolute deviation and perplexity 30. Gene set enrichment and pathway analyses were performed using Metascape (version 3.5)32 and gene set enrichment analysis (version 4.1.0)33,34 as previously described.16
WGS
WGS data were processed as described in our previous study.22 Sequencing reads were mapped to the GRCh37 human genome reference by Burrows-Wheeler Alignment (version 0.7.12).35 For the detection of mutations, we used the combination of GenomonFisher (version 0.2.1), GenomonMutationFilter (version 0.2.9), and EBFilter (version 0.2.1; https://genomon-project.github.io/GenomonPagesR/). For the detection of structural variants (SVs), 3 SV callers were implemented including GenomonSV (version 0.8.0), Delly (version 0.7.7),36 and Manta (version 1.6.0).37 All variant calls were further manually reviewed for read depth and to remove artifacts using the Integrated Genome Viewer38 as described in our previous studies.16,22 CNVkit (version 0.9.1)39 was used for copy number alteration detection from WGS data. The prediction of cryptic recombination signal sequences (RSS) was screened using RSS database (https://www.itb.cnr.it/rss/analyze.html).40
HiChIP and ChIP-seq
HiChIP41 data were processed and analyzed using MAPS software (version 2.0)42 as described in our previous study.22 Super-enhancers ranked by H3K27ac enrichment were identified using the Ranking of Super Enhancers43 software with default parameters. Chromatin immunoprecipitation (ChIP)-seq data of GM12878 cell line for UBTF were downloaded from the Gene Expression Omnibus using the following accession (GSE91779; Moore et al44) and analyzed as described previously.22
Statistical analysis
Categorical variables were examined with the use of 2-sided Fisher’s exact test and the Holm-Bonferroni method was used for multiple testing. Statistical significance of gene expression level and variant allele frequency (VAF) were assessed using the Wilcoxon rank-sum test. Analyses were performed using Prism version 7.0 (GraphPad Software) and R version 3.6.1 (http://www.r-project.org).
Results
Identification of the high-risk B-ALL subtype with unique characteristics
We analyzed RNA-seq data from 1988 B-ALL cases from our previous study16 to explore new subtypes without a known leukemic driver (B-other). Among 125 cases (6.4%) of B-other, we identified 6 cases, which by transcriptome analysis clustered together with a distinct expression profile compared with other B-ALL cases.16 In contrast, the remaining B-other cases did not have a specific expression pattern and clustered with other known subtypes. We next collected and analyzed additional RNA-seq data from children and adults from independent studies and institutions. In total, we analyzed 3398 cases and identified an additional 16 cases (total 22 cases) that clustered with the distinct expression group, including 6 out of 731 (0.82%) from ECOG-ACRIN, 3 out of 92 (3.3%) from INO-VATE study (relapse cohort; supplemental Figure 1),24 2 out of 862 (0.23%) from the Children's Oncology Group, 4 out of 280 (1.4%) from MLL, and 7 from other cohorts (Table 1; supplemental Table 1). All patients were older than 10 years (median age 35, range 12-70), mostly adolescents and young adults (AYA) (54.5%) and female (77.3%) (supplemental Figure 2; Table 1). Incidence was highest in AYA (1.4%), especially females (2.2%), not common in adults (0.33%), and rare in children (0.17%, P = .011; supplemental Figure 2; supplemental Table 2). There was enrichment (2.8%; 5/177, P = .015) of the subtype in relapse cases compared with diagnosis cases (0.53%; 17/3221) and a high incidence of relapse or minimal residual disease (MRD) >1% at the end of induction therapy (56.3%; 9/16; Table 1).
Clinical characteristics of CDX2/UBTF B-ALL
| Immunophenotype | |||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Sample ID | Sample status | Gender | Age group | Outcome | Relapse | MRD >1% at EOI | Karyotype | CD10 | IgM | CD19 | CD20 | CD22 | CD34 |
| ADU_0788_ 201543 | Diagnosis | Female | Adult | Alive (CR1) | No | NA | G-banding: failure. | Dim | NA | Posi | Posi | Posi | Posi |
| ADI_0252 | Diagnosis | Female | Adult | Dead | Yes | NA | 46,XX,t(1;7)(q25;p12),add (6)(q13),der(11)add(11)(p?13) add(11)(q?22), del(13) (q12q22),?add(18)(q23)[23] | Neg | NA | Posi | Neg | Posi | Posi |
| MLL_13382 | Diagnosis | Female | Adult | Alive (CR1) | No | NA | 46,XX[20] | Neg | NA | Posi | Neg | Neg | Posi |
| MLL_137117 | Diagnosis | Female | Adult | Alive (CR1) | No | NA | 46,XX[20] | Neg | NA | Posi | Neg | Dim | Posi |
| SJALL060150 | Diagnosis | Female | Adult | Alive (Refractory post cycle 2 of Induction) | Yes | NA | NA | Neg | Posi | Posi | Neg | Dim | Posi |
| AYAII_0107 | Diagnosis | Female | AYA | Alive (CR1) | No | NA | NA | Posi | NA | Posi | Neg | Neg | Posi |
| AYAII_0369 | Diagnosis | Female | AYA | Alive (CR1) | No | Positive | Hyperdiploid. 82-86 (near tetraploid) gain of 1q in 7/20 | Neg | NA | Posi | NA | NA | Posi |
| MLL_13360 | Diagnosis | Female | AYA | Dead (CR1; hepatorenal syndrome with acute renal failure, sepsis) | No | NA | 46,XX[15] | Neg | NA | Posi | Neg | Dim | Posi |
| MLL_202248- M228 | Diagnosis | Female | AYA | Alive (CR1) | No | NA | 46,XX[22] | Neg | NA | Posi | Neg | Dim | Posi |
| SJALL054476 | Diagnosis | Male | AYA | Alive | Yes | NA | 46,XY,t(12;13)(p11.2;q14)[30]/ 46,idem,del(6)(q21q25)[7]/ 46,XY[14];abnormal;del(6q) | Neg | Posi | Posi | Neg | Posi | Posi |
| SJALL054518 | Diagnosis | Male | AYA | NA | NA | NA | Inv(9)(p13q13) | Neg | Posi | Posi | Neg | Dim | Posi |
| SJALL066367 | Diagnosis | Male | AYA | Alive (CR1) | Yes | NA | NA | Dim | Neg | Weak | Neg | Posi | Posi |
| SJBALL016287 | Diagnosis | Female | AYA | Alive (CR1) | No | Positive | 46,XX[20] | Neg | Posi | NA | NA | NA | NA |
| SJBALL020165 | Diagnosis | Female | AYA | NA | No | Positive | 46,XX,del(9)(q13q22) [9]/46,XX[11] | Neg | Posi | Posi | Neg | Posi | Posi |
| SJBALL020169 | Diagnosis | Female | AYA | NA | No | Positive | 46,XX,del(9)(q13q22) [6]/46,XX[14] | Neg | Posi | Posi | Neg | NA | Posi |
| SJBALL020889 | Diagnosis | Female | Childhood HR | Alive (CR1) | No | Positive | 46,XX,del(9)(q12q32) [10]/92,idemx2,+ add(1)(p11),-20[8]/46,XX[20] | Neg | Posi | NA | NA | NA | NA |
| SJBALL022051 | Diagnosis | Male | Childhood HR | Alive (CR1) | No | NA | 46,XY,der(6)t(1;6)(q21.1;p25.3), add(14)(q32.3)[13]/46,XY[7] | Dim | Posi | Posi | Dim | Posi | NA |
| SJALL058818_R1 | Relapse | Male | Adult | Dead | Yes | Positive | Complex, near tetraploidy | Neg | Posi | Posi | NA | Posi | Posi |
| SJALL073397_R1 | Relapse | Female | Adult | Refractory after INO | Yes | Positive | NA | Neg | NA | Posi | NA | Posi | Posi |
| SJBALL042249_R1 | Relapse | Female | Adult | Dead (refractory disease) | Yes | Positive | 46,XX,dup(1)(q21q42),der (?2)t(2;13)(p11.2;q14), -3,add(6)(p23),?inv (7)(p13p22),der(?8)t (8;13)(q22;q22), del(9) (q13q22),add(12) (q24.3),add(13)(q14),del (13)(q12),del(?15)(q15q24), add(20)(q13.1),+mar | Dim | NA | Posi | Neg | Neg | Posi |
| SJALL073380_R1 | Relapse | Female | AYA | Alive (CR2) | Yes | NA | NA | Neg | NA | Posi | NA | Posi | Posi |
| SJALL073389_R1 | Relapse | Female | AYA | Alive (CR2) | Yes | NA | NA | Neg | NA | Posi | NA | Dim | Posi |
| Immunophenotype | |||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Sample ID | Sample status | Gender | Age group | Outcome | Relapse | MRD >1% at EOI | Karyotype | CD10 | IgM | CD19 | CD20 | CD22 | CD34 |
| ADU_0788_ 201543 | Diagnosis | Female | Adult | Alive (CR1) | No | NA | G-banding: failure. | Dim | NA | Posi | Posi | Posi | Posi |
| ADI_0252 | Diagnosis | Female | Adult | Dead | Yes | NA | 46,XX,t(1;7)(q25;p12),add (6)(q13),der(11)add(11)(p?13) add(11)(q?22), del(13) (q12q22),?add(18)(q23)[23] | Neg | NA | Posi | Neg | Posi | Posi |
| MLL_13382 | Diagnosis | Female | Adult | Alive (CR1) | No | NA | 46,XX[20] | Neg | NA | Posi | Neg | Neg | Posi |
| MLL_137117 | Diagnosis | Female | Adult | Alive (CR1) | No | NA | 46,XX[20] | Neg | NA | Posi | Neg | Dim | Posi |
| SJALL060150 | Diagnosis | Female | Adult | Alive (Refractory post cycle 2 of Induction) | Yes | NA | NA | Neg | Posi | Posi | Neg | Dim | Posi |
| AYAII_0107 | Diagnosis | Female | AYA | Alive (CR1) | No | NA | NA | Posi | NA | Posi | Neg | Neg | Posi |
| AYAII_0369 | Diagnosis | Female | AYA | Alive (CR1) | No | Positive | Hyperdiploid. 82-86 (near tetraploid) gain of 1q in 7/20 | Neg | NA | Posi | NA | NA | Posi |
| MLL_13360 | Diagnosis | Female | AYA | Dead (CR1; hepatorenal syndrome with acute renal failure, sepsis) | No | NA | 46,XX[15] | Neg | NA | Posi | Neg | Dim | Posi |
| MLL_202248- M228 | Diagnosis | Female | AYA | Alive (CR1) | No | NA | 46,XX[22] | Neg | NA | Posi | Neg | Dim | Posi |
| SJALL054476 | Diagnosis | Male | AYA | Alive | Yes | NA | 46,XY,t(12;13)(p11.2;q14)[30]/ 46,idem,del(6)(q21q25)[7]/ 46,XY[14];abnormal;del(6q) | Neg | Posi | Posi | Neg | Posi | Posi |
| SJALL054518 | Diagnosis | Male | AYA | NA | NA | NA | Inv(9)(p13q13) | Neg | Posi | Posi | Neg | Dim | Posi |
| SJALL066367 | Diagnosis | Male | AYA | Alive (CR1) | Yes | NA | NA | Dim | Neg | Weak | Neg | Posi | Posi |
| SJBALL016287 | Diagnosis | Female | AYA | Alive (CR1) | No | Positive | 46,XX[20] | Neg | Posi | NA | NA | NA | NA |
| SJBALL020165 | Diagnosis | Female | AYA | NA | No | Positive | 46,XX,del(9)(q13q22) [9]/46,XX[11] | Neg | Posi | Posi | Neg | Posi | Posi |
| SJBALL020169 | Diagnosis | Female | AYA | NA | No | Positive | 46,XX,del(9)(q13q22) [6]/46,XX[14] | Neg | Posi | Posi | Neg | NA | Posi |
| SJBALL020889 | Diagnosis | Female | Childhood HR | Alive (CR1) | No | Positive | 46,XX,del(9)(q12q32) [10]/92,idemx2,+ add(1)(p11),-20[8]/46,XX[20] | Neg | Posi | NA | NA | NA | NA |
| SJBALL022051 | Diagnosis | Male | Childhood HR | Alive (CR1) | No | NA | 46,XY,der(6)t(1;6)(q21.1;p25.3), add(14)(q32.3)[13]/46,XY[7] | Dim | Posi | Posi | Dim | Posi | NA |
| SJALL058818_R1 | Relapse | Male | Adult | Dead | Yes | Positive | Complex, near tetraploidy | Neg | Posi | Posi | NA | Posi | Posi |
| SJALL073397_R1 | Relapse | Female | Adult | Refractory after INO | Yes | Positive | NA | Neg | NA | Posi | NA | Posi | Posi |
| SJBALL042249_R1 | Relapse | Female | Adult | Dead (refractory disease) | Yes | Positive | 46,XX,dup(1)(q21q42),der (?2)t(2;13)(p11.2;q14), -3,add(6)(p23),?inv (7)(p13p22),der(?8)t (8;13)(q22;q22), del(9) (q13q22),add(12) (q24.3),add(13)(q14),del (13)(q12),del(?15)(q15q24), add(20)(q13.1),+mar | Dim | NA | Posi | Neg | Neg | Posi |
| SJALL073380_R1 | Relapse | Female | AYA | Alive (CR2) | Yes | NA | NA | Neg | NA | Posi | NA | Posi | Posi |
| SJALL073389_R1 | Relapse | Female | AYA | Alive (CR2) | Yes | NA | NA | Neg | NA | Posi | NA | Dim | Posi |
CR, complete remission; EOI, end of induction; HR, high-risk; NA, not available; Neg, negative; Posi, positive.
RNA-seq–based gene expression profile data from our previous study (1988 cases)16 and additional 16 cases were analyzed using hierarchical clustering and tSNE analysis, confirming the distinct expression profile of this group (Figure 1A; supplemental Figure 3). Gene expression analysis identified 1216 differentially expressed genes (fold change >2, adjusted P < .001) including CDX2, NTRK3, PREX2, and LRRC4C and low expression of FLT3 and MME (CD10) (Figure 1B; supplemental Table 3). Immunophenotyping showed negativity of CD10 for most cases (78.9%) and positivity for cytoplasmic immunoglobulin M (IgM) (Figure 1C; supplemental Table 1). High expression of NTRK3 was observed in almost half of cases (45.5%, 10/22; Figure 1D), which is rare in B-ALL. We also observed high expression of histone genes, with enrichment of histone and ribosomal RNA–related pathways (Figure 1E; supplemental Table 4).
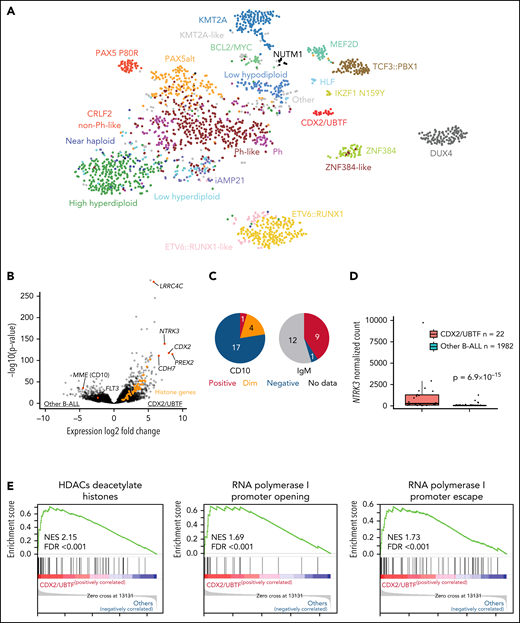
Identification of a distinct subtype in B-ALL. (A) Gene expression profiling of 2004 B-ALL cases including 22 cases of CDX2/UBTF subtype (red) shown in a 2-dimensional tSNE plot. Each dot represents a sample. The top 1000 most variable genes (on the basis of median absolute deviation) were selected and processed by the tSNE algorithm with a perplexity score of 30. Major B-ALL subtypes are highlighted in different colors. (B) Differentially expressed genes in CDX2/UBTF cases compared with other B-ALL cases are shown in the volcano plot. Genes exclusively expressed in CDX2/UBTF B-ALL are colored in red and annotated. Significantly low expression of MME (CD10) and FLT3 in CDX2/UBTF B-ALL is also shown. Histone cluster genes are colored in orange showing upregulation in CDX2/UBTF B-ALL. (C) Immunophenotype of CD10 and cytoplasmic IgM in CDX2/UBTF B-ALL at diagnosis or relapse. Most cases are negative for CD10 and positive for IgM. (D) Expression of NTRK3 gene by normalized read counts comparing CDX2/UBTF and other B-ALL cases is shown. (E) Pathway analysis using top 216 differentially expressed genes (fold change >2, adjusted P < 1 × 10−30) in CDX2/UBTF B-ALL revealed enrichment of histone and ribosomal RNA related pathways.
Identification of a distinct subtype in B-ALL. (A) Gene expression profiling of 2004 B-ALL cases including 22 cases of CDX2/UBTF subtype (red) shown in a 2-dimensional tSNE plot. Each dot represents a sample. The top 1000 most variable genes (on the basis of median absolute deviation) were selected and processed by the tSNE algorithm with a perplexity score of 30. Major B-ALL subtypes are highlighted in different colors. (B) Differentially expressed genes in CDX2/UBTF cases compared with other B-ALL cases are shown in the volcano plot. Genes exclusively expressed in CDX2/UBTF B-ALL are colored in red and annotated. Significantly low expression of MME (CD10) and FLT3 in CDX2/UBTF B-ALL is also shown. Histone cluster genes are colored in orange showing upregulation in CDX2/UBTF B-ALL. (C) Immunophenotype of CD10 and cytoplasmic IgM in CDX2/UBTF B-ALL at diagnosis or relapse. Most cases are negative for CD10 and positive for IgM. (D) Expression of NTRK3 gene by normalized read counts comparing CDX2/UBTF and other B-ALL cases is shown. (E) Pathway analysis using top 216 differentially expressed genes (fold change >2, adjusted P < 1 × 10−30) in CDX2/UBTF B-ALL revealed enrichment of histone and ribosomal RNA related pathways.
Deletion of UBTF resulting in UBTF::ATXN7L3 fusion
We next examined the genomic features of this subtype with integrated analysis of RNA-seq (n = 22) and WGS (n = 11; 5 tumor-germline paired cases and 6 tumor-only cases). All 22 cases exhibited expression of a chimeric fusion involving the C-terminus of UBTF and the downstream gene ataxin 7 like 3 (ATXN7L3) (Figure 2A; supplemental Tables 1 and 5). The fusions involved juxtaposition of UBTF exon 15 (n = 1), exon 17 (n = 19), exon 18 (n = 1), or exon 20 (n = 1) to the 5′ untranslated region of ATXN7L3 exon 1 on chromosome 17q21.31 (supplemental Figure 4A; supplemental Table 1). All fusions encoded an in-frame chimeric fusion protein utilizing 60 nucleotides upstream of the canonical translation start site of ATXN7L3 (Figure 2A), which was confirmed by immunoblotting in transfected human embryonic kidney 293T cells (Figure 2B). Both UBTF and ATXN7L3 are on chromosome 17q21.31, ~7000 bases apart. Analysis of WGS data of 11 available UBTF::ATXN7L3-rearranged cases identified the presence of a genomic deletion of ~10 kb between the 2 genes (Figure 2C; supplemental Table 1) with genomic breakpoints corresponding to the UBTF fusion junction (introns 15, 17, 18, or 20) and upstream of the ATXN7L3 coding sequence (Figure 2C; supplemental Figure 4B; supplemental Table 1). Notably, the breakpoints in 9 out of 11 cases were observed at approximately the same location (chr17:42,276,135 ± 6 and chr17:42,278,627 ± 3; GRCh37), and cryptic RSS were identified at these sites, with addition of nontemplated nucleotides, suggesting that the deletion may result from aberrant recombination-activating gene (RAG)-mediated recombination (Figure 2D; supplemental Table 6) as previously described for deletions observed for other genes in B-ALL.45-48 Collectively, the structural variants involving UBTF resulted in the generation of an in-frame chimeric UBTF::ATXN7L3 fusion protein, which is a universal genomic feature of this subtype.
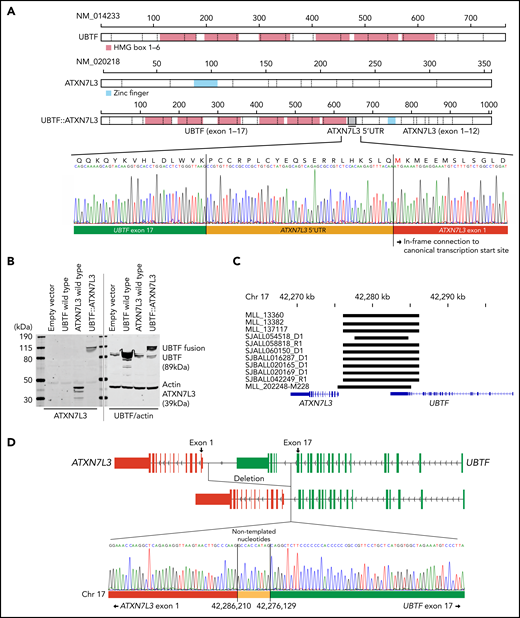
Detection of novel UBTF::ATXN7L3 fusion gene. (A) Protein structures of wild-type UBTF, ATXN7L3, and UBTF::ATXN7L3 fusion (exon 17 type) are shown. UBTF exon 17 is rearranged to the 5′ untranslated region (UTR) of ATXN7L3 and encodes an in-frame chimeric fusion protein. Direct sequencing of a representative case (SJALL060150) is shown. (B) Immunoblotting showing wild-type UBTF (right second lane), ATXN7L3 (left third lane), and UBTF::ATXN7L3 fusion (exon 17 type, 4th lane) proteins. Protein lysate was collected from human embryonic kidney 293T cells with transient transfection of wild-type UBTF and ATXN7L3, and UBTF::ATXN7L3 fusion containing plasmids. All samples were run in the same gel. After transferring, the membrane was cut, probed with 2 different antibodies, and then the 2 halves scanned together. (C) The deleted UBTF regions detected by WGS are shown in black bars. (D) The schema of UBTF deletion and direct sequencing of a representative case (SJBALL020169) is shown. Nontemplated nucleotides are inserted at the breakpoint.
Detection of novel UBTF::ATXN7L3 fusion gene. (A) Protein structures of wild-type UBTF, ATXN7L3, and UBTF::ATXN7L3 fusion (exon 17 type) are shown. UBTF exon 17 is rearranged to the 5′ untranslated region (UTR) of ATXN7L3 and encodes an in-frame chimeric fusion protein. Direct sequencing of a representative case (SJALL060150) is shown. (B) Immunoblotting showing wild-type UBTF (right second lane), ATXN7L3 (left third lane), and UBTF::ATXN7L3 fusion (exon 17 type, 4th lane) proteins. Protein lysate was collected from human embryonic kidney 293T cells with transient transfection of wild-type UBTF and ATXN7L3, and UBTF::ATXN7L3 fusion containing plasmids. All samples were run in the same gel. After transferring, the membrane was cut, probed with 2 different antibodies, and then the 2 halves scanned together. (C) The deleted UBTF regions detected by WGS are shown in black bars. (D) The schema of UBTF deletion and direct sequencing of a representative case (SJBALL020169) is shown. Nontemplated nucleotides are inserted at the breakpoint.
Deletion proximal of FLT3 results in enhancer retargeting and deregulation of CDX2
In addition to UBTF alterations, recurrent 13q12.2 (FLT3/PAN3) deletions were detected by WGS analysis from all analyzed cases in this subtype, in which the involvement of aberrant RAG-mediated recombination was also estimated (Figure 3A; supplemental Tables 1 and 6). Deletions in this region have been recently described in B-ALL with breakpoints occurring upstream of the FLT3 coding sequence, but sparing the FLT3 promoter, and associated with increased expression of FLT3 (type I deletion; Figure 3A).49 In contrast, in UBTF-rearranged cases, all 13q12.2 deletions included the promoter region and exon 1 of the FLT3 gene and the promoter of the adjacent gene PAN3 (type II deletions), which is oriented in head-to-head configuration with FLT3 (Figure 3A). In samples with type I deletion, upregulation of FLT3 is caused by an enhancer hijacking mechanism, in which an enhancer in the PAN3 gene interacts with the FLT3 promoter instead of the PAN3 promoter because the latter is deleted (Figure 3A).49 In contrast, in UBTF-rearranged cases with type II deletion, we observed low FLT3 expression but ectopic upregulation of CDX2 (Figures 1B and 3B), a transcription factor normally expressed in intestinal tissue, which is located ~30 kb downstream of FLT3. Furthermore, allele-specific expression of CDX2 and FLT3 was confirmed (Figure 3C; supplemental Figure 5), suggesting that the type II deletion at 13q12.2 deregulates CDX2 expression through hijacking and retargeting of the PAN3 coding enhancer. Based on the unique and universal UBTF rearrangements and CDX2 overexpression in this subtype, we herein termed this new subtype “CDX2/UBTF.” Neither of the 2 types of SVs, UBTF and 13q12.2 type II deletion, were observed in 2754 samples of leukemia with WGS/Single Nucleotide Polymorphism array DNA copy number data or leukemia cell lines (https://www.stjude.cloud/); however, high CDX2 expression and amplification of the region including the enhancer in PAN3 were observed in the hypodiploid cell line Nalm-16 (Figure 3A; supplemental Figure 6).
![Enhancer retargeting of FLT3-PAN3 regions drives deregulation of CDX2 in CDX2/UBTF subtype. (A) The deleted FLT3/PAN3 regions detected by WGS (type II deletion; top) and previously reported49 deleted 13q12.2 regions (type I deletion; bottom) are shown in black bars. Amplification of 13q12.2 found in Nalm-16 (hypodiploid B-ALL) is shown in a red bar. Type II deletion includes the promoter of FLT3 but not in type I deletion. Type II deletion does not affect the enhancer in PAN3. (B) Expression of FLT3 and CDX2 genes by normalized read counts comparing CDX2/UBTF and other B-ALL cases are shown. CDX2/UBTF B-ALL cases exhibited lower FLT3 expression and higher CDX2 expression. (C) Allele-specific expression of CDX2 is confirmed by comparison of allele frequencies at bases of heterozygous single-nucleotide polymorphism among RNA-seq (red), WGS (blue), and HiChIP (green). (D) H3K27ac HiChIP data are shown for representative patient samples (n = 3, SJALL060150 and SJBALL020169 [CDX2/UBTF B-ALL with type II deletion] and SJBALL113 [B-other with type I deletion]) and cell lines (n = 2, Nalm-16 [hypodiploid with PAN3 genic enhancer amplification] and Reh [ETV6::RUNX1 with no type I or type II deletion]). The heatmap shows raw read counts for all pairwise 5 kb genomic bins in the viewing window (chr13:28,457,037-28,911,626) for sample SJALL060150. The black box outlines read-pairs mapping to the CDX2 gene (highlighted in gold) and PAN3 genic enhancers (black dotted outline). Below the heatmap are RNA-seq and 1-dimensional HiChIP coverage tracks for all 5 samples. The corresponding heatmap for each sample can be found in supplemental Figure 8. Black bars indicate sample-specific deletions, whereas the Nalm-16 amplification is shown in red. Significant (false discovery rate < 0.01) loops called with MAPS42 are shown as blue arcs. Significant loops linking the PAN3 genic enhancers to CDX2 are shown in red.](https://ash.silverchair-cdn.com/ash/content_public/journal/blood/139/24/10.1182_blood.2022015444/3/m_bloodbld2022015444f3.png?Expires=1769080234&Signature=q-Tc0iXKNj7F0zi4wyq3HqCxr2Si5dipoOmfqeNn2YiQHLMH3FzA9HBvCXbBLf0H9n7zvgT2tqd7291-uDVgnEwSycTukCZKqnnieYHSm1xyNsCYzKQpetxQoJ31zpCfDbT13sBOjtAlqxLmH~fazuKt57zMf4qsEzbKNeo0IcykVLhKB8X2HDahwjvbLCEdsxSfzhE51bIcmbHdAkPmT4ZRzY60KnqUEv6BeAq6KnZf4ANtHJZHPuxVIFatu2CQOA12UN5yr6Qq3jJLobno2sLGogVqhUw1zB3CdZQPhhTxtvQDUOnD0XT6XD6yd6e7ze4wEm-RSXCfcL~j~yTTlw__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
Enhancer retargeting of FLT3-PAN3 regions drives deregulation of CDX2 in CDX2/UBTF subtype. (A) The deleted FLT3/PAN3 regions detected by WGS (type II deletion; top) and previously reported49 deleted 13q12.2 regions (type I deletion; bottom) are shown in black bars. Amplification of 13q12.2 found in Nalm-16 (hypodiploid B-ALL) is shown in a red bar. Type II deletion includes the promoter of FLT3 but not in type I deletion. Type II deletion does not affect the enhancer in PAN3. (B) Expression of FLT3 and CDX2 genes by normalized read counts comparing CDX2/UBTF and other B-ALL cases are shown. CDX2/UBTF B-ALL cases exhibited lower FLT3 expression and higher CDX2 expression. (C) Allele-specific expression of CDX2 is confirmed by comparison of allele frequencies at bases of heterozygous single-nucleotide polymorphism among RNA-seq (red), WGS (blue), and HiChIP (green). (D) H3K27ac HiChIP data are shown for representative patient samples (n = 3, SJALL060150 and SJBALL020169 [CDX2/UBTF B-ALL with type II deletion] and SJBALL113 [B-other with type I deletion]) and cell lines (n = 2, Nalm-16 [hypodiploid with PAN3 genic enhancer amplification] and Reh [ETV6::RUNX1 with no type I or type II deletion]). The heatmap shows raw read counts for all pairwise 5 kb genomic bins in the viewing window (chr13:28,457,037-28,911,626) for sample SJALL060150. The black box outlines read-pairs mapping to the CDX2 gene (highlighted in gold) and PAN3 genic enhancers (black dotted outline). Below the heatmap are RNA-seq and 1-dimensional HiChIP coverage tracks for all 5 samples. The corresponding heatmap for each sample can be found in supplemental Figure 8. Black bars indicate sample-specific deletions, whereas the Nalm-16 amplification is shown in red. Significant (false discovery rate < 0.01) loops called with MAPS42 are shown as blue arcs. Significant loops linking the PAN3 genic enhancers to CDX2 are shown in red.
Enhancer retargeting of FLT3-PAN3 regions drives deregulation of CDX2 in CDX2/UBTF subtype. (A) The deleted FLT3/PAN3 regions detected by WGS (type II deletion; top) and previously reported49 deleted 13q12.2 regions (type I deletion; bottom) are shown in black bars. Amplification of 13q12.2 found in Nalm-16 (hypodiploid B-ALL) is shown in a red bar. Type II deletion includes the promoter of FLT3 but not in type I deletion. Type II deletion does not affect the enhancer in PAN3. (B) Expression of FLT3 and CDX2 genes by normalized read counts comparing CDX2/UBTF and other B-ALL cases are shown. CDX2/UBTF B-ALL cases exhibited lower FLT3 expression and higher CDX2 expression. (C) Allele-specific expression of CDX2 is confirmed by comparison of allele frequencies at bases of heterozygous single-nucleotide polymorphism among RNA-seq (red), WGS (blue), and HiChIP (green). (D) H3K27ac HiChIP data are shown for representative patient samples (n = 3, SJALL060150 and SJBALL020169 [CDX2/UBTF B-ALL with type II deletion] and SJBALL113 [B-other with type I deletion]) and cell lines (n = 2, Nalm-16 [hypodiploid with PAN3 genic enhancer amplification] and Reh [ETV6::RUNX1 with no type I or type II deletion]). The heatmap shows raw read counts for all pairwise 5 kb genomic bins in the viewing window (chr13:28,457,037-28,911,626) for sample SJALL060150. The black box outlines read-pairs mapping to the CDX2 gene (highlighted in gold) and PAN3 genic enhancers (black dotted outline). Below the heatmap are RNA-seq and 1-dimensional HiChIP coverage tracks for all 5 samples. The corresponding heatmap for each sample can be found in supplemental Figure 8. Black bars indicate sample-specific deletions, whereas the Nalm-16 amplification is shown in red. Significant (false discovery rate < 0.01) loops called with MAPS42 are shown as blue arcs. Significant loops linking the PAN3 genic enhancers to CDX2 are shown in red.
The enhancer region in PAN3 is ubiquitously active in hematopoietic and immune cell types but not in other cell types or tissues (supplemental Figure 7A-D), indicating that the enhancer itself is not aberrantly activated in B-ALL and suggesting that the type II deletion in CDX2/UBTF B-ALL may result in CDX2 deregulation through enhancer retargeting as type I deletions have been described to do for FLT3. To investigate this, we performed HiChIP to profile acetylation of histone H3 at lysine 27 (H3K27ac) as a mark of enhancers, simultaneously with 3-dimensional chromatin architecture in 2 CDX2/UBTF samples with type II deletions, 1 B-other ALL sample with type I deletion leading to FLT3 overexpression (SJBALL113), the hypodiploid cell line Nalm-16 with amplification of the enhancer in PAN3, and the ETV6::RUNX1 cell line Reh with no alteration in this region (Figure 3D). The CDX2/UBTF B-ALL cases (SJALL060150 and SJBALL020169) harboring the type II deletion, which removes both the PAN3 and FLT3 promoters, showed evidence of chromatin looping between the enhancer in PAN3 and the CDX2 gene located ~280 kb away (Figure 3D; supplemental Figure 8A), supporting an enhancer retargeting mechanism for CDX2 expression. In contrast, in the B-other case SJBALL113 that harbors a type I deletion that removes the promoter of PAN3 but not FLT3, no interactions were observed between the enhancer in PAN3 and CDX2 (Figure 3D; supplemental Figure 8B); rather, multiple significant chromatin loops were identified between the PAN3 coding enhancers and the FLT3 gene, consistent with previous reports that type I deletions result in FLT3 overexpression.49 H3K27ac HiChIP data from Nalm-16 cells, which harbor amplification of the region containing the enhancer in PAN3, showed long-range interactions with the CDX2 gene that did not reach statistical significance (Figure 3D; supplemental Figure 8C; supplemental Table 7). Finally, Reh cells that lack FLT3/PAN3 deletions showed interactions between the PAN3 and FLT3 promoters, albeit to a lesser extent compared with the case with a type I deletion (SJBALL113; Figure 3D; supplemental Figure 8D). Collectively, these data functionally connect different classes of FLT3-PAN3 SVs with changes in local gene expression and provide a mechanistic basis for high CDX2 expression in the CDX2/UBTF B-ALL subtype.
Other genomic abnormalities in CDX2/UBTF B-ALL
We identified 2 unique and universal SVs involving UBTF and the FLT3/PAN3 region in all analyzed CDX2/UBTF B-ALL cases. We estimated the order of acquiring these SVs during clonal leukemic evolution by VAF of each SV. Most cases showed similar VAFs for both SVs, whereas 2 cases (SJBALL020169 and SJBALL042249) had higher VAFs for FLT3-PAN3 deletion (VAF above 0.35) than UBTF deletion (VAF below 0.3), suggesting aberrant CDX2 expression is the founder alteration (supplemental Figure 9; supplemental Table 8).
Additional co-alterations detected by WGS (n = 11) included the heterozygous deletion of ETV6 (36.3%, 4/11) and rearrangements of PAX5/ZCCHC7 (36.3%; 3 cases with genomic deletion and 1 due to inversion between the 2 genes) (Figure 4A; supplemental Tables 9 and 10). Aberrant RAG activity was suggested in PAX5/ZCCHC7 rearrangements (supplemental Table 6). The rearrangements of PAX5 included exon 1a of PAX5 gene but spared the alternate first exon of PAX5, exon 1b (Figure 4A). Although the use of alternative exon 1b was observed in cases without PAX5 rearrangements, these PAX5 rearranged cases showed higher exon 1b utilization compared with CDX2/UBTF cases without PAX5/ZCCHC7 rearrangements, leading to extremely high PAX5 expression, especially for the case with inversion (Figure 4B-C).
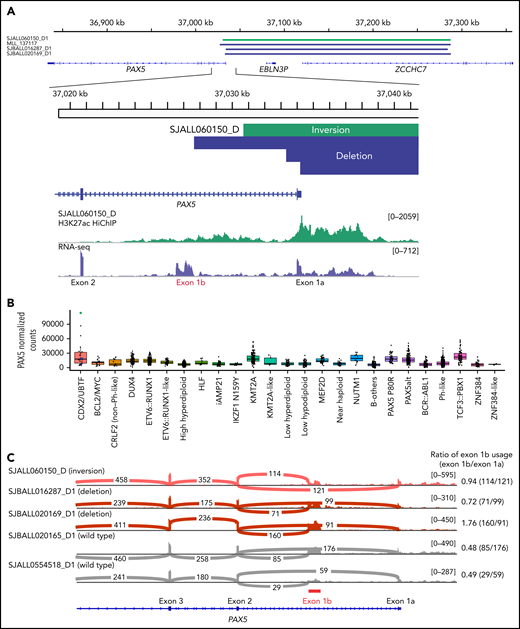
PAX5 deregulation in a subset of CDX2/UBTF subtype induced by the structural variants at PAX5-ZCCHC7 region. (A) The SVs at PAX5/ZCCHC7 region detected by WGS were shown in green (inversion) and blue (deletion) bars. Below the coverage tracks of 1-dimensional HiChIP and RNA-seq for representative CDX2/UBTF inversion case (SJALL060150_D) are shown. SVs include the promoter and exon 1 of PAX5 (exon 1a) but do not affect the alternative PAX5 exon 1b. (B) Expression of PAX5 by normalized read counts in each B-ALL subtype is shown. CDX2/UBTF cases with PAX5 rearrangements are shown in green (inversion) and blue (deletion). PAX5 is expressed highly in CDX2/UBTF subtype, especially for the case with PAX5/ZCCHC7 inversion. (C) Sashimi plot showing the junctions with the number of reads split across the junction (junction depth) in the PAX5 gene for representative CDX2/UBTF samples: inversion (n = 1, SJALL060150_D), deletion (n = 2, SJBALL016287_D1 and SJBALL020169_D1), and without PAX5 rearrangements (n = 2, SJBALL020165_D1 and SJALL0554518_D1). PAX5 exon 1b is shown in red bar. The ratio of exon 1b usage to exon 1a is shown in the right of plot. The cases with PAX5 rearrangements exhibited higher exon 1b usage ratio than cases without rearrangements.
PAX5 deregulation in a subset of CDX2/UBTF subtype induced by the structural variants at PAX5-ZCCHC7 region. (A) The SVs at PAX5/ZCCHC7 region detected by WGS were shown in green (inversion) and blue (deletion) bars. Below the coverage tracks of 1-dimensional HiChIP and RNA-seq for representative CDX2/UBTF inversion case (SJALL060150_D) are shown. SVs include the promoter and exon 1 of PAX5 (exon 1a) but do not affect the alternative PAX5 exon 1b. (B) Expression of PAX5 by normalized read counts in each B-ALL subtype is shown. CDX2/UBTF cases with PAX5 rearrangements are shown in green (inversion) and blue (deletion). PAX5 is expressed highly in CDX2/UBTF subtype, especially for the case with PAX5/ZCCHC7 inversion. (C) Sashimi plot showing the junctions with the number of reads split across the junction (junction depth) in the PAX5 gene for representative CDX2/UBTF samples: inversion (n = 1, SJALL060150_D), deletion (n = 2, SJBALL016287_D1 and SJBALL020169_D1), and without PAX5 rearrangements (n = 2, SJBALL020165_D1 and SJALL0554518_D1). PAX5 exon 1b is shown in red bar. The ratio of exon 1b usage to exon 1a is shown in the right of plot. The cases with PAX5 rearrangements exhibited higher exon 1b usage ratio than cases without rearrangements.
Gain of chromosome 1q (>20 Mb; n = 6, 54.5%) was common in CDX2/UBTF B-ALL cases (supplemental Tables 1 and 10). Notably, frequent gain of the histone gene cluster region (chr1: 149,260,000-149,900,000) was observed in >80% (9/11) of CDX2/UBTF cases with gains of 1q of any size, which were otherwise rare in B-ALL (1.83%, 22/1202) except for MEF2D-rearranged cases (25.9%, 7/27) (supplemental Figure 10A-C; supplemental Table 11). Histone genes were overexpressed in CDX2/UBTF cases, and notably, ChIP-seq analysis has shown UBTF to bind at histone gene promoters in the Epstein-Barr virus B lymphoid cell line GM12878 (ref. 44; supplemental Figure 10D). Finally, ranking of super-enhancers (as identified by H3K27ac HiChiP data) demonstrated stronger super-enhancer signals for the histone cluster on chromosome 6 and PAX5 regions in CDX2/UBTF cases compared with non-CDX2/UBTF B-ALL (supplemental Figure 10E; supplemental Table 12). These data support a role for upregulation of histone genes and PAX5 in CDX2/UBTF B-ALL in the case of histone gene deregulation either by chromosome 1 gain and/or the direct action of UBTF::ATXN7L3 (Figure 1B; supplemental Figure 10C).
Discussion
This study describes the entity and driver genomic alterations that define a high-risk B-ALL subtype with deregulation of CDX2 and UBTF. Increased expression of CDX2 has been described in B-ALL,50 but the mechanistic basis has been unknown. Here, we show the power of integrated RNA-seq and WGS to identify 2 new genomic alterations involving intergenic/noncoding regions that are not directly identifiable by RNA-seq,22 and by coupling these analyses with H3K27ac HiChIP analysis, we have directly demonstrated an enhancer retargeting mechanism at 13q12.2 leading to aberrant expression of CDX2 by HiChiP. Importantly, we demonstrated the different interaction of the same enhancer with nearby genes in 3 genomic alterations found in this region, type I deletion (FLT3 upregulation), type II deletion (unique in CDX2/UBTF B-ALL; CDX2 upregulation), and amplification (FLT3 and CDX2 upregulation). These different functional engagements between the enhancer and its target promoters can be explained by the recently described concept of enhancer release and retargeting, in which functional loss of a preferred promoter results in release of its normal enhancer partner, resulting in looping to, and activation of, an alternative promoter(s) within the same topologically associated domain.51 This was also observed in the PAX5 and ZCCHC7 region,51 one of the genomic features of CDX2/UBTF B-ALL that may drive the aberrant PAX5 expression observed in these cases. Notably, all these genomic deletions or inversions were estimated to have cryptic RSS sites at breakpoints, suggesting the involvement of aberrant RAG activity in the etiology of CDX2/UBTF B-ALL subtype.
The 2 distinct but universal genomic events observed in all CDX2/UBTF B-ALL cases are striking and reminiscent of DUX4-rearranged B-ALL. In that subtype, enhancer hijacking events drive deregulation of DUX4 that is followed by DUX4 binding to ERG, resulting in its deregulation and deletion, with expression of truncated, functionally aberrant ERG proteins.6 We estimated that the CDX2 abnormality arose first by comparing the VAFs of the 2 structural variants. Ectopic expression of CDX2 in stem and progenitor hematopoietic cells results in self-renewal, myeloid lineage skewing, and myelodysplasia and acute monoblastic leukemia, with suppression of lymphoid pathways.52,53 Additional alterations are likely required for the leukemogenesis of B-ALL, and accordingly, Ikzf1 alterations are associated with lymphoid phenotype in these mouse models. UBTF is one of the main downstream targets of MYC and regulates ribosome biogenesis by binding ribosomal RNA genes54 and deregulates gene expression, including those regulating B-cell differentiation, by reorganizing ribosomal DNA-genome contacts in a MYC-driven model of lymphoma.55 Thus, we hypothesize that aberrant expression of CDX2 may directly drive the genesis of the UBTF::ATXN7L3 deletion/rearrangement, and cooperativity of these alterations skews the phenotype of transformed cells to a B lymphoid lineage.
Identification of the CDX2/UBTF subtype of B-ALL at diagnosis is clinically important in that while uncommon in B-ALL overall (0.53%), this subtype was most frequently identified in AYA patients (1.4%), especially females (2.2%), and exhibits high-risk features including high incidence of relapse or refractory disease and elevated levels of MRD. Although the outcome of AYA with B-ALL has improved with the use of intensive, pediatric-inspired therapy,56-58 outcomes remain suboptimal. Several distinctive clinical and genomic features may be used to detect CDX2/UBTF B-ALL in clinical practice: (1) the distinctive immunophenotype, negativity for CD10, and positivity for cytoplasmic IgM; (2) the detection of UBTF::ATXN7L3 fusion by reverse transcription polymerase chain reaction, as the recurrent breakpoints (95%, 21/22) involve UBTF exon 17 or later and ATXN7L3 exon 1; (3) the use of genomic polymerase chain reaction to detect UBTF (17q21.31) deletions (10 kb) as the breakpoints were at approximately the same location (chr17:42,276,135 ± 6 and chr17:42,278,627 ± 3; GRCh37) in most cases (82%, 9/11); (4) the use of RNA-seq to confirm UBTF::ATXN7L3 fusion and high expression of CDX2; or (5) the use of WGS to detect the 2 universal genomic deletions on FLT3/PAN3 (13q12.2) and UBTF (17q21.31). In conclusion, the unique genomic and clinical characteristics of CDX2/UBTF B-ALL described here may help to guide refined risk stratification at diagnosis for improved outcome of AYA leukemia patients.
Acknowledgments
The authors thank the Haematology Tissue Bank (Peter MacCallum Cancer Centre/Royal Melbourne Hospital) and Children’s Cancer Centre Tissue Bank (Murdoch Children’s Research Institute) for assistance with sample collection and the Genomics Core Facility and Genomics Platform Group (University of Melbourne Centre for Cancer Research) for sequencing and analysis support.
The authors are supported by the American and Lebanese Syrian Associated Charities of St. Jude Children’s Research Hospital; the St. Jude Children’s Research Hospital Chromatin Collaborative (C.G.M.); National Institutes of Health National Cancer Institute (NCI) grants P30 CA021765, R35 CA197695 (C.G.M.), U10 CA180820 (ECOG-ACRIN), UG1 CA232760 (M.L.), and UG1 CA189859 (E.M.P.); the Henry Schueler 41&9 Foundation (C.G.M.); a St. Baldrick’s Foundation Robert J. Arceci Innovation Award (to C.G.M.); a Garwood Postdoctoral Fellowship of the Hematological Malignancies Program of the St Jude Children’s Research Hospital Comprehensive Cancer Center (to S.K.); grants from the Wilson Centre for Blood Cancer Genomics (P.B.); the Snowdome Foundation (P.B.); the Peter MacCallum Cancer Foundation (G.L.R.); a SCOR Grant (7015-18); the Lymphoma and Leukemia Society (P.G.E.); the Perpetual Trustees and the Samuel Nissen Foundation (P.G.E.); and the National Health and Medical Research Council of Australia (A.O., GNT1140626).
Authorship
Contribution: S.K., L.M., I.I., and C.G.M. were responsible for conception and design of the study; S.K. and L.M. performed genomic data analysis; S.K. and P.E.M. performed experiments; Y.Z., Q.G., Z.G., G.L.R., M.M., W.W., S.B., J.Z., and K.G.R. assembled data; E.M.P., C.H., A.D.L., W.S., M.L., J.M.R., S.M.L., S.P.H., G.L.R., B.S., P.G.E., A.O., S.M.G., D.L.W., J.B., I.A., E.J.J., C.-H.P., W.K., T.H., and P.B. collected samples; S.K., L.M., I.I., and C.G.M. wrote the manuscript; and all authors critically revised the article and approved the final version.
Conflict-of-interest disclosure: I.I. received honoraria from Amgen and Mission Bio. P.G.E. received royalty distributions through Commercial Income Distribution from the Walter and Eliza Hall Institute. C.G.M. received research funding from Loxo Oncology, Pfizer, and AbbVie; honoraria from Amgen and Illumina; and holds stock in Amgen. There are no conflicts of interest with the work presented in this manuscript. A.D.L reports he is an employee of and has stock and/or other ownership interests in Pfizer. M.M. and W.W. report employment by Munich Leukemia Laboratory (MLL). C.H., W.K., and T.H. report equity ownership of MLL. The remaining authors declare no competing financial interests.
The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Correspondence: Charles G. Mullighan, Department of Pathology, Hematological Malignancies Program, St. Jude Children's Research Hospital, 262 Danny Thomas Place, Mail Stop 342, Memphis, TN 38105; e-mail: charles.mullighan@stjude.org.
RNA-seq, WGS, and HiChIP data have been deposited at the EGAS00001005863 (human primary samples), GSE190690 (cell lines), and housed in a protected Cloud environment in accordance with European General Data Protection regulations. Other legacy data used in this study have been deposited in the European Genome-Phenome Archive (EGA) in previous projects under accession numbers EGAS00001003266, EGAS00001000654, EGAS00001001952, EGAS00001001923, EGAS00001002217, and EGAS00001000447. The TARGET genomic data used in this study are available through the TARGET website (https://ocg.cancer.gov/programs/target) and also in the database of Genotypes and Phenotypes (dbGaP; http://www.ncbi.nlm.nih.gov/gap) under accession number phs000218 (TARGET).
Requests for data sharing may be submitted to Charles G. Mullighan (charles.mullighan@stjude.org).
The online version of this article contains a data supplement.
There is a Blood Commentary on this article in this issue.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal