

Key Points
A Bcl-2 inhibitor, venetoclax, increases the effector activity of antileukemic T cells without inducing T-cell apoptosis.
Venetoclax increases T-cell effector function in a reactive oxygen species–dependent manner.

Abstract
Venetoclax, a Bcl-2 inhibitor, in combination with the hypomethylating agent azacytidine, achieves complete remission with or without count recovery in ∼70% of treatment-naive elderly patients unfit for conventional intensive chemotherapy. However, the mechanism of action of this drug combination is not fully understood. We discovered that venetoclax directly activated T cells to increase their cytotoxicity against acute myeloid leukemia (AML) in vitro and in vivo. Venetoclax enhanced T-cell effector function by increasing reactive oxygen species generation through inhibition of respiratory chain supercomplexes formation. In addition, azacytidine induced a viral mimicry response in AML cells by activating the STING/cGAS pathway, thereby rendering the AML cells more susceptible to T cell–mediated cytotoxicity. Similar findings were seen in patients treated with venetoclax, as this treatment increased reactive oxygen species generation and activated T cells. Collectively, this study presents a new immune-mediated mechanism of action for venetoclax and azacytidine in the treatment of AML and highlights a potential combination of venetoclax and adoptive cell therapy for patients with AML.
Introduction
Adoptive T-cell therapy, particularly chimeric antigen receptor T-cell therapy, has achieved remarkable success in treating B-cell malignancies.1,2 Despite various approaches investigated, similar success has not been translated for patients with acute myeloid leukemia (AML), contributed to by inadequate target antigen, low mutational rate, and high disease heterogeneity.3
Recently, venetoclax, a Bcl-2 inhibitor, combined with the hypomethylating agent azacytidine, achieved complete remission or complete remission with incomplete count recovery in ∼70% of treatment-naive elderly patients unfit for conventional intensive chemotherapy.4-6 The impressive clinical outcome led to accelerated US Food and Drug Administration approval of the treatment of this patient population. However, the mechanism of action of this drug combination is not fully understood. Although highly effective in the de novo setting, it is less active in patients who relapse after induction chemotherapy7,8 ; the reasons for a reduced response rate in relapsed or refractory patients are not fully understood.
CD3+CD4–CD8– double-negative T cell (DNT) is a rare subset of mature T cells that comprises 1% to 5% of circulating leukocytes.3,9-11 We recently developed a method to ex vivo expand the clinical quality and quantity of these unconventional immune cells from healthy donors. We further showed that ex vivo expanded allogeneic DNTs can selectively target AML without alloreactivity and off-tumor toxicity. DNTs also fulfill the requirements for an “off-the-shelf” cell therapy, including scalability, donor-independent activity, and lack of graft-versus-host disease and host-versus-graft rejection.11 Clinical trials to assess the safety and efficacy of allogeneic DNT therapy as a treatment of high-risk AML patients are in progress.
In the current study, we discovered that venetoclax effectively enhances the effector function of DNT and CD8+ T cells by increasing reactive oxygen species (ROS) generation. We also show that azacytidine renders AML cells more susceptible to T cells via induction of viral mimicry. These data provide an important mechanism to explain the clinical activity of venetoclax and azacytidine in AML and highlight the potential of combining adoptive T-cell therapy with venetoclax for the treatment of this disease.
Materials and methods
Primary AML and normal peripheral blood mononuclear cells
Human blood was collected from healthy adult donors after obtaining written informed consent and was used according to protocols approved by the University Health Network (UHN) Research Ethics Board (05-0221-T) and the National Heart, Lung, and Blood Institute. The AML samples were collected with informed consent and viably frozen in the Leukemia Tissue Bank at Princess Margaret Cancer Centre according to procedures approved by the UHN Research Ethics Board (UHN REB# 01-0573). Primary human AML samples were obtained from peripheral blood or bone marrow of AML patients with a malignant cell frequency of 80% among mononuclear cells, determined according to morphology. In addition, the frequency of malignant cells in each AML patient sample was determined by flow cytometry by using a combination of SSClow CD45low CD3– CD33+ with or without CD34 and CD117 expression. Differential density centrifugation was used to isolate AML cells and healthy donor–derived peripheral blood mononuclear cells (PBMCs). Primary AML cells were frozen in Minimum Essential Medium α (Thermo Fisher Scientific) + 5% fetal bovine serum or 90% fetal bovine serum + 15 U/mL of heparin + 10% dimethyl sulfoxide.
The UHN institutional review boards approved the collection and use of human tissue for this study (Research Ethics Board protocol #15-9324). All specimens were deidentified, and each experiment was performed by using a single aliquot from a donor. Information about the patients who were the source of the cells is provided in supplemental Tables 1 and 2 (available on the Blood Web site).
Ex vivo expansion of DNTs and venetoclax treatment
DNT expansions were performed as previously described.11 Briefly, CD4+ and CD8+ cell-depleted healthy donor–derived PBMCs were cultured on anti-CD3 antibody-coated plates (OKT3; BioLegend) for 3 days in AIM-V (Thermo Fisher Scientific) with 250 IU/mL of interleukin-2 (IL-2) (Proleukin; Novartis Pharmaceuticals); soluble anti-CD3 antibody and IL-2 were added to the cultures every 2 to 4 days. The purity of DNTs was assessed by staining cells with fluorochrome-conjugated anti-human CD3, -CD4, -CD8, and -CD56 antibodies and flow cytometry analysis. DNTs were used between days 10 and 20 of culture. For venetoclax treatment, 25 to 400 nM venetoclax (ABT-199; TargetMol) was added to DNT culture 4 hours to 3 days before use. DNTs were washed twice with phosphate-buffered saline and counted before use for each experiment.
Flow cytometry–based in vitro killing assay
DNTs were cocultured with target cells for 2 to 4 hours, and cells were then stained with anti-human CD3 (HIT3a), CD33 (WM53), CD45 (HI30), and CD34 (561) antibodies, Annexin V, and 7AAD (all from BioLegend), and analyzed by using flow cytometry. Specific killing was calculated by as previously described10
Single-cell cytokine analysis
Statistical analysis
All graphs and statistical analysis were generated by using GraphPad Prism 5 (GraphPad Software). Student t test, 1-way analysis of variance, 2-way analysis of variance, and linear regression tests were used for analyses. *P < .05, **P < .01, ***P < .001, and ****P < .0001 indicate significance between experimental and control values. Error bars represent ± standard error of the mean (SEM) or standard deviation (SD) as indicated.
Results
Venetoclax enhances T cell–mediated cytotoxicity against AML
To identify drugs that increase the potency of T cell–mediated cytotoxicity against AML, we used ex vivo expanded DNTs as surrogates because DNTs can be expanded ex vivo and allow investigation of T-cell and AML-cell interaction without alloreactivity or xenogeneic graft-versus-host disease in vitro and in the xenograft model.10 We then validated the findings with conventional T (Tconv) cells, although our studies were not designed to compare the superiority of DNTs vs Tconv cells.
DNTs were treated with an in-house library of 189 on- and off-patent drugs for 18 hours and then used as effectors against the human AML cell line OCI-AML2. From this screen, we identified venetoclax as the top hit that increased the cytotoxicity of DNTs (Figure 1A). The effects of venetoclax on DNTs were tested in dose-dependent studies against 3 AML cell lines (OCI-AML2, OCI-AML3, and KG1a). In all 3 tested cell lines, venetoclax increased the DNT-mediated killing of AML cells (Figure 1B) without reducing the viability of the DNTs (supplemental Figure 1). Increased antileukemic activity in venetoclax-treated DNTs was seen in DNTs derived from all 11 tested DNT donors with an average increase of 61.25 ± 31% (Figure 1C).
![Venetoclax increases antileukemic activity of T cells. (A) A total of 189 drugs were added to ex vivo expanded DNTs (50 000 cells per well) in 96 well plates at a final concentration of 400 nM for 18 hours. The compound-treated DNTs were washed and incubated with OCI-AML2 at a 2:1 ratio for 2 hours. AML cell viability was determined by Annexin V using flow cytometry. Data represent the change in cytotoxicity relative to untreated DNT control. (B) DNTs were treated with increasing concentrations of venetoclax for 18 hours followed by coculture for 2 hours with OCI-AML2 and OCI-AML3 at a 2:1 and KG1a at an 8:1 DNT to AML ratio. The viability of AML cells (CD3– CD33+ or CD34+) was determined as described in panel A. Each experiment was done in triplicate, and data represent the mean ± SD specific killing from 1 of 5 independent experiments conducted by using DNTs from different donors. (C) DNTs expanded from 11 donors were untreated or treated with 400 nM venetoclax for 18 hours. Subsequently, they were cultured with OCI-AML2 at a 1:1, 2:1, or 4:1 DNT:AML ratio, and the viability of AML cells was measured by Annexin V staining and flow cytometry. Each paired symbol represents DNTs from an individual donor. (D) Venetoclax 400 nM treated or untreated DNTs were cocultured with primary AML samples (n = 17) at a 2:1 ratio for 2 hours. After incubation, cell viability was measured as described in panel A. Each sample was measured in triplicate, and data represent the changes in AML cell death in the presence of venetoclax-treated DNTs relative to untreated DNTs. DNTs from 6 different donors were used for the screening. (E) OCI-AML2, KG1a, or primary AML cells (100857) were treated with DNT or venetoclax-treated (Ven-treated) DNTs for 18 hours. Equal volumes (105 cells per mL per dish) of cells were seeded in colony-forming assays, and the colonies were counted. Data represent mean ± SD of number of colonies formed. The results for OCI-AML2 and KG1a are representative of 3 independent experiments conducted by using DNTs from 2 different donors. (F) Primary AML cells (ID: 130607) untreated or treated with DNTs or venetoclax-treated DNTs for 2 hours at a 2:1 DNT:AML ratio were injected intrafemorally into NOD/SCID mice (1.6 × 106 cells per mouse; n = 6 per group). Six weeks after injection, the percentage of AML engraftment (human CD45+ CD33+ cells) in the bone marrow from each group was determined by flow cytometry. (G) OCI-AML2, autologous PBMCs, or allogeneic PBMCs (n = 4) were used as targets and cultured with DNTs or venetoclax-treated DNTs for 2 hours at a 2:1 DNT to AML ratio or 8:1 DNT to PBMC ratio. The viability of AML cells and PBMCs was determined by Annexin V. Data represent the mean ± SD specific killing. The results are representative of 2 independent experiments. (H) Ex vivo expanded polyclonally activated CD4+/CD8+ Tconv cells were treated with increasing concentrations of venetoclax for 18 hours. Tconv cells were added as effectors against OCI-AML2, OCI-AML3, and KG1a at a 4:1 target ratio. Two hours after incubation, AML cell viability was determined by Annexin V. Data represent the mean ± SD specific killing from 1 of 5 independent experiments. (I) Sublethally irradiated (250 cGy) NSG mice were injected intravenously with KG1a cells (2 × 106 cells per mouse). Two weeks post-KG1a infusion, mice were treated with 3 infusions of vehicle control (phosphate-buffered saline [PBS]) or 1.5 to 2 × 107 cells per infusion of DNTs or venetoclax-treated DNTs 3 to 4 days apart. Five weeks post–AML injection, bone marrow engraftment of KG1a (human CD45+ CD34+) was determined by using flow cytometry. The result shown is representative of 2 independent experiments conducted by using 2 different donor-derived DNTs. (J) Sublethally irradiated NSG mice were intravenously injected with primary AML cells (n = 4; 2-5 × 106 per mouse). Two weeks later, mice were treated with 3 infusions of vehicle control or 1.5 to 2 × 107 cells per infusion of DNTs or venetoclax-treated DNTs, 3 to 4 days apart. Five weeks post–AML injection, bone marrow engraftment of primary AML cells (human CD45low CD33+ with or without CD34 expression) was determined by flow cytometry. Left: representative contour plot of bone marrow cells from each group stained with CD45 and CD33. Right: summarized results from patient-derived xenograft experiments performed by using 4 different primary AML patient samples. Horizontal bar represents the mean of bone marrow AML engraftment level normalized to vehicle control group; each symbol represents an individual mouse, and error bars represent SD. Data represent the mean ± SEM reduction in bone marrow leukemia level relative to the PBS group. Student t test or 1-way analysis of variance was used for statistics. *P < .05; **P < .01; ***P < .001; ****P < .0001. ns, not significant.](https://ash.silverchair-cdn.com/ash/content_public/journal/blood/138/3/10.1182_blood.2020009081/4/m_bloodbld2020009081r2f1.png?Expires=1770797782&Signature=o8a4gyb~39pZF~0wVMCagrE5Hb~3qjDK0o6gUSr0nCX2a95B3ryYMj9RZe68xCIYtnGl2YkV07uo-k9D01dyGIMhRqD654KG2248Pj5LKaq1w7cxIlhlo7FnqaGrIwMUhtVW-FhLp~mrQSVViR8uC2Ryb4zlDilybTvKDSUm8e3ZAXdV1Ue~Z1VxRoiiYCT-4EaDJKp3-kcxeJJLa~HIlvRVM99Nj5rlDtzhgWmAb3lPiGYjTkwlWEt-KeQscw3NB~THaBxnUANhpLKqMhmkSiI2DoqJmHI371bfWOYD4P45oWxw5Chv3rSDABRehCVqjGw-61~RKWGa1wnNYo0Y8w__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
Venetoclax increases antileukemic activity of T cells. (A) A total of 189 drugs were added to ex vivo expanded DNTs (50 000 cells per well) in 96 well plates at a final concentration of 400 nM for 18 hours. The compound-treated DNTs were washed and incubated with OCI-AML2 at a 2:1 ratio for 2 hours. AML cell viability was determined by Annexin V using flow cytometry. Data represent the change in cytotoxicity relative to untreated DNT control. (B) DNTs were treated with increasing concentrations of venetoclax for 18 hours followed by coculture for 2 hours with OCI-AML2 and OCI-AML3 at a 2:1 and KG1a at an 8:1 DNT to AML ratio. The viability of AML cells (CD3– CD33+ or CD34+) was determined as described in panel A. Each experiment was done in triplicate, and data represent the mean ± SD specific killing from 1 of 5 independent experiments conducted by using DNTs from different donors. (C) DNTs expanded from 11 donors were untreated or treated with 400 nM venetoclax for 18 hours. Subsequently, they were cultured with OCI-AML2 at a 1:1, 2:1, or 4:1 DNT:AML ratio, and the viability of AML cells was measured by Annexin V staining and flow cytometry. Each paired symbol represents DNTs from an individual donor. (D) Venetoclax 400 nM treated or untreated DNTs were cocultured with primary AML samples (n = 17) at a 2:1 ratio for 2 hours. After incubation, cell viability was measured as described in panel A. Each sample was measured in triplicate, and data represent the changes in AML cell death in the presence of venetoclax-treated DNTs relative to untreated DNTs. DNTs from 6 different donors were used for the screening. (E) OCI-AML2, KG1a, or primary AML cells (100857) were treated with DNT or venetoclax-treated (Ven-treated) DNTs for 18 hours. Equal volumes (105 cells per mL per dish) of cells were seeded in colony-forming assays, and the colonies were counted. Data represent mean ± SD of number of colonies formed. The results for OCI-AML2 and KG1a are representative of 3 independent experiments conducted by using DNTs from 2 different donors. (F) Primary AML cells (ID: 130607) untreated or treated with DNTs or venetoclax-treated DNTs for 2 hours at a 2:1 DNT:AML ratio were injected intrafemorally into NOD/SCID mice (1.6 × 106 cells per mouse; n = 6 per group). Six weeks after injection, the percentage of AML engraftment (human CD45+ CD33+ cells) in the bone marrow from each group was determined by flow cytometry. (G) OCI-AML2, autologous PBMCs, or allogeneic PBMCs (n = 4) were used as targets and cultured with DNTs or venetoclax-treated DNTs for 2 hours at a 2:1 DNT to AML ratio or 8:1 DNT to PBMC ratio. The viability of AML cells and PBMCs was determined by Annexin V. Data represent the mean ± SD specific killing. The results are representative of 2 independent experiments. (H) Ex vivo expanded polyclonally activated CD4+/CD8+ Tconv cells were treated with increasing concentrations of venetoclax for 18 hours. Tconv cells were added as effectors against OCI-AML2, OCI-AML3, and KG1a at a 4:1 target ratio. Two hours after incubation, AML cell viability was determined by Annexin V. Data represent the mean ± SD specific killing from 1 of 5 independent experiments. (I) Sublethally irradiated (250 cGy) NSG mice were injected intravenously with KG1a cells (2 × 106 cells per mouse). Two weeks post-KG1a infusion, mice were treated with 3 infusions of vehicle control (phosphate-buffered saline [PBS]) or 1.5 to 2 × 107 cells per infusion of DNTs or venetoclax-treated DNTs 3 to 4 days apart. Five weeks post–AML injection, bone marrow engraftment of KG1a (human CD45+ CD34+) was determined by using flow cytometry. The result shown is representative of 2 independent experiments conducted by using 2 different donor-derived DNTs. (J) Sublethally irradiated NSG mice were intravenously injected with primary AML cells (n = 4; 2-5 × 106 per mouse). Two weeks later, mice were treated with 3 infusions of vehicle control or 1.5 to 2 × 107 cells per infusion of DNTs or venetoclax-treated DNTs, 3 to 4 days apart. Five weeks post–AML injection, bone marrow engraftment of primary AML cells (human CD45low CD33+ with or without CD34 expression) was determined by flow cytometry. Left: representative contour plot of bone marrow cells from each group stained with CD45 and CD33. Right: summarized results from patient-derived xenograft experiments performed by using 4 different primary AML patient samples. Horizontal bar represents the mean of bone marrow AML engraftment level normalized to vehicle control group; each symbol represents an individual mouse, and error bars represent SD. Data represent the mean ± SEM reduction in bone marrow leukemia level relative to the PBS group. Student t test or 1-way analysis of variance was used for statistics. *P < .05; **P < .01; ***P < .001; ****P < .0001. ns, not significant.
Venetoclax increases antileukemic activity of T cells. (A) A total of 189 drugs were added to ex vivo expanded DNTs (50 000 cells per well) in 96 well plates at a final concentration of 400 nM for 18 hours. The compound-treated DNTs were washed and incubated with OCI-AML2 at a 2:1 ratio for 2 hours. AML cell viability was determined by Annexin V using flow cytometry. Data represent the change in cytotoxicity relative to untreated DNT control. (B) DNTs were treated with increasing concentrations of venetoclax for 18 hours followed by coculture for 2 hours with OCI-AML2 and OCI-AML3 at a 2:1 and KG1a at an 8:1 DNT to AML ratio. The viability of AML cells (CD3– CD33+ or CD34+) was determined as described in panel A. Each experiment was done in triplicate, and data represent the mean ± SD specific killing from 1 of 5 independent experiments conducted by using DNTs from different donors. (C) DNTs expanded from 11 donors were untreated or treated with 400 nM venetoclax for 18 hours. Subsequently, they were cultured with OCI-AML2 at a 1:1, 2:1, or 4:1 DNT:AML ratio, and the viability of AML cells was measured by Annexin V staining and flow cytometry. Each paired symbol represents DNTs from an individual donor. (D) Venetoclax 400 nM treated or untreated DNTs were cocultured with primary AML samples (n = 17) at a 2:1 ratio for 2 hours. After incubation, cell viability was measured as described in panel A. Each sample was measured in triplicate, and data represent the changes in AML cell death in the presence of venetoclax-treated DNTs relative to untreated DNTs. DNTs from 6 different donors were used for the screening. (E) OCI-AML2, KG1a, or primary AML cells (100857) were treated with DNT or venetoclax-treated (Ven-treated) DNTs for 18 hours. Equal volumes (105 cells per mL per dish) of cells were seeded in colony-forming assays, and the colonies were counted. Data represent mean ± SD of number of colonies formed. The results for OCI-AML2 and KG1a are representative of 3 independent experiments conducted by using DNTs from 2 different donors. (F) Primary AML cells (ID: 130607) untreated or treated with DNTs or venetoclax-treated DNTs for 2 hours at a 2:1 DNT:AML ratio were injected intrafemorally into NOD/SCID mice (1.6 × 106 cells per mouse; n = 6 per group). Six weeks after injection, the percentage of AML engraftment (human CD45+ CD33+ cells) in the bone marrow from each group was determined by flow cytometry. (G) OCI-AML2, autologous PBMCs, or allogeneic PBMCs (n = 4) were used as targets and cultured with DNTs or venetoclax-treated DNTs for 2 hours at a 2:1 DNT to AML ratio or 8:1 DNT to PBMC ratio. The viability of AML cells and PBMCs was determined by Annexin V. Data represent the mean ± SD specific killing. The results are representative of 2 independent experiments. (H) Ex vivo expanded polyclonally activated CD4+/CD8+ Tconv cells were treated with increasing concentrations of venetoclax for 18 hours. Tconv cells were added as effectors against OCI-AML2, OCI-AML3, and KG1a at a 4:1 target ratio. Two hours after incubation, AML cell viability was determined by Annexin V. Data represent the mean ± SD specific killing from 1 of 5 independent experiments. (I) Sublethally irradiated (250 cGy) NSG mice were injected intravenously with KG1a cells (2 × 106 cells per mouse). Two weeks post-KG1a infusion, mice were treated with 3 infusions of vehicle control (phosphate-buffered saline [PBS]) or 1.5 to 2 × 107 cells per infusion of DNTs or venetoclax-treated DNTs 3 to 4 days apart. Five weeks post–AML injection, bone marrow engraftment of KG1a (human CD45+ CD34+) was determined by using flow cytometry. The result shown is representative of 2 independent experiments conducted by using 2 different donor-derived DNTs. (J) Sublethally irradiated NSG mice were intravenously injected with primary AML cells (n = 4; 2-5 × 106 per mouse). Two weeks later, mice were treated with 3 infusions of vehicle control or 1.5 to 2 × 107 cells per infusion of DNTs or venetoclax-treated DNTs, 3 to 4 days apart. Five weeks post–AML injection, bone marrow engraftment of primary AML cells (human CD45low CD33+ with or without CD34 expression) was determined by flow cytometry. Left: representative contour plot of bone marrow cells from each group stained with CD45 and CD33. Right: summarized results from patient-derived xenograft experiments performed by using 4 different primary AML patient samples. Horizontal bar represents the mean of bone marrow AML engraftment level normalized to vehicle control group; each symbol represents an individual mouse, and error bars represent SD. Data represent the mean ± SEM reduction in bone marrow leukemia level relative to the PBS group. Student t test or 1-way analysis of variance was used for statistics. *P < .05; **P < .01; ***P < .001; ****P < .0001. ns, not significant.
We also tested the effects of venetoclax-treated DNTs on primary AML samples. Venetoclax-treated DNTs showed superior cytotoxicity against 16 of 17 primary AML samples compared with untreated DNTs (Figure 1D); patient information is provided in supplemental Table 1. Notably, 4 samples (090271, 080043, 290985, and 150099) that were resistant to DNTs were effectively killed by venetoclax-treated DNTs. An inverse correlation between the susceptibility of AML to DNTs and the degree of increase in DNT-mediated cytotoxicity by venetoclax treatment was observed (supplemental Figure 2). Venetoclax-treated DNTs were equally effective at killing AML cells from patients at diagnosis and relapsed/refractory after induction chemotherapy (supplemental Figure 3). Venetoclax-treated DNTs also effectively reduced the colony formation of AML cell lines, OCI-AML2 and KG1a, and primary AML cells (Figure 1E). Similarly, a primary AML sample treated ex vivo with venetoclax-treated DNT engrafted fewer than the same cells treated with DNTs alone (Figure 1F).
To determine if venetoclax increases the cytotoxicity of DNTs against normal blood cells, autologous and allogeneic PBMCs from healthy donors were used as targets. In contrast to AML cell lines and primary samples, insignificant cytotoxicity was seen with both DNTs and venetoclax-treated DNTs against autologous and allogeneic PBMCs (Figure 1G), showing that venetoclax selectively increases the cytotoxic activity of DNTs against AML.
We next assessed the effect of venetoclax on Tconv cell–mediated cytotoxicity against AML. Ex vivo expanded Tconv cells were pretreated with venetoclax and used as effectors in cytotoxicity assays. As with DNTs, venetoclax pretreatment significantly increased Tconv cell cytotoxicity against AML cells in a dose-dependent manner (Figure 1H).
To determine the antileukemic activity of DNTs in the presence of venetoclax, KG1a and OCI-AML2 were treated with venetoclax, DNTs, or both. Treating AML cells with both DNTs and venetoclax resulted in a lower number of viable AML cells than either treatment alone (supplemental Figure 4).
Venetoclax enhances T cell–mediated cytotoxicity against AML in vivo
To test whether venetoclax-treated DNTs are effective in vivo, mice xenografted with KG1a were treated with vehicle control, DNT, or venetoclax-treated DNT. Consistent with our previous report,9 DNTs alone had minimal effect on the KG1a xenograft. However, venetoclax-treated DNTs significantly lowered the KG1a engraftment level compared with vehicle control or DNT-treated groups (Figure 1I).
In addition, we examined the effects of venetoclax-treated DNTs on the engraftment of primary AML samples. Mice were injected intravenously with primary AML cells and then treated with DNTs or venetoclax-treated DNTs. Treatment of mice with venetoclax-treated DNTs decreased AML engraftment and counts compared with mice treated with vehicle control or DNTs (Figure 1J; supplemental Figure 5). Importantly, no notable toxicity was observed from these treatments (supplemental Figure 6). Collectively, these results show the superior antileukemic activity of T cells treated with venetoclax.
Venetoclax directly activates effector T cells to increase their cytotoxicity
Increased T-cell cytotoxicity after venetoclax treatment may be due to direct activation of effector T cells or depletion of inhibitory naive and regulatory T cells, thereby enriching effector T-cell populations. Therefore, we tested whether venetoclax directly activates effector T cells or alters the abundance of T-cell subsets. We treated DNTs and Tconv cells with venetoclax for increasing times and measured T-cell activation and T-cell subsets. Treatment of DNTs and Tconv cells with venetoclax for as little as 4 hours and up to 3 days increased T-cell cytotoxicity against AML (Figure 2A) with increased expression of T-cell activation markers (CD69 and CD25) (Figure 2B) and activating receptors (NKG2D and DNAM-1) (Figure 2C) without changing the T-cell viability (Figure 2D) or abundance of T-cell subsets (Figure 2E). Thus, venetoclax directly activates effector T cells to increase their cytotoxicity without depleting naive or inhibitory T-cell subsets.
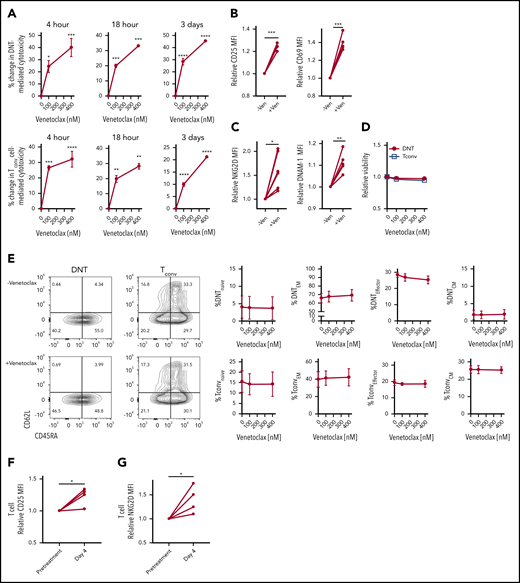
Venetoclax rapidly and directly increases cytotoxicity of T cells against AML. (A) DNT (top panels) and Tconv cells (bottom panels) were untreated or treated with venetoclax (100 nM and 400 nM) for 4 hours, 18 hours, and 3 days. Subsequently, their cytotoxicity against OCI-AML2 was determined. Data represent the mean ± standard error of the mean of results from 4 different donor T cells. (B-C) Median fluorescence intensity (MFI) of T-cell activation markers, CD25 and CD69 (B), activation molecules, NKG2D and DNAM-1 (C) measured on DNTs untreated or treated with venetoclax (400 nM) for 18 hours. (D-E) DNT and Tconv cells untreated or treated with venetoclax (100 nM or 400 nM) for 4 hours. Subsequently, their viability (D) and frequency of T-cell memory subsets (E) were determined. Data represent the mean ± SEM of results from 4 different donor T cells. (F-G) Expression of CD25 (F) and NKG2D (G) of T cells obtained from 4 patients with AML before and at day 4 of venetoclax and azacytidine treatment. Student t test or 1-way analysis of variance was used for statistics. *P < .05; **P < .01; ***P < .001; ****P < .0001.
Venetoclax rapidly and directly increases cytotoxicity of T cells against AML. (A) DNT (top panels) and Tconv cells (bottom panels) were untreated or treated with venetoclax (100 nM and 400 nM) for 4 hours, 18 hours, and 3 days. Subsequently, their cytotoxicity against OCI-AML2 was determined. Data represent the mean ± standard error of the mean of results from 4 different donor T cells. (B-C) Median fluorescence intensity (MFI) of T-cell activation markers, CD25 and CD69 (B), activation molecules, NKG2D and DNAM-1 (C) measured on DNTs untreated or treated with venetoclax (400 nM) for 18 hours. (D-E) DNT and Tconv cells untreated or treated with venetoclax (100 nM or 400 nM) for 4 hours. Subsequently, their viability (D) and frequency of T-cell memory subsets (E) were determined. Data represent the mean ± SEM of results from 4 different donor T cells. (F-G) Expression of CD25 (F) and NKG2D (G) of T cells obtained from 4 patients with AML before and at day 4 of venetoclax and azacytidine treatment. Student t test or 1-way analysis of variance was used for statistics. *P < .05; **P < .01; ***P < .001; ****P < .0001.
Finally, we tested the effects of venetoclax on T cells in patients with AML receiving this therapy. We isolated T cells from patients with AML before and on day 4 of their first cycle of venetoclax and azacytidine. Compared with the pretreatment sample, T cells obtained during treatment showed increased expression of a T-cell activation marker, CD25 (Figure 2F), and a T-cell activating receptor, NKG2D (Figure 2G). Thus, in vitro, in vivo, and in patients, venetoclax directly and rapidly augments the cytotoxicity of T cells against AML.
Venetoclax increases ROS by impairing respiratory chain supercomplex formation to increase T-cell effector function
Venetoclax increases ROS generation in malignant cells,6,13 and ROS plays an important role in the T-cell activation and differentiation.14-17 Therefore, to understand the mechanism by which venetoclax activates T cells, we measured ROS generation in DNT and Tconv cells treated with venetoclax. Venetoclax increased cellular and mitochondrial ROS in DNT and Tconv cells at concentrations and times associated with increased T-cell effector function (Figure 3A-D).
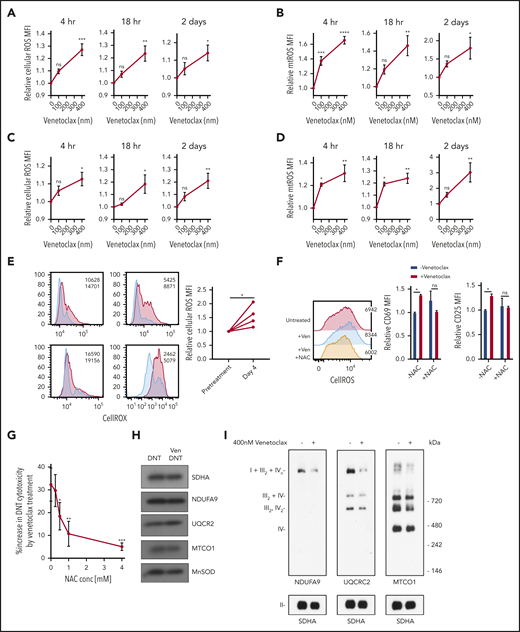
Venetoclax increases ROS level to enhance T-cell effector function. (A-D) DNT (A-B) or Tconv cells (C-D) were treated with 0 nM, 100 nM, or 400 nM venetoclax for 4 hours, 18 hours, and 2 days. Cells were stained with CellROX (A,C) or MitoSOX (B,D). Median fluorescence intensity (MFI) of cellular or mitochondrial (mt) ROS was measured by using flow cytometry. Data represent the mean ± SEM of results from 4 different donor T cells. (E) Cellular ROS level in T cells obtained from 4 patients with AML before and during venetoclax and azacytidine treatment were determined by using flow cytometry. Flow histogram shows the CellROX staining for each patient’s T cells. Blue represents cellular ROS level in T cells from pretreatment samples and red represents those from day 4 of treatment. (F) DNTs treated with 400 nM venetoclax with or without 2 mM NAC for 18 hours. Flow histogram shows the cellular ROS level measured by using flow cytometry. MFI of CD25 and CD69 were measured by flow cytometry. Experiments were done in triplicate, and the data shown are representative of 2 independent experiments conducted by using DNTs from 2 donors. (G) DNTs treated with 400 nM venetoclax in the presence of increasing concentrations of NAC for 18 hours followed by coculture with OCI-AML2 for 2 hours at a 2:1 DNT:AML ratio. Experiments were done in triplicate, and data represent the percent increase in DNT-mediated cytotoxicity by venetoclax ± SD specific killing from 1 of 3 independent experiments done using DNTs from 3 different donors. (H) DNT cells were treated with 400 nM venetoclax for 18 hours. After treatment, mitochondria were isolated and levels of respiratory chain complex subunits were measured by sodium dodecyl sulfate–polyacrylamide gel electrophoresis gels and immunoblotting with antibodies against NDUFB8 (complex I), SDHA (complex II), UQCRC2 (complex III), and MTCO1 (complex IV). (I) Mitochondrial fractions were isolated after DNTs were treated with 400 nM venetoclax for 18 hours. Complex and respiratory chain supercomplex assembly were measured by blue native–polyacrylamide gel electrophoresis with antibodies against NDUFB8 (complex I), SDHA (complex II), UQCRC2 (complex III), and MTCO1 (complex IV). The results shown are representative of 3 independent experiments conducted by using DNTs from 3 different donors. Student t test or 1-way analysis of variance was used for statistics. *P < .05; **P < .01; ***P < .001; ****P < .0001.
Venetoclax increases ROS level to enhance T-cell effector function. (A-D) DNT (A-B) or Tconv cells (C-D) were treated with 0 nM, 100 nM, or 400 nM venetoclax for 4 hours, 18 hours, and 2 days. Cells were stained with CellROX (A,C) or MitoSOX (B,D). Median fluorescence intensity (MFI) of cellular or mitochondrial (mt) ROS was measured by using flow cytometry. Data represent the mean ± SEM of results from 4 different donor T cells. (E) Cellular ROS level in T cells obtained from 4 patients with AML before and during venetoclax and azacytidine treatment were determined by using flow cytometry. Flow histogram shows the CellROX staining for each patient’s T cells. Blue represents cellular ROS level in T cells from pretreatment samples and red represents those from day 4 of treatment. (F) DNTs treated with 400 nM venetoclax with or without 2 mM NAC for 18 hours. Flow histogram shows the cellular ROS level measured by using flow cytometry. MFI of CD25 and CD69 were measured by flow cytometry. Experiments were done in triplicate, and the data shown are representative of 2 independent experiments conducted by using DNTs from 2 donors. (G) DNTs treated with 400 nM venetoclax in the presence of increasing concentrations of NAC for 18 hours followed by coculture with OCI-AML2 for 2 hours at a 2:1 DNT:AML ratio. Experiments were done in triplicate, and data represent the percent increase in DNT-mediated cytotoxicity by venetoclax ± SD specific killing from 1 of 3 independent experiments done using DNTs from 3 different donors. (H) DNT cells were treated with 400 nM venetoclax for 18 hours. After treatment, mitochondria were isolated and levels of respiratory chain complex subunits were measured by sodium dodecyl sulfate–polyacrylamide gel electrophoresis gels and immunoblotting with antibodies against NDUFB8 (complex I), SDHA (complex II), UQCRC2 (complex III), and MTCO1 (complex IV). (I) Mitochondrial fractions were isolated after DNTs were treated with 400 nM venetoclax for 18 hours. Complex and respiratory chain supercomplex assembly were measured by blue native–polyacrylamide gel electrophoresis with antibodies against NDUFB8 (complex I), SDHA (complex II), UQCRC2 (complex III), and MTCO1 (complex IV). The results shown are representative of 3 independent experiments conducted by using DNTs from 3 different donors. Student t test or 1-way analysis of variance was used for statistics. *P < .05; **P < .01; ***P < .001; ****P < .0001.
We also measured ROS levels in T cells isolated from patients receiving venetoclax and azacytidine. Compared with pretreatment levels, T cells isolated on day 4 of the first cycle of venetoclax and azacytidine had higher levels of cellular ROS (Figure 3E).
To determine the functional relevance of increased ROS in venetoclax-treated DNTs, DNTs were cotreated with venetoclax and increasing concentrations of N-acetylcysteine (NAC), a ROS scavenger. Treatment with NAC abrogated venetoclax-induced ROS generation and blocked the upregulation of activation markers (Figure 3F). NAC also blocked venetoclax-mediated increases in DNT-mediated cytotoxicity against AML (Figure 3G). Thus, the upregulation of ROS is a functionally important mechanism by which venetoclax activates T cells. To determine the effect of other ROS-inducing agents on DNT-mediated cytotoxicity, DNTs were treated with increasing concentrations of cytarabine, daunorubicin, and antimycin. We observed increased ROS levels in DNTs treated with cytarabine and antimycin in a dose-dependent manner, with no to little loss of viability (supplemental Figure 7A-B). Daunorubicin-treated DNTs had lower ROS levels with a large reduction in viability. Unlike venetoclax treatment, cytarabine and antimycin did not enhance the cytotoxicity of DNTs despite the increase in cellular ROS level, and daunorubicin reduced DNT-mediated cytotoxicity against AML (supplemental Figure 7C). These data show that the ROS-dependent increase in DNT-mediated cytotoxicity is unique for venetoclax.
To understand the mechanism by which venetoclax increased ROS production, we measured levels of respiratory chain proteins. No change was observed in NDUFA9, UQCRC2, and MTCO1, subunits of electron transport chain complex I, III, and IV, respectively (Figure 3H; supplemental Figure 8). We also observed no changes in basal oxygen consumption, glycolysis, glycolytic capacity, or glutathione levels after venetoclax treatment (supplemental Figure 9).
ROS production is also regulated by formation of respiratory chain supercomplexes, higher order quaternary structures containing respiratory chain complexes I, III, and IV.18,19 Decreased respiratory chain supercomplex formation can increase mitochondrial ROS production without altering other metabolic or mitochondrial parameters.20-22 We therefore measured respiratory chain supercomplex formation in DNTs after venetoclax treatment. Venetoclax reduced formation of respiratory chain supercomplexes (Figure 3I).
Azacytidine sensitizes AML to T cell–mediated cytotoxicity by activating the STING/cGAS pathway and induction of viral mimicry
Recent reports indicate that the combination of venetoclax and azacytidine produces high clinical response rates with low treatment-associated toxicities in patients with newly diagnosed AML.5,6 Azacytidine did not affect the viability or cytotoxic activity of untreated DNTs (supplemental Figure 10A-B). Azacytidine also did not hinder the impact of venetoclax on the cytotoxic activity of DNTs (supplemental Figure 10C). We hypothesized that azacytidine-treated AML cells may have an increased susceptibility to DNT-mediated cytotoxicity, which is further enhanced by venetoclax, leading to a superior T cell–mediated antileukemic efficacy. Indeed, treating AML cell lines with azacytidine increased their susceptibility to DNT-mediated cytotoxicity in a dose-dependent manner (Figure 4A; supplemental Figure 11). Similarly, superior cytotoxicity was seen against 5 of 7 primary AML samples pretreated with azacytidine (Figure 4B). Moreover, we observed that the killing of azacytidine-treated AML was further augmented by treating DNTs with venetoclax (Figure 4C), suggesting that azacytidine and venetoclax may have a synergistic role to increase the potency of T-cell antileukemic function in patients. In contrast, DNTs or venetoclax-treated DNTs did not kill normal hematopoietic cells, and treatment of normal hematopoietic cells with azacytidine did not increase their susceptibility to DNTs or venetoclax-treated DNTs (supplemental Figure 12).
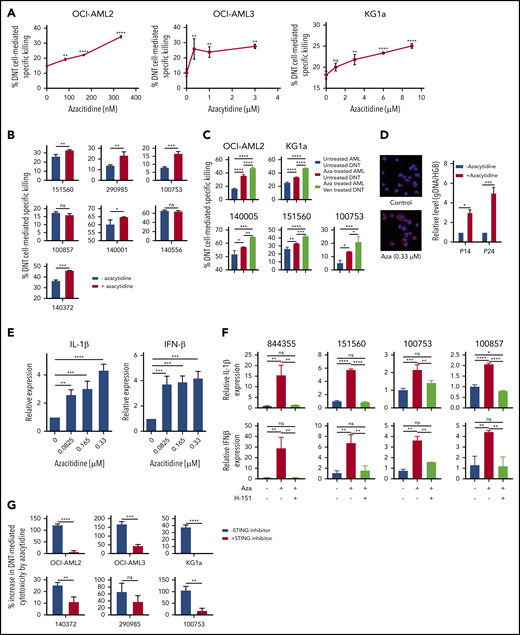
Azacytidine sensitizes AML to DNT-mediated cytotoxicity and produces a viral mimicry response. (A-B) AML cell lines (A; OCI-AML2, OCI-AML3, and KG1a) and primary AML samples (B; n = 7) were treated with azacytidine for 5 days followed by coculture with DNTs at a 2:1 DNT:AML ratio for OCI-AML2, OCI-AML3, and primary AML samples or an 8:1 DNT:AML ratio for KG1a. Two hours after coincubation, percent specific killing of AML cells by DNTs was determined by flow cytometry. (C) AML cell lines (OCI-AML2 and KG1a) and primary AML cells (n = 3) were untreated or treated with azacytidine (0.3-9 μM) for 5 days, followed by coculture with untreated or venetoclax-treated (400 nM for 18 hours) DNTs for 2 hours. AML cell viability was measured by Annexin V and flow cytometry. (D) Left panel: OCI-AML2 cells were untreated or treated with azacytidine (0.33 μM) for 5 days. After the treatment period, cells were cytospun, fixed with 4% paraformaldehyde, and immunostained with anti-dsDNA antibody. DNA was stained by 4′,6-diamidino-2-phenylindole. Right panel: DNA was isolated from the cytoplasmic fraction of the OCI-AML2 cells treated with or without azacytidine. The relative level of cytosolic genomic DNA of transposon origin was analyzed by quantitative polymerase chain reaction by using transposon DNA 14 (P14) and 24 (P24) specific primers and nuclear human globulin gene (HGB). Data represent the mean ± SD relative to untreated cells. (E) OCI-AML2 cells were untreated or treated with azacytidine (0.08-0.33 μM) for 5 days. The relative expression of IL-1β and IFN-1β was analyzed by quantitative reverse transcription polymerase chain reaction. Data are relative mean ± SD (n = 3; untreated = 1.0). (F) Primary AML cells were untreated or treated with azacytidine (3 or 9 μM) for 5 days in the presence or absence of the STING inhibitor H-151. The relative expression of 1L-1β (top) and IFN-1β (bottom) were analyzed by quantitative reverse transcription polymerase chain reaction. The experiments were conducted by using DNTs from 4 different donors. (G) AML cell lines (OCI-AML2, OCI-AML3, and KG1a) and primary AML cells (n = 3) were treated with azacytidine (0.3-3 μM) for 5 days with or without the STING inhibitor (H-151). Data represent the mean ± SD increase in DNT-mediated cytotoxicity by azacytidine-treated AML relative to the untreated control. Student t test or 1-way analysis of variance was used for statistics. *P < .05; **P < .01; ***P < .001; ****P < .0001.
Azacytidine sensitizes AML to DNT-mediated cytotoxicity and produces a viral mimicry response. (A-B) AML cell lines (A; OCI-AML2, OCI-AML3, and KG1a) and primary AML samples (B; n = 7) were treated with azacytidine for 5 days followed by coculture with DNTs at a 2:1 DNT:AML ratio for OCI-AML2, OCI-AML3, and primary AML samples or an 8:1 DNT:AML ratio for KG1a. Two hours after coincubation, percent specific killing of AML cells by DNTs was determined by flow cytometry. (C) AML cell lines (OCI-AML2 and KG1a) and primary AML cells (n = 3) were untreated or treated with azacytidine (0.3-9 μM) for 5 days, followed by coculture with untreated or venetoclax-treated (400 nM for 18 hours) DNTs for 2 hours. AML cell viability was measured by Annexin V and flow cytometry. (D) Left panel: OCI-AML2 cells were untreated or treated with azacytidine (0.33 μM) for 5 days. After the treatment period, cells were cytospun, fixed with 4% paraformaldehyde, and immunostained with anti-dsDNA antibody. DNA was stained by 4′,6-diamidino-2-phenylindole. Right panel: DNA was isolated from the cytoplasmic fraction of the OCI-AML2 cells treated with or without azacytidine. The relative level of cytosolic genomic DNA of transposon origin was analyzed by quantitative polymerase chain reaction by using transposon DNA 14 (P14) and 24 (P24) specific primers and nuclear human globulin gene (HGB). Data represent the mean ± SD relative to untreated cells. (E) OCI-AML2 cells were untreated or treated with azacytidine (0.08-0.33 μM) for 5 days. The relative expression of IL-1β and IFN-1β was analyzed by quantitative reverse transcription polymerase chain reaction. Data are relative mean ± SD (n = 3; untreated = 1.0). (F) Primary AML cells were untreated or treated with azacytidine (3 or 9 μM) for 5 days in the presence or absence of the STING inhibitor H-151. The relative expression of 1L-1β (top) and IFN-1β (bottom) were analyzed by quantitative reverse transcription polymerase chain reaction. The experiments were conducted by using DNTs from 4 different donors. (G) AML cell lines (OCI-AML2, OCI-AML3, and KG1a) and primary AML cells (n = 3) were treated with azacytidine (0.3-3 μM) for 5 days with or without the STING inhibitor (H-151). Data represent the mean ± SD increase in DNT-mediated cytotoxicity by azacytidine-treated AML relative to the untreated control. Student t test or 1-way analysis of variance was used for statistics. *P < .05; **P < .01; ***P < .001; ****P < .0001.
Previously, azacytidine has been reported to evoke a viral mimicry response in colon and ovarian cancer cells by increasing levels of double-stranded DNA(dsDNA)/double-stranded RNA in the cytoplasm.23,24 To determine whether a similar mechanism renders AML more susceptible to T cell–mediated killing, we measured cytoplasmic levels of dsDNA, transposon DNA 14 (P14) and 24 (P24), after azacytidine treatment.25,26 Azacytidine-treated AML cells had higher dsDNA levels in the cytoplasm (Figure 4D) and increased expression of type I interferons, interferon-β (IFN-β) and IL-1β (Figure 4E).
Next, we queried whether the induction of viral mimicry after azacytidine treatment was functionally important to explain the increased susceptibility of AML to DNT-mediated cytotoxicity. Primary AML cells were treated with azacytidine in the presence of the STING inhibitor N-(4-ethylphenyl)-N′-1H-indol-3-yl-urea (H-151). The addition of the STING inhibitor abrogated azacytidine-induced increases in IFN-β and IL-1β expression (Figure 4F). Importantly, cotreatment with the STING inhibitor abrogated azacytidine-mediated increases in susceptibility to DNTs in 5 of 6 primary AML samples tested (Figure 4G). Thus, these data show that azacytidine sensitizes AML to T cell–mediated cytotoxicity, at least partially, through the activation of the STING/cGAS pathway and induction of viral mimicry.
Patient T cells enhance the antileukemic effect of venetoclax and azacytidine
Although the combination of venetoclax and azacytidine is highly effective in treatment-naive patients, it is less effective in patients who received prior induction chemotherapy.7,8 We hypothesized that the function of T cells may differ in samples from patients with AML after relapse/no response to induction chemotherapy compared with samples from newly diagnosed patients. Using single-cell analysis of secreted cytokines,12,27 the cytokine profile and pSI of CD8+ T cells from patients with newly diagnosed and relapsed AML stimulated ex vivo with IL-2 were determined as a marker of T-cell functionality. The cytokine profile and pSI of CD8+ T cells from patients with newly diagnosed AML and healthy control subjects were similar. In contrast, CD8+ T cells from patients with relapsed AML were dysfunctional, with decreased pSI and lower levels of effector and stimulatory cytokines (supplemental Figure 13).
To determine the role of T cells in venetoclax and azacytidine combination therapy, PBMCs obtained from patients with AML were depleted or enriched for autologous CD3+ T cells (supplemental Figure 14) and then treated with venetoclax and azacytidine. We observed a greater reduction in viable AML cell counts in T cell–enriched samples compared with T cell–depleted samples (Figure 5A), supporting the role of patient T cells in the antileukemic effect of venetoclax and azacytidine.
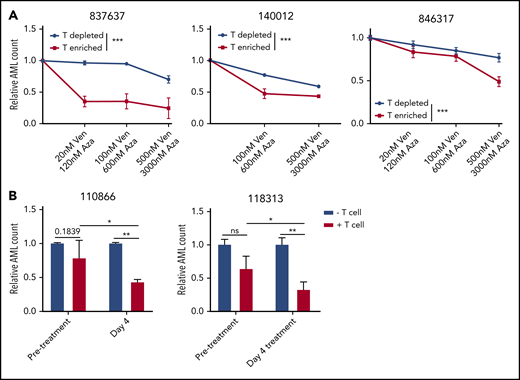
T cells influence cytotoxicity of venetoclax and azacytidine in AML. (A) PBMCs obtained from 3 patients with AML (837637, 140012, and 846317) were depleted or enriched for autologous T cells, followed by treatment with increasing concentrations of venetoclax (0-500 nM) and azacytidine (0-3000 nM) (1 day for 140012 or 2 days for 837637 and 846317). The cells were stained with Annexin V, and the relative number of viable AML cells was determined by flow cytometry. (B) AML cells obtained from patients (110866 and 118313) before venetoclax and azacytidine treatment were cultured with or without autologous T cells isolated from the same patient with AML before or on day 4 of venetoclax and azacytidine treatment (day 4). The cells were stained with Annexin V, and the relative numbers of viable AML cells was determined by flow cytometry. Student t test or 2-way analysis of variance was used for statistics. *P < .05; **P < .01; ***P < .001.
T cells influence cytotoxicity of venetoclax and azacytidine in AML. (A) PBMCs obtained from 3 patients with AML (837637, 140012, and 846317) were depleted or enriched for autologous T cells, followed by treatment with increasing concentrations of venetoclax (0-500 nM) and azacytidine (0-3000 nM) (1 day for 140012 or 2 days for 837637 and 846317). The cells were stained with Annexin V, and the relative number of viable AML cells was determined by flow cytometry. (B) AML cells obtained from patients (110866 and 118313) before venetoclax and azacytidine treatment were cultured with or without autologous T cells isolated from the same patient with AML before or on day 4 of venetoclax and azacytidine treatment (day 4). The cells were stained with Annexin V, and the relative numbers of viable AML cells was determined by flow cytometry. Student t test or 2-way analysis of variance was used for statistics. *P < .05; **P < .01; ***P < .001.
To test whether the presence of T cells is important for the effectiveness of venetoclax and azacytidine in vivo, A20 murine leukemia/lymphoma cells were injected subcutaneously into immunocompetent BALB/c mice. Mice were then treated with venetoclax and azacytidine with or without preceding CD4+/CD8+ T-cell depletion. Treatment with venetoclax and azacytidine reduced tumor growth more effectively in mice with intact T cells than that of T cell–depleted mice (supplemental Figure 15). Collectively, these results suggest the role of endogenous T cells in the antileukemic effect, which can be enhanced by venetoclax and azacytidine.
Finally, T cells were isolated from patients with AML before and on day 4 of venetoclax and azacytidine treatment. Subsequently, patient autologous blasts were cocultured with the isolated T cells in the presence of IL-2 to stimulate T cells. The number of viable AML cells were measured after coculture. T cells obtained during venetoclax and azacytidine treatment were more cytotoxic to the leukemic cells compared with T cells isolated from the pretreatment samples of the same patients (Figure 5B). Collectively, these results support the importance of T-cell immunity in facilitating the antileukemic effect induced by the combination therapy of venetoclax and azacytidine.
Discussion
The combination of venetoclax and azacytidine produces responses in up to 80% of newly diagnosed patients with AML. The response is rapid, with a median time to best response of 2.1 months,5 suggesting a unique mechanism of action for the combination therapy. However, the mechanism of action of the drug combination is not yet fully understood. We discovered that venetoclax directly increased the antileukemic effector function of T cells, whereas azacytidine renders AML cells more susceptible to T cells by inducing a viral mimicry response in leukemic cells.
In this study, DNTs were used as a surrogate for leukemia-specific T cells. Although DNTs share similar effector function as Tconv cells, such as perforin-granzyme mediated cytotoxicity and release of IFN-γ and TNF-α upon activation, these cells are not alloreactive. As such, they provide a good model to study the effects of venetoclax and azacytidine on T-cell function. However, we recognize that extrapolating the findings of DNTs to Tconv cells should be done with caution. Because DNTs can be expanded ex vivo and have been safely administered to patients with relapsed and refractory AML,28 there may be an opportunity to combine DNT therapy with venetoclax and azacytidine to further enhance their antileukemic activity.
DNTs selectively target AML partly through NKG2D and DNAM-110 and synergize with conventional chemotherapy, which induces expression of NKG2D and DNAM-1 ligands on AML.9 In this study, we observed increased NKG2D and DNAM-1 expression on DNTs and CD8+ T cells post–venetoclax treatment. Given the involvement of NKG2D and DNAM-1 in functioning of various effector immune cell subsets,29-31 our results suggest that venetoclax may also enhance the antileukemic activity of other T-cell or natural killer–cell therapy products.
Venetoclax increased ROS generation in T cells, leading to increased effector activity. Although high levels of ROS can be toxic, moderate levels of ROS act as signaling messengers in T-cell activation and differentiation.32,33 We showed that therapeutically relevant concentrations of venetoclax increased T-cell effector function without decreasing T-cell viability. We also showed that T cells isolated from patients receiving venetoclax also had increased levels of ROS. ROS enhances the localization of nuclear factor of activated T cells to increase expression of genes associated with T-cell activation,32 thus potentially linking the increased ROS to enhanced T-cell activity.
Metabolic reprogramming toward oxidative phosphorylation is a common source of ROS production. However, venetoclax did not alter basal levels of oxygen consumption, glycolysis, levels of respiratory chain complex proteins, or levels of antioxidants in T cells. Instead, venetoclax disrupted respiratory chain supercomplex formation. The function of respiratory supercomplexes is not fully understood, but previous studies have reported that these complexes have multiple roles, including ensuring efficient oxidative phosphorylation19 and preventing ROS formation.18,20,21 Recently, we showed that inhibiting respiratory chain supercomplex formation decreases oxidative phosphorylation in AML cells without increasing ROS production.34 Our current results indicate that respiratory chain supercomplexes have different functions in different cells or tissues. Future studies will be required to understand how Bcl-2 controls the formation of respiratory chain supercomplexes, but the effect of venetoclax suggests that supercomplex formation in T cells may depend on the structural integrity of the inner mitochondrial membrane.
Azacytidine induces viral mimicry in tumors and makes them more immunogenic.23,24 Despite the routine use of azacytidine to treat AML, the effects of azacytidine on the viral mimicry response in AML has not been reported. Here, we showed that azacytidine primes AML cells for killing by T cells by inducing a viral mimicry response. Azacytidine elevated cytoplasmic dsDNA/double-stranded RNA levels and activated the STING-cGAS pathway to induce a type I IFN response, similar to the response to a viral infection. Due to pleotropic functions of type I IFN,35 how the viral mimicry response renders AML more susceptible to T cells remains to be elucidated.
A recent study by Pollyea et al4 indicated that venetoclax and azacytidine synergize to inhibit respiratory chain complex II activity and impair oxidative phosphorylation. As AML cells and stem cells have increased reliance on oxidative phosphorylation, targeting mitochondrial pathways and respiratory chain complex activity may lead to preferential killing of AML cells.4,6 Our data do not contradict these previous findings but offer an additional mechanism of action for the combination therapy.
Although the combination of venetoclax and azacytidine is highly effective in patients newly diagnosed with AML, it is less effective in relapsed and refractory patients.7,8 Induction chemotherapy lowers the number of T cells in patients.36-39 Furthermore, increased T-cell exhaustion has been reported in nonresponders to chemotherapy.40 Thus, the T-cell dysfunction in patients with AML who relapse after chemotherapy may explain why these patients are less responsive to venetoclax and azacytidine. Although our data provide a new and intriguing mechanism of action for venetoclax and azacytidine, we recognize the limitations to our study, including the limited number of patient samples studied and the majority of experiments conducted either in vitro or ex vivo. In future studies, characterizing T cells from a large number of patients who are responders and nonresponders to venetoclax and azacytidine treatment may help identify biomarkers that could be used stratifying those most and least likely to respond to this therapy.
In summary, our data highlight a new immune-mediated mechanism of action for venetoclax in promoting T-cell effector function. These data shed light on how venetoclax could be used to increase antileukemic activity of T-cell therapies and provide new insights to explain the antileukemic activity of the combination of venetoclax and azacytidine.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
E-mail the corresponding authors for data sharing requests.
The full-text version of this article contains a data supplement.
Acknowledgments
The authors thank all the donors and patients who participated in this study. They also thank Jill Flewelling (Princess Margaret Cancer Centre) for administrative assistance and the Leukemia Tissue Bank (Princess Margaret Cancer Centre) for providing the primary AML samples, and IsoPlexis for assistance in generating the single-cell cytokine data.
This work was supported by Canadian Cancer Society Impact Grant 704121, Canadian Institutes of Health Research Grant 419699, and Maria H. Bacardi Chair in Transplantation (all to L.Z.); the Ontario Institute for Cancer Research with funding provided by the Ontario Ministry of Research and Innovation, the Princess Margaret Cancer Centre Foundation, and the Ministry of Long Term Health and Planning in the Province of Ontario (all to A.D.S.); and the National Institutes of Health, National Cancer Institute (P30 CA16672), Cancer Prevention & Research Institute of Texas (CPRIT) RP160693, and Haas Chair in Genetics (all to M.A.).
Authorship
Contribution: J.B.L., D.H.K., L.Z., and A.D.S. conceived and designed the study; J.B.L., D.H.K., R.H., M.X., Y.N., H.K., S.M., X.W., M.G., Y.J., N.M., and Z.A. performed the experiments; J.B.L., D.H.K., M.X., S.M., and M.G. analyzed and interpreted the data; M.Y.K., M.D.M., and A.A. provided AML patient samples for the study; Z.A. and M.A. performed single-cell cytokine analysis; J.B.L. wrote the manuscript; D.H.K., M.G., M.D.M., L.Z., and A.D.S. provided feedback and edited the manuscript; M.A., L.Z., and A.D.S. provided funding support; and all authors read and approved the final manuscript.
Conflict-of-interest disclosure: A.D.S. has received research funding from Takeda Pharmaceuticals and Medivir AB; has consulting fees/honorarium from Takeda, Novartis, Jazz, and Otsuka Pharmaceuticals; and holds stock in AbbVie. M.D.M. is a consultant for Astellas, AbbVie, and Celgene. L.Z. received funding and consulting fee/honorarium from WYZE Biotech Co Ltd; and holds stock in AbbVie. A.D.S., J.B.L., and L.Z. are inventors on patent applications claiming the use of DNTs for the treatment of AML. J.B.L. and L.Z. are cofounders of Denote Therapeutics. The remaining authors declare no competing financial interests.
Correspondence: Aaron D. Schimmer, University Health Network, Princess Margaret Cancer Research Tower, 101 College St, Room 8-706, Toronto, ON M5G 1L7, Canada; e-mail: aaron.schimmer@utoronto.ca; or Li Zhang, University Health Network, Princess Margaret Cancer Research Tower, 101 College St, Room 2-207, Toronto, ON M5G 1L7, Canada; e-mail: li.zhang@uhnresearch.ca.
