

Key Points
Chd8 gene targeting in mice causes HSPC apoptosis and hematopoiesis failure.
CHD8 organizes chromatin to maintain proper ATM-P53 cell survival signaling in HSPCs.

Abstract
The Chd8 gene encodes a member of the chromodomain helicase DNA-binding (CHD) family of SNF2H-like adenosine triphosphate (ATP)-dependent chromatin remodeler, the mutations of which define a subtype of autism spectrum disorders. Increasing evidence from recent studies indicates that ATP-dependent chromatin-remodeling genes are involved in the control of crucial gene-expression programs in hematopoietic stem/progenitor cell (HSPC) regulation. In this study, we identified CHD8 as a specific and essential regulator of normal hematopoiesis. Loss of Chd8 leads to severe anemia, pancytopenia, bone marrow failure, and engraftment failure related to a drastic depletion of HSPCs. CHD8 forms a complex with ATM and its deficiency increases chromatin accessibility and drives genomic instability in HSPCs causing an activation of ATM kinase that further stabilizes P53 protein by phosphorylation and leads to increased HSPC apoptosis. Deletion of P53 rescues the apoptotic defects of HSPCs and restores overall hematopoiesis in Chd8−/− mice. Our findings demonstrate that chromatin organization by CHD8 is uniquely necessary for the maintenance of hematopoiesis by integrating the ATM-P53–mediated survival of HSPCs.
Introduction
Hematopoiesis is a tightly regulated process that constantly generates a large number of mature blood cells through a cascade differentiation of hematopoietic stem/progenitor cells (HSPCs). The longevity and integrity of the stem cell pool governs the lifelong homeostasis of the hematopoietic system.1 The adult HSPCs are maintained in the bone marrow (BM) and are capable of dramatic expansion and contraction to ensure proper homeostatic replacement of blood cells upon stress signals.2,3 At the cellular level, well-maintained chromatin organization is critical to HSPC health and function, as defective chromatin structure can be susceptible to physiological stress resulting in DNA damage and genomic instability. Mice lacking components and regulators of the chromosomes and genomic stability display severe hematopoietic phenotypes and HSPC defects.4-10
Adenosine triphosphate (ATP)-dependent chromatin remodeling enzymes play an essential role in stem cell biology.11 These enzymes use the energy from ATP hydrolysis to regulate DNA accessibility, which enables transcription regulators to bind, to activate or repress gene expression.12 Among the evolutionarily conserved SWI-like ATP-dependent chromatin remodeling complexes, the CHD remodeling enzymes are defined by the presence of 2 chromodomains: a helicase/ATPase domain and a DNA-binding domain.11,13 Genetic, biochemical, and structural studies demonstrate that CHD proteins are important regulators of transcription and are indispensable during developmental processes.14,15 Several CHD proteins, including CHD3, CHD4, and CHD7, have been found to be involved in hematopoiesis.16-20
CHD8 was first identified as a negative regulator of the Wnt/β-catenin signaling pathway and is one of the most frequently mutated genes involved in the autism spectrum disorders.21-23 Recent studies have focused on its role in neurogenesis and brain development.24,25 CHD8 appears to be essential for B acute lymphoblastic leukemia cell survival and correlates with Brd4-NSD3 across the genome in acute myeloid leukemia cells.26,27 In the current study, we conditionally deleted Chd8 in hematopoietic cells using the Mx1-Cre driver and found CHD8 essential for the maintenance of normal hematopoiesis, different from its role in most other tissues. Loss of CHD8 led to severe pancytopenia and BM failure because of its essential role in regulating HSPC survival. Although CHD8 is not a DNA damage response and repair cue, it binds directly to P53 and ATM to restrict their function, and loss of CHD8 causes a change in chromatin integrity resulting in ATM activation and consequent P53 activation, leading to cell apoptosis. We implicate the chromatin remodeling factor CHD8 as an essential controller of HSPC survival and hematopoiesis.
Materials and Methods
Mice
Chd8Flox mice and their genotyping were reported previously,24 and P53Flox mice were purchased from The Jackson Laboratory (#008462). To induce hematopoietic cell–specific deletion, Chd8Flox and P53Flox mice were crossed with Mx1-Cre mice. All animal experiments were performed in accordance with the protocols from the Institutional Animal Care and Use Committee at Cincinnati Children’s Hospital Medical Center. For Cre-mediated recombination, 6- to 8-week-old control and Cre-expressing animals were injected with polyinosinic/polycytidylic acid (pI/pC) (15 mg/kg; GE Healthcare) 4 times, and BM cells were harvested for analysis 6 days, 10 days, and 8 weeks after the last injection. pI/pC-treated Chd8F/F mice were considered wild type (WT), and pI/pC-treated Chd8F/F;Mx1-Cre mice that lost Chd8 alleles were considered to be Chd8−/−.
BM transplantation
For direct transplantation, 2 × 106 fresh, isolated BM cells from Chd8F/F and Chd8−/− mice (CD45.2) were transplanted into lethally irradiated wild-type mice (CD45.1). For competitive transplantation, 1 × 106 fresh, isolated BM cells from donors (CD45.2) were mixed 1:1 with WT (CD45.1) cells and transplanted into lethally irradiated WT recipients (CD45.1). BM chimerism was monitored by fluorescence-activated cell sorting in peripheral blood (PB) at 4 weeks, and then the mice were injected 4 times with pI/pC (GE Healthcare) at a dose of 15 mg/kg. Chimerism was assessed every 4 weeks for 16 weeks by cell sorting. For a secondary transplant, 2 × 106 BM cells from primary recipient mice were transplanted into lethally irradiated WT recipients (CD45.1). The mice were euthanized 16 weeks after transplantation and used for flow cytometry.
HSPCs in vitro culture
Low-density BM cells were harvested from the Chd8F/F and Chd8−/− mice using Histopaque-1083 and then cKit+ cells were further enriched with CD117 microbeads (Miltenyi Biotec). The number of cells was determined, and the cells were cultured in Iscove’s modified Dulbecco’s medium (IMDM; Thermo Fisher Scientific) supplemented with a final concentration of 10% fetal bovine serum (FBS), 100 ng/mL stem cell factor (PeproTech), 100 ng/mL troponin (PeproTech), and 100 ng/mL granulocyte colony?stem cell factor (G-CSF; PeproTech), with or without specific inhibitors or stress treatment. The final concentration of chemicals was 10 μM MG132 (#S2619; Selleck Chemicals), 10 μg/mL cycloheximide (#01810; MilliporeSigma), 1 μM and 10 μM KU-55933 (#SML1109; MilliporeSigma), and 5 μM etoposide (#33419-42-0; Cayman Chemical). The dose of irradiation was 3 Gy. After culture, the cells were further harvested for analysis.
Ubiquitination assay
The ubiquitination assay was performed according to a published protocol.28 The low-density BMs were cultured in Iscove’s modified Dulbecco’s medium for 4 hours with 10 M MG132 and harvested by centrifuge. The cells were lysed in cell lysis buffer (2% sodium dodecyl sulfate, 150 mM NaCl, and 10 mM Tris-HCl [pH 8.0]) with inhibitors, and the protein was harvested by centrifuge. Protein concentration was determined with Quick Start Bradford (Bio-Rad) and similar amounts of proteins across samples were used for P53 immunoprecipitation. The elution was further blotted with a ubiquitin antibody to detect ubiquitinated P53.
Statistical analysis
Statistical analyses were performed with GraphPad Prism software. All data are shown as means ± standard error of the mean. The 2-tailed Student t test was used to determine statistical significance.
Results
Conditional deletion of Chd8 leads to acute BM failure and pancytopenia
Chd8 is found expressed in all subpopulations of HSPCs by reverse transcription-polymerase chain reaction (RT-PCR) (supplemental Figure 1A, available on the Blood Web site). To determine whether Chd8 plays a role in hematopoiesis, we crossed Chd8Flox mice with an Mx1-Cre driver and conditionally deleted Chd8 by pI/pC induction. After pI/pC injection, Chd8F/F mice were considered to be WT and Chd8F/F;Mx1-Cre mice were considered to be Chd8−/− (Figure 1A). In Chd8−/− mice, Chd8 messenger RNA (mRNA) in whole-BM cells and CHD8 protein in the sorted hematopoietic progenitors were found to be efficiently deleted 6 days after pI/pC administration (Figure 1B; supplemental Figure 1B). PB cell analyses showed that Chd8 depletion led to pancytopenia (Figure 1C). Hematoxylin and eosin staining of femur sections showed that Chd8−/− mice had BM failure, with the total BM cellularity remarkably reduced compared with the WT control (Figure 1D-E). Consistently, we observed a decreased number of mature hematopoietic cells in the BM of Chd8−/− mice (Figure 1F). As the likely result of severe anemia, Chd8−/− mice showed deficient survival (supplemental Figure 1C). The spleen weight was also reduced in Chd8−/− mice, but the white pulp structure in the spleen appeared normal (supplemental Figure 1D). These data indicate that CHD8 is critical for hematopoiesis and an acute loss of CHD8 results in rapid BM failure.
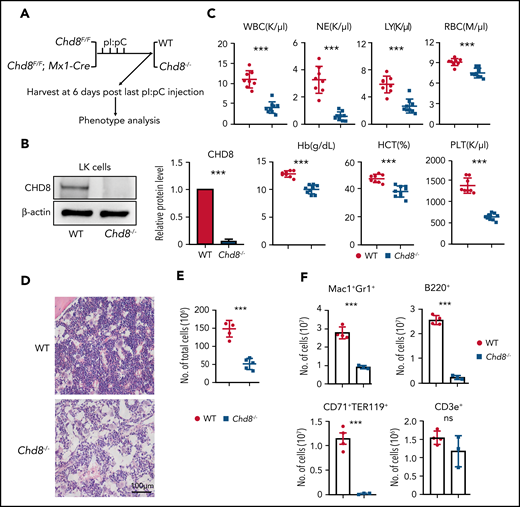
Conditional loss of Chd8 in hematopoietic cells leads to hematopoiesis and BM failure. (A) Schematic model of CHD8 deletion in hematopoietic cells. Chd8Flox mice were crossed with Mx1-Cre mice, and deletion was induced by 4 pI/pC injections. (B) Western blot confirmation of CHD8 protein loss after 6 days of pI/pC injections in BM progenitors. The quantification represents data from 3 repeat experiments. (C) PB counts of Chd8Flox and Chd8Flox;Mx1-Cre mice after 6 days of pI/pC induction by Hemavet. ***P < .001. (D) Representative H&E staining of femurs from WT and Chd8−/− mice which were harvested at 6 days after deletion. WT indicates Chd8F/F mice. (E) Total BM cells count with Hemavet after 6 days of pI/pC injections. (F) Number of mature lineage cells in WT vs Chd8−/− mice. WT vs Chd8−/− (E-F). ***P < .001.
Conditional loss of Chd8 in hematopoietic cells leads to hematopoiesis and BM failure. (A) Schematic model of CHD8 deletion in hematopoietic cells. Chd8Flox mice were crossed with Mx1-Cre mice, and deletion was induced by 4 pI/pC injections. (B) Western blot confirmation of CHD8 protein loss after 6 days of pI/pC injections in BM progenitors. The quantification represents data from 3 repeat experiments. (C) PB counts of Chd8Flox and Chd8Flox;Mx1-Cre mice after 6 days of pI/pC induction by Hemavet. ***P < .001. (D) Representative H&E staining of femurs from WT and Chd8−/− mice which were harvested at 6 days after deletion. WT indicates Chd8F/F mice. (E) Total BM cells count with Hemavet after 6 days of pI/pC injections. (F) Number of mature lineage cells in WT vs Chd8−/− mice. WT vs Chd8−/− (E-F). ***P < .001.
Depletion of CHD8 causes a drastic loss of HSPCs and hematopoiesis failure
To examine how the rapid BM failure in Chd8−/− mice occurs, we used flow cytometry to analyze hematopoietic progenitors. We found that CHD8-deficient mice had drastically fewer Lineage−cKit+Sca1+ (LSK) and Lineage−cKit+ (LK) cell populations (Figure 2A-B). Moreover, LSK cells combined with CD150 and CD48 analysis showed that SLAM HSCs (LSK CD150+CD48−) and multipotent progenitors MPP2 (LSK CD150+CD48+) and MPP3 (LSK CD150−CD48+) were profoundly depleted after CHD8 loss (Figure 2D-E). The common myeloid progenitors (CMPs, LK CD34+CD16/32−), granulocyte-macrophage progenitors (GMPs; LK CD34+CD16/32+), and megakaryocyte-erythroid progenitors (MEPs; LK CD34−CD16/32−) were nearly absent from Chd8−/− BM (supplemental Figure 2A-B). Consistent with the loss of HSPCs in Chd8−/−, we detected little colony-forming activity after Chd8 depletion (Figure 2C; supplemental Figure 2C). These results indicate that CHD8 is essential for the maintenance of HSPCs.
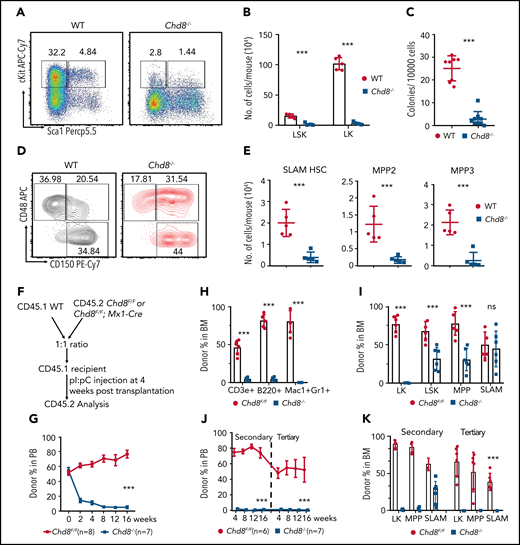
Depletion of CHD8 causes a drastic loss of HSPCs. (A) Flow cytometry of LSK and LK cells from WT and Chd8−/− mice after 6 days of pI/pC injection. Representative dot plots and frequencies of the HSPC subpopulations in lineage− cells are shown. (B) WT indicates Chd8F/F mice. Absolute number of LSK and LK cells from WT and Chd8−/− mice after 6 days of pI/pC injections. ***P < .001. (C) Number of colonies after 7-day plating. Total BM cells were harvested 6 days after pI/pC injection and plated in MethoCult medium. ***P < .001. (D) Immunophenotypic analysis of SLAM HSC (LSK CD150+CD48−), MPP2 (LSK CD150+CD48+), and MPP3 (LSK CD150−CD48+) cells from WT and Chd8−/− BM. Averaged frequencies of HSPC subpopulations in LSKs are shown. (E) Absolute number of SLAM HSC, MPP2, and MPP3 cells from WT and Chd8−/− mice. **P < .01; ***P < .001. (F) The experimental scheme of the native competitive transplant into CD45.1 WT recipient. (G) Percentage of CD45.2 cells at different time points in the PB after pI/pC injection in the successfully reconstituted mice. Statistical analysis was performed at 16 weeks after deletion. ***P < .001. (H) Percentage of donor-derived mature lineage cells after 4 months of deletion. ***P < .001. (I) Percentage of CD45.2 BM progenitors in the transplant-recipient mice after 4 months of pI/pC injections. **P < .01; ***P < .001. ns, not significant. (J) The proportion of CD45.2-derived cells in PB in the secondary and tertiary transplants. Statistical analysis was performed at 16 weeks after transplantation. ***P < .001. (K) The proportion of CD45.2-derived BM progenitors at 4 months in the secondary and tertiary transplants. ***P < .001.
Depletion of CHD8 causes a drastic loss of HSPCs. (A) Flow cytometry of LSK and LK cells from WT and Chd8−/− mice after 6 days of pI/pC injection. Representative dot plots and frequencies of the HSPC subpopulations in lineage− cells are shown. (B) WT indicates Chd8F/F mice. Absolute number of LSK and LK cells from WT and Chd8−/− mice after 6 days of pI/pC injections. ***P < .001. (C) Number of colonies after 7-day plating. Total BM cells were harvested 6 days after pI/pC injection and plated in MethoCult medium. ***P < .001. (D) Immunophenotypic analysis of SLAM HSC (LSK CD150+CD48−), MPP2 (LSK CD150+CD48+), and MPP3 (LSK CD150−CD48+) cells from WT and Chd8−/− BM. Averaged frequencies of HSPC subpopulations in LSKs are shown. (E) Absolute number of SLAM HSC, MPP2, and MPP3 cells from WT and Chd8−/− mice. **P < .01; ***P < .001. (F) The experimental scheme of the native competitive transplant into CD45.1 WT recipient. (G) Percentage of CD45.2 cells at different time points in the PB after pI/pC injection in the successfully reconstituted mice. Statistical analysis was performed at 16 weeks after deletion. ***P < .001. (H) Percentage of donor-derived mature lineage cells after 4 months of deletion. ***P < .001. (I) Percentage of CD45.2 BM progenitors in the transplant-recipient mice after 4 months of pI/pC injections. **P < .01; ***P < .001. ns, not significant. (J) The proportion of CD45.2-derived cells in PB in the secondary and tertiary transplants. Statistical analysis was performed at 16 weeks after transplantation. ***P < .001. (K) The proportion of CD45.2-derived BM progenitors at 4 months in the secondary and tertiary transplants. ***P < .001.
To determine the blood cell–intrinsic effects of CHD8 depletion, we harvested Chd8F/F and Chd8−/− BM cells 6 days after pI/pC injection and performed BM transplantation. Lethally irradiated recipients receiving CHD8-deficient BM cells died within 3 weeks (supplemental Figure 2D), manifesting a failure in HSPC engraftment and BM repopulation in the absence of CHD8. This is further evidenced by the observation that no donor CD45.2+Chd8−/− cells were detected in 1:1 WT vs Chd8−/−-competitive BM transplantations (supplemental Figure 2E-F). When a competitive BM transplantation was performed with an equal number of BM cells from CD45.2+Chd8Flox;Mx1-Cre mice, Chd8Flox littermates, and CD45.1 WT BM cells, to fully engraft in the lethally irradiated recipients, followed by pI/pC injection (Figure 2F), it is clear that CD45.2+ CHD8-deficient cells were rapidly outcompeted by CD45.1 WT cells over a 16-week period (Figure 2G). Analyses of the mature blood cells from these mice showed a dramatic loss of CHD8-deficient donor cells compared with Chd8F/F cells (Figure 2H). In addition, the LK, LSK, and MPP subpopulations of the CD45.2+, CHD8-deficient cells were drastically reduced (Figure 2I). In the secondary and tertiary transplants, all Chd8−/− cells were outcompeted by the WT competitors (Figure 2J-K). Moreover, we performed reciprocal transplantation by transplanting CD45.1 WT cells into lethally irradiated Chd8Flox or Chd8Flox;Mx1-Cre mice, followed by pI/pC injection. No detectable phenotypes from PB or BM cells were observed (supplemental Figure 2G-I). These results show that the hematopoietic failure caused by CHD8 loss is related to the intrinsic defects of CHD8-deficient HSPCs.
In mice, CHD8 haploinsufficiency could present autisticlike phenotypes. However, CHD8 haploinsufficiency in hematopoietic cells did not yield detectable phenotypes in the PB (supplemental Figure 3A). Transplantation or competitive transplantation of Chd8+/− BM cells into WT recipients resulted in no significant changes in hematopoiesis compared with the Chd8F/+ control (supplemental Figure B-G), indicating that CHD8 heterozygosity is insufficient to cause blood defects in mice.

CHD8 deletion causes increased HSPC apoptosis. (A) Immunophenotypic of SLAM HSC apoptosis by annexin V and 7-AAD staining at 6 days after deletion. Averaged frequencies of cells are shown. WT indicates Chd8F/F mice. (B) Graph showing the percentage of annexin V+ and apoptotic (annexin V+ 7-AAD−) cells in SLAM HSCs. *P < .05; ***P < .001. (C) Ratio of annexin V+ cells in the LK population. ***P < .001. (D) Western blot of cleaved caspase 3 in sorted LK cells. The quantification represents data from 3 repeat experiments. (E) Cell cycle analysis by flow cytometry of Ki67 and 4′,6-diamidino-2-phenylindole in sorted LSK cells. Averaged frequencies of subpopulations are shown. (F) Percentage of LSK cells in G0, G1, and S-G2-M phase in WT and Chd8−/− mice. ***P < .001, **P < .01. (G) Cell cycle analysis by flow cytometry of Ki67 and 7-AAD in sorted SLAM-HSCs. Averaged frequencies of subpopulations are shown. (H) Percentages of SLAM HSC cells in G0, G1, and S-G2-M phase in WT and Chd8−/− mice are shown. **P < .01; *P < .05.
CHD8 deletion causes increased HSPC apoptosis. (A) Immunophenotypic of SLAM HSC apoptosis by annexin V and 7-AAD staining at 6 days after deletion. Averaged frequencies of cells are shown. WT indicates Chd8F/F mice. (B) Graph showing the percentage of annexin V+ and apoptotic (annexin V+ 7-AAD−) cells in SLAM HSCs. *P < .05; ***P < .001. (C) Ratio of annexin V+ cells in the LK population. ***P < .001. (D) Western blot of cleaved caspase 3 in sorted LK cells. The quantification represents data from 3 repeat experiments. (E) Cell cycle analysis by flow cytometry of Ki67 and 4′,6-diamidino-2-phenylindole in sorted LSK cells. Averaged frequencies of subpopulations are shown. (F) Percentage of LSK cells in G0, G1, and S-G2-M phase in WT and Chd8−/− mice. ***P < .001, **P < .01. (G) Cell cycle analysis by flow cytometry of Ki67 and 7-AAD in sorted SLAM-HSCs. Averaged frequencies of subpopulations are shown. (H) Percentages of SLAM HSC cells in G0, G1, and S-G2-M phase in WT and Chd8−/− mice are shown. **P < .01; *P < .05.
CHD8 is essential for HSPC survival and cell cycle progression
To examine the underlying mechanism of HSPC loss in Chd8−/− mice, we performed annexin V staining of the BM cells. There were significantly elevated annexin V+ SLAM HSC and LK cells after deletion of CHD8, which accounted for up to 80% of dying cells (Figure 3A-C). Western blot of cleaved caspase 3 in sorted LK cells showed dramatically increased apoptosis in Chd8−/− cells compared with WT cells (Figure 3D). In addition, we observed increased apoptosis in mature Mac1+Gr1+ cells, but not in B220+ cells (supplemental Figure 4A-B). These results show that CHD8 is essential for the survival of HSPCs.
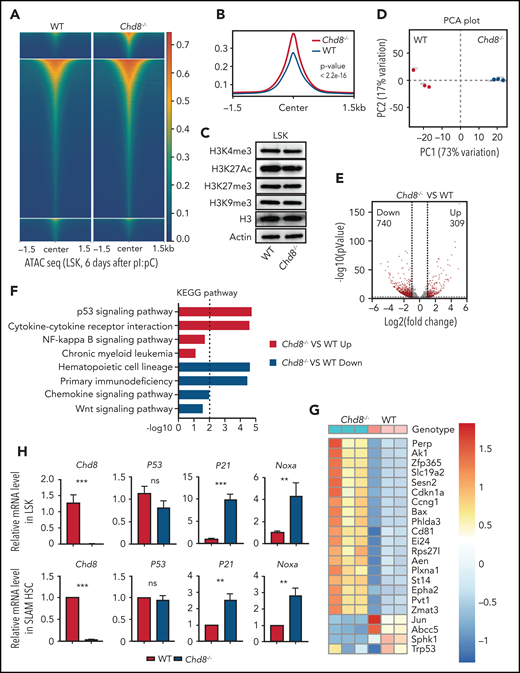
CHD8 is essential for suppressing P53 signaling in HSPCs. (A) Heat maps for ATAC-seq peaks from WT and Chd8−/− LSKs showing ±1.5 kb around the ATAC-seq peak center. Results are from 2 biological replicates. WT represents Chd8F/F mice. (B) Tag enrichment of accessible genomic regions relative to ATAC-seq peaks. Results are from 2 biological replicates. (C) Western blot of histone modification markers in WT and Chd8−/− HSPCs. (D) PCA plot of RNA-seq data from WT and Chd8−/− LSKs. (E) Differential expression of transcripts between WT and Chd8−/− LSKs. Log2-fold change >1.0; P < .05. (F) KEGG enrichment of DEGs from RNA-seq showed increased P53 signaling after loss of CHD8, using DAVID (Database for Annotation, Visualization, and Integrated Discovery). (G) Heat map of differentially expressed P53 downstream genes between WT and Chd8−/− LSKs. (H) RT-PCR of selective P53 target genes in WT and Chd8−/− LSKs and SLAM HSCs. The data represent 3 biological replicates. **P < .01; ***P < .001. ns, not significant.
CHD8 is essential for suppressing P53 signaling in HSPCs. (A) Heat maps for ATAC-seq peaks from WT and Chd8−/− LSKs showing ±1.5 kb around the ATAC-seq peak center. Results are from 2 biological replicates. WT represents Chd8F/F mice. (B) Tag enrichment of accessible genomic regions relative to ATAC-seq peaks. Results are from 2 biological replicates. (C) Western blot of histone modification markers in WT and Chd8−/− HSPCs. (D) PCA plot of RNA-seq data from WT and Chd8−/− LSKs. (E) Differential expression of transcripts between WT and Chd8−/− LSKs. Log2-fold change >1.0; P < .05. (F) KEGG enrichment of DEGs from RNA-seq showed increased P53 signaling after loss of CHD8, using DAVID (Database for Annotation, Visualization, and Integrated Discovery). (G) Heat map of differentially expressed P53 downstream genes between WT and Chd8−/− LSKs. (H) RT-PCR of selective P53 target genes in WT and Chd8−/− LSKs and SLAM HSCs. The data represent 3 biological replicates. **P < .01; ***P < .001. ns, not significant.
Next, we examined the proliferation of the Chd8−/− HSPCs by Ki67 staining. CHD8 depletion led to a loss of quiescence in LSK cells, as there are more Chd8−/− cells in the G1 and S-G2-M phases compared with WT control (Figure 3E-F). Ki67 staining of SLAM HSCs and MPPs further showed that CHD8 is necessary to maintain HSC and MPP quiescence (Figure 3G-H; supplemental Figure 4C), which is probably caused by a compensatory response to the Chd8−/− caused by BM failure and pancytopenia.
To rule out a possible effect of pI/pC on HSPC survival, we examined the mice at a later time points after pI/pC injections. At 10 days, the hematopoietic phenotypes were similar to those observed at 6 days after pI/pC (supplemental Figure D-F), and the Chd8−/− HSPCs still showed dramatic apoptosis (supplemental Figure 4G). The pancytopenia of Chd8−/− mice extend to 8 weeks after pI/pC injection as the cell death effect in LK cells (supplemental Figure 4H-I). The surviving HSPCs showed a differentiation block and cell cycle progression block (supplemental Figure 4J-K). In addition, in vitro knockdown or Cre transduction assays found a significant increase of apoptosis and/or reduction in colony-forming activity in Chd8-deficient LSKs (supplemental Figure 5A-E). Administration of an IFNAR1 mAb to the cell culture did not restore the colony-forming unit–defects in the Chd8−/− BM cells (supplemental Figure 5F).
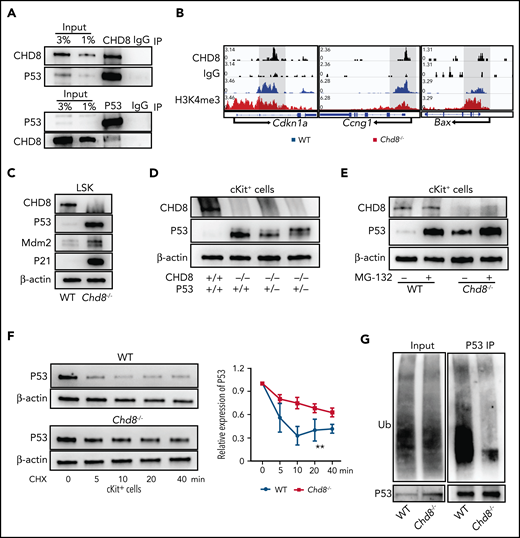
CHD8 depletion increases P53 protein stability. (A) CHD8 interacts with P53 protein in hematopoietic progenitor–like HPC7 cells. IgG immunoprecipitation as a negative control. (B) Representative CHD8 and H3K4me3 CUT&RUN tracks of Cdkn1a, Ccng1, and Bax in WT and Chd8−/− LSK cells. WT represents Chd8F/F mice. (C) Protein level of P53 and its targets in sorted LSKs. β-Actin was the loading control. (D) Western blot of P53 in cKit+ cells of WT, Chd8−/−, and Chd8−/−P53+/− mice. β-Actin was the loading control. (E) Comparison of P53 protein level in WT and Chd8−/− cKit+ cells after 4 hours of MG132 treatment. Final MG132 concentration, 10 µM. (F) CHX treatment of cKit+ cells to determine the degradation rate of P53 protein in WT and Chd8−/− mice. β-Actin was the loading control. Ratio of P53 protein level compared with 0 minutes of treatment with CHX. Final CHX concentration, 10 µg/mL. The exposure times of P53 between WT and Chd8−/− cells varied because of different P53 expression levels and were normalized to that before CHX treatment. Statistical analysis was made with 3 biological replicates. **P < .01. (G) Western blot of ubiquitination of P53 in WT and Chd8−/− low-density BM cells after 10 µM MG132 block for 4 hours.
CHD8 depletion increases P53 protein stability. (A) CHD8 interacts with P53 protein in hematopoietic progenitor–like HPC7 cells. IgG immunoprecipitation as a negative control. (B) Representative CHD8 and H3K4me3 CUT&RUN tracks of Cdkn1a, Ccng1, and Bax in WT and Chd8−/− LSK cells. WT represents Chd8F/F mice. (C) Protein level of P53 and its targets in sorted LSKs. β-Actin was the loading control. (D) Western blot of P53 in cKit+ cells of WT, Chd8−/−, and Chd8−/−P53+/− mice. β-Actin was the loading control. (E) Comparison of P53 protein level in WT and Chd8−/− cKit+ cells after 4 hours of MG132 treatment. Final MG132 concentration, 10 µM. (F) CHX treatment of cKit+ cells to determine the degradation rate of P53 protein in WT and Chd8−/− mice. β-Actin was the loading control. Ratio of P53 protein level compared with 0 minutes of treatment with CHX. Final CHX concentration, 10 µg/mL. The exposure times of P53 between WT and Chd8−/− cells varied because of different P53 expression levels and were normalized to that before CHX treatment. Statistical analysis was made with 3 biological replicates. **P < .01. (G) Western blot of ubiquitination of P53 in WT and Chd8−/− low-density BM cells after 10 µM MG132 block for 4 hours.
Loss of CHD8 leads to increased chromatin accessibility and P53 activation in HSPCs
CHD8 is an ATP-dependent chromatin remodeling enzyme that has roles in regulating chromatin accessibility. To determine whether CHD8 regulates chromatin accessibility in HSPCs, we performed assay for transposase-accessible chromatin sequencing (ATAC-Seq) with sorted LSK cells from Chd8F/F and Chd8−/− mice. We found a globally increased genomic accessibility after CHD8 loss (Figure 4A-B). We also performed CHD8 CUT&RUN (cleavage under targets & release using nuclease) assay in WT LSK cells and found that most of the peaks were in the gene body regions (supplemental Figure 6A-B). Chd8−/− LSK cells showed increased chromatin accessibility at CHD8-binding sites compared with the Chd8F/F cells (supplemental Figure 6C-D). Whereas previous work has found that CHD8 could recruit H3K4 histone methyltransferase to orchestrate oligodendrocyte lineage progression,24 we did not detect any significant change in H3K4me3 or H3K27me3 modification in CHD8-depleted LSK cells in a CUT&RUN assay (supplemental Figure 6E-F). Consistent with these observations, we did not observe any changes at the protein level in the H3K4me3, H3K27Ac, and H3K27me3 chromatin markers caused by Chd8 depletion (Figure 4C; supplemental Figure 7A).
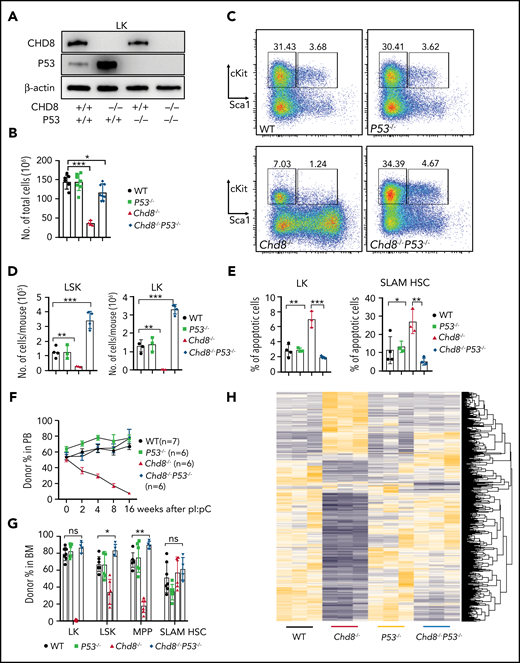
P53 loss fully rescues HSPC survival and hematopoiesis defects caused by CHD8 deletion. (A) Efficient deletion of CHD8 or P53 shown by western blot analysis in Chd8−/−, P53−/−, and Chd8−/−P53−/− BM progenitors. (B) Total cellularity of BM cells in WT, Chd8−/−, P53−/−, and Chd8−/−P53−/− mice after 6 days of pI/pC injections. *P < .05; ***P < .001. WT represents Chd8F/FP53F/F mice. (C) Flow cytometry of LSK and LK cells in WT, Chd8−/−, P53−/−, and Chd8−/−P53−/− BM. Representative dot plots and frequencies of the HSPC subpopulations in lineage− cells are shown. (D) Absolute number of LSK and LK cells from WT, Chd8−/−, P53−/−, and Chd8−/−P53−/− mice after 6 days of pI/pC injections. **P < .01; ***P < .001. (E) Percentage of apoptotic (annexin V+; 4′,6-diamidino-2-phenylindole−) SLAM HSCs and LKs in WT, Chd8−/−, P53−/−, and Chd8−/−P53−/− mice. ***P < .001; **P < .01; *P < .05. (F) Percentage of CD45.2 cells at different time points in the PB after pI/pC injection in successfully reconstituted mice. (G) Percentage of CD45.2 BM progenitors in the transplant-recipient mice after 4 months of pI/pC injections. *P < .05; **P < .01; ns, not significant. (H) Heat map of all differentially expressed transcripts in WT, Chd8−/−, P53−/−, and Chd8−/−P53−/− LSK RNA sequencing.
P53 loss fully rescues HSPC survival and hematopoiesis defects caused by CHD8 deletion. (A) Efficient deletion of CHD8 or P53 shown by western blot analysis in Chd8−/−, P53−/−, and Chd8−/−P53−/− BM progenitors. (B) Total cellularity of BM cells in WT, Chd8−/−, P53−/−, and Chd8−/−P53−/− mice after 6 days of pI/pC injections. *P < .05; ***P < .001. WT represents Chd8F/FP53F/F mice. (C) Flow cytometry of LSK and LK cells in WT, Chd8−/−, P53−/−, and Chd8−/−P53−/− BM. Representative dot plots and frequencies of the HSPC subpopulations in lineage− cells are shown. (D) Absolute number of LSK and LK cells from WT, Chd8−/−, P53−/−, and Chd8−/−P53−/− mice after 6 days of pI/pC injections. **P < .01; ***P < .001. (E) Percentage of apoptotic (annexin V+; 4′,6-diamidino-2-phenylindole−) SLAM HSCs and LKs in WT, Chd8−/−, P53−/−, and Chd8−/−P53−/− mice. ***P < .001; **P < .01; *P < .05. (F) Percentage of CD45.2 cells at different time points in the PB after pI/pC injection in successfully reconstituted mice. (G) Percentage of CD45.2 BM progenitors in the transplant-recipient mice after 4 months of pI/pC injections. *P < .05; **P < .01; ns, not significant. (H) Heat map of all differentially expressed transcripts in WT, Chd8−/−, P53−/−, and Chd8−/−P53−/− LSK RNA sequencing.
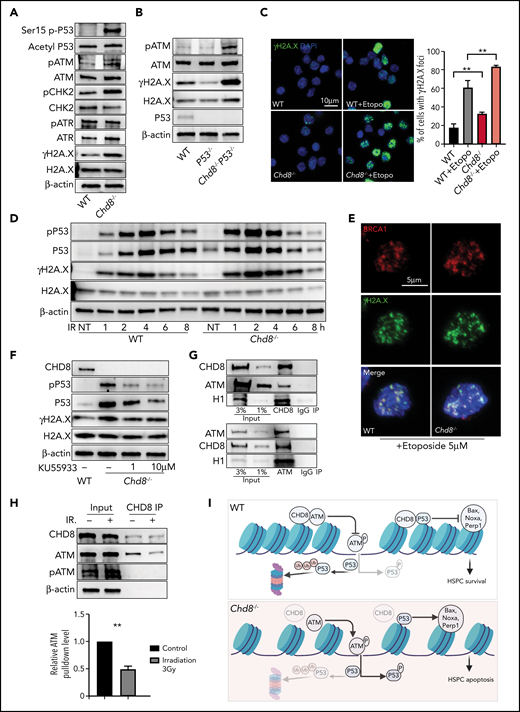
CHD8 regulates genomic stability of HSPCs by restricting the ATM-p53 pathway. (A) Western blot of p-P53 S15, acetyl-P53 K379, and damage-response proteins in Lin−cKit+ cells of WT and Chd8−/− mice. (B) Increased DNA damage response cannot be rescued by additional loss of P53. (C) Immunofluorescence staining of γH2A.X showed increased DNA damage foci in LSK cells after loss of CHD8. Etoposide treatment for 1 hour. The graph shows the mean ± standard error of the mean of 3 biological replicates. **P < .01. (D) CHD8-deficient cells are more sensitive to irradiation-induced DNA damage. Lin−cKit+ cells were irradiated at a dose of 3 Gy and harvested after various recovery times. NT, no treatment. (E) Colocalization of BRCA1 and γH2A.X in WT and Chd8−/− LSK cells. Etoposide treatment for 1 hour. (F) ATM inhibitor KU55933 treatment decreased the P53 and p-P53 in CHD8-depleted HSPCs. Cells were harvested after 3 hours of treatment. (G) CHD8 interacts with ATM in HPC7 cells. (H) CHD8 immunoprecipitation under irradiation in HPC7 cells. The relative ATM pulldown level is normalized to that of the CHD8 pulldown level and represents data from 3 biological repeats. **P < .01. (I) Model of CHD8’s function in restricting P53 signaling and maintaining genomic integrity in HSPCs. The figure was created with BioRender (https://biorender.com/).
CHD8 regulates genomic stability of HSPCs by restricting the ATM-p53 pathway. (A) Western blot of p-P53 S15, acetyl-P53 K379, and damage-response proteins in Lin−cKit+ cells of WT and Chd8−/− mice. (B) Increased DNA damage response cannot be rescued by additional loss of P53. (C) Immunofluorescence staining of γH2A.X showed increased DNA damage foci in LSK cells after loss of CHD8. Etoposide treatment for 1 hour. The graph shows the mean ± standard error of the mean of 3 biological replicates. **P < .01. (D) CHD8-deficient cells are more sensitive to irradiation-induced DNA damage. Lin−cKit+ cells were irradiated at a dose of 3 Gy and harvested after various recovery times. NT, no treatment. (E) Colocalization of BRCA1 and γH2A.X in WT and Chd8−/− LSK cells. Etoposide treatment for 1 hour. (F) ATM inhibitor KU55933 treatment decreased the P53 and p-P53 in CHD8-depleted HSPCs. Cells were harvested after 3 hours of treatment. (G) CHD8 interacts with ATM in HPC7 cells. (H) CHD8 immunoprecipitation under irradiation in HPC7 cells. The relative ATM pulldown level is normalized to that of the CHD8 pulldown level and represents data from 3 biological repeats. **P < .01. (I) Model of CHD8’s function in restricting P53 signaling and maintaining genomic integrity in HSPCs. The figure was created with BioRender (https://biorender.com/).
To elucidate the mechanism driving the HSPC loss, we performed RNA-seq in LSK cells of WT and Chd8−/− mice. Triplicate samples from each genotype are well correlated (Figure 4D): more than 1000 genes are differentially expressed between WT and Chd8−/− cells (Figure 4E). A decreased HSC signature and increased interferon α response pathway in Chd8−/− mice were detected, but Wnt signaling appeared normal (Figure 4F; supplemental Figure 6G). Among the differential pathways seen in a Kyoto Encyclopedia Genes and Genomes (KEGG) analysis, P53 signaling was highly enriched and significantly increased in the Chd8−/− HSPCs (Figure 4F; supplemental Figure 6G). The heat map of P53 target genes shows a clear pattern of the p53 signatures (Figure 4G). Further reverse transcription polymerase chain reaction (RT-PCR) analysis of LSK cells and SLAM HSCs found highly elevated P53 target genes, including P21 and Noxa but no change in P53 mRNA level (Figure 4H).
CHD8 depletion increases P53 protein stability
CHD8 could interact with P53 and suppress its transactivation during embryonic development.29 To examine whether CHD8 plays a similar role in hematopoietic cells, we performed CHD8 immunoprecipitation in HPC7 cells and found that CHD8 formed a complex with P53 (Figure 5A). The CUT&RUN analysis of primary LSK cells shows that CHD8 binds directly to the gene body of Cdkn1a and a promotor region of Ccng1 (Figure 5B), suggesting that CHD8 plays a role in repressing P53 transactivation of its targets in HSPCs.
Interestingly, we found that the P53 protein level in CHD8-deficient LSK cells increased massively (Figure 5C; supplemental Figure 7B). Deletion of 1 allele of the P53 gene was not sufficient to restore the elevated P53 protein level in the Chd8−/− cells (Figure 5D; supplemental Figure 7C). Alternatively, the excessive P53 protein in Chd8−/− HSPCs could have been due to increased protein stability. To examine this possibility, we treated BM progenitors with MG132, a proteasome inhibitor and found that protease inhibition could significantly stabilize P53 in WT progenitors, but with a reduced effect on Chd8−/− cells (Figure 5E; supplemental Figure 7D). In addition, cycloheximide (CHX) blockage of P53 translation showed that P53 protein degradation in Chd8−/− progenitors was significantly slower (Figure 5F; supplemental Figure 7E). Consistently, the ubiquitinated P53 was also significantly reduced in Chd8−/− low-density BM cells (Figure 5G; supplemental Figure 7F). We also examined the effect of CHD8 deletion in CD45−CD31+ BM endothelial cells and liver cells and found that CHD8 loss did not affect cell survival, the P53 protein level, or genomic stability of these cells from the same Mx1-Cre;Chd8F/F mice used for the hematopoiesis studies (supplemental Figure 7G-L). Together, these results demonstrate that CHD8 loss leads to increased P53 protein stability and elevated P53 in HSPCs.
P53 loss readily rescues survival defects of Chd8−/− HSPCs and BM failure of Chd8−/− mice
To determine the role of P53 in HSPCs in the Chd8-controlled hematopoiesis, we crossed P53Flox mice to Chd8Flox;Mx1-Cre mice and generated P53;Chd8 compounding deletion by pI/pC injection. CHD8 and P53 protein can be efficiently deleted in these mice (Figure 6A). Whereas deletion of 1 allele of P53 did not rescue the apoptosis of HSPCs (data not shown), deletion of both alleles of P53 readily restored cell counts of Mac1+Gr1+ and B220+ cells because of CHD8 loss (supplemental Figure 8B). In addition, total BM cellularity in the CHD8-depleted mice was significantly restored (Figure 6B; supplemental Figure 8A).
Next, we analyzed hematopoietic progenitors of the mice by flow cytometry. The numbers of both LK and LSK cells were restored in Chd8−/−P53−/− BM (Figure 6C), whereas the total cell counts of these populations showed an overcompensation compared with WT in Chd8−/−P53−/− mice (Figure 6D). The counts of committed progenitors and HSCs were also restored by P53 deletion (supplemental Figure 8D-F), even though the percentages of CMP and GMP in LK cells remained relatively lower. Consistent with these genotypic observations, the colony-forming unit activity of the in Chd8−/− BM cells also recovered in the Chd8−/−P53−/− mice (supplemental Figure 8C). Functionally, the survival defects of SLAM HSC and LK cells were fully rescued by P53 deletion (Figure 6E). Competitive BM transplantations showed that the deletion of P53 in CD45.2+Chd8Flox;Mx1-Cre mice enabled an effective engraftment of the CHD8-depleted HSPCs with the chimerism in PB and progenitors restored to that of WT (Figure 6F-G). Moreover, RNA-seq analysis of Chd8−/−P53−/− LSK cells showed that the P53 deletion mostly restored global gene expression in the Chd8−/− cells (Figure 6H; supplemental Figure 8G). However, there remain ∽200 differentially expressed genes between WT and Chd8−/−P53−/− LSK cells, which may contribute to the overcompensation of some progenitors (supplemental Figure 9A-B). Thus, P53 regulated cell survival plays a central role in CHD8-mediated HSPC function and hematopoiesis.
CHD8 regulates HSPC genomic stability by restricting ATM and P53 activities
Posttranslational regulation of P53 is important for p53 protein stability. Phosphorylation and acetylation of P53 could interrupt its binding to MDM2 and suppress P53 degradation. In the Chd8−/− cells, we observed a significant increase of p-P53 at serine 15, whereas acetyl P53 K379 remained unchanged (Figure 7A; supplemental Figure 9C). In parallel, we saw an increased p-ATM, p-CHK2, and γH2A.X, but not p-ATR or reactive oxygen species (Figure 7A, supplemental Figure 10A-C), indicating an activated ATM signaling pathway in Chd8−/− cells. Moreover, the increased p-ATM and DNA damage marker γH2A.X in Chd8−/− cells was not rescued by compounding P53 deletion (Figure 7B; supplemental Figure 9D), suggesting that CHD8 suppresses genomic instability independent of p53. Consistent with increased γH2A.X expression, we observed increased γH2A.X foci in Chd8−/− progenitors (Figure 7C). During chemostress, this phenomenon was exaggerated, producing a more intense γH2A.X immunofluorescence in Chd8−/− progenitors (Figure 7C). To determine if CHD8 is involved in DNA damage response, we irradiated WT and CHD8-null cells at a 3-Gy dose and harvested cells after various times of recovery. The DNA damage response in CHD8 deleted cells, as indicated by p-P53 and γH2A.X expression, appears to be similar to that of WT cells in a 6-hour recovery period (Figure 7D). In addition, we found that the recruitment of BRCA1 to γH2A.X foci (Figure 7E), along with several other DNA damage repair factors, were not affected in Chd8−/− progenitors (supplemental Figure 10D-F). The γH2A.X level was also repaired in Chd8−/−P53−/− progenitors (supplemental Figure 10G). These data show that, although CHD8 is required maintain genomic integrity, it is not involved in DNA damage repair.
To examine whether activated ATM contributes to the increased P53 protein level, we administered an ATM inhibitor, KU55933, to BM progenitors. KU55933 significantly decreases p-P53 and P53 levels dose dependently in the Chd8−/− cells (Figure 7F; supplemental Figure 9E), suggesting that the elevated ATM activity in CHD8-cKO cells is responsible for P53 elevation. To better understand how ATM is involved, we performed a coimmunoprecipitation test with HPC7 cells and found that CHD8 and ATM directly interact with each other in either a CHD8 immunoprecipitation or an ATM immunoprecipitation (Figure 7G). Moreover, the activated p-ATM appeared to lose interaction with CHD8 upon irradiation (Figure 7H). A knockdown of CHD8 in these cells mimicked the DNA damage response effects with increased pP53 and γH2A.X expression as in Chd8−/− HSPCs (supplemental Figure 10H). Thus, CHD8 forms a complex with both ATM and p53 and maintains genomic stability by integrating the ATM-p53 signaling.
Discussion
CHD8 is an ATP-dependent chromatin remodeling enzyme that is essential for brain development, and its mutations represent a subclass of autism in humans,22,24 but its role in hematopoiesis remains unknown. In this study, we found that loss of CHD8 led to dramatic loss of HSPCs, because of increased apoptosis driven by P53 activation, and loss of p53 rescued the survival defects in Chd8−/− HSPCs. Moreover, our findings revealed that CHD8 is essential for maintaining the genomic integrity of HSPCs by forming a complex with ATM and p53 and regulating ATM-p53 mediated signaling and cell survival. We demonstrated that CHD8 is essential for normal hematopoiesis.
Aside from neurodevelopmental defects, autism is associated with anemia and other blood disorders.30,31 To date, no information is available for the underlying mechanism of autism-associated blood abnormalities beyond disease-related food intake/iron deficiencies.32 In a subpopulation of patients with autism, heterozygous Chd8 mutations, mostly loss of function changes, are considered functionally significant. Although we observed severe BM failure in Chd8−/− mice, we did not detect significant hematopoietic phenotypes in mice with a 1-allele deletion of Chd8, perhaps because of mouse vs human differences or involvement of other compounding genetic factors in autism. Nonetheless, our finding of an essential function of CHD8 in hematopoiesis raises a possible mechanism of the blood cell intrinsic role of Chd8 in autism beyond defective food intake and iron deficiencies.
HSPC survival is a critical function for the maintenance of the stem/progenitor cell pool, and in this study, CHD8 was found to be essential for this process. One of the well-known functions of CHD8 is as a negative regulator of P53.29 Indeed, our results showed overactivation of P53 signaling in Chd8−/− mice. Consistent with previous findings, we observed a direct binding between P53 and CHD8 in HSPCs and found that CHD8 was recruited to P53 target promotors, such as Cdkn1a and Ccng1. More interestingly, we observed dramatic accumulation of P53 protein in Chd8−/− HSPCs, without a detectable mRNA change. Further analysis showed that CHD8 also interacted with ATM, and loss of CHD8 led to activation of the ATM signaling pathway, which resulted in increased P53 phosphorylation and decreased P53 ubiquitination. The role of CHD8 in P53 protein stabilization has not been seen in other cell types, including U2OS osteosarcoma and endothelial cells. We propose a model in which, during normal hematopoiesis, CHD8 maintains the HSPC cell survival by integrating ATM and p53 to restrict P53 protein stability and regulate P53 transactivation (Figure 7I). Consistent with what we observed in HSPCs, CHD8 contributes to B-ALL cell survival,27 suggesting an essential role of CHD8 in maintaining hematopoietic cell survival. CHD8 is also a negative regulator of the Wnt-β catenin signaling pathway in multiple cell types other than blood lineages,33,34 but we did not observe significant changes of this pathway in HSPCs, suggesting that CHD8 involvement in restricting Wnt pathway does not apply to HSPCs.
Chromatin structure and dynamics are key factors in the maintenance of genomic integrity,35 and the CHD family chromatin remodeling enzymes are emerging players.36 Unlike the function of CHD8 in chromatin opening in oligodendrocyte progenitors,24 we detected globally increased chromatin accessibility after CHD8 loss in HSPCs by ATAC-seq, suggesting that CHD8 is important for chromatin compaction rather than opening. The relaxation of chromatin may be causal to the increased sensitivity to a DNA damage response in Chd8−/− HSPCs. Multiple CHD proteins have been found to act in regulating DNA damage response and repair.37-41 In our study, however, unlike other CHD proteins, the DNA damage repair response appears normal in Chd8−/− HSPCs as indicated by decreased γH2A.X after irradiation and the timely recovery of DNA damaged foci in both Chd8−/− and Chd8−/−P53−/− HSPCs. This suggests that CHD8 is mainly required for chromatin integrity but is not involved in DNA damage repair per se. Our and other studies have established that the biological role and the p53 stabilization/ATM sequestration mechanism of CHD8 is specific to HSPCs as CHD8 loss in BM endothelial cells, and liver cells, and other lineages do not yield similar effects, as in blood cells.42-44
Overall, our study discovered an HSPC-specific role of CHD8 in regulating hematopoiesis by maintaining chromatin integrity and genomic stability through a CHD8-ATM-p53 complex.
All the next generation sequencing data have been deposited in the NCBI Gene Expression Omnibus (accession number GEO:GSE161613).
Investigators can access materials and protocols by contacting the corresponding author.
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
There is a Blood Commentary on this article in this issue.
Acknowledgments
The authors thank James Johnson for offering technical assistance; the Research Flow Cytometry Core, the Comprehensive Mouse and Cancer Core, Biomedical Informatics Core, DNA Core, and the Pathology Core at Cincinnati Children’s Hospital Medical Center for their services and technical support.
This work was partially supported by National Institutes of Health, National Cancer Institute grants R01CA234038 and R01CA204895.
Authorship
Contribution: Z.T., Q.R.L., and Y.Z. designed the study; Z.T. performed the experiments, interpreted the data, and generated the figures; C.W., A.K.D., M.H., C.Z., and M.X. performed the experiments; Z.T. and Y.Z. wrote the manuscript; and Y.Z. provided funding support.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Yi Zheng, Cincinnati Children‘s Hospital Medical Center, 3333 Burnet Ave, Cincinnati, OH 45229; e-mail: yi.zheng@cchmc.org.
