

Key Points
nm-PMBLsig+ tumors represent a distinct group of DLBCLs that share molecular features with bf-PMBL.
Mutational patterns and anatomic presentations suggest distinct evolutionary modes between nm-PMBLsig+ tumors and bf-PMBL.

Abstract
Primary mediastinal large B-cell lymphoma (PMBL) is a type of aggressive B-cell lymphoma that typically affects young adults, characterized by presence of a bulky anterior mediastinal mass. Lymphomas with gene expression features of PMBL have been described in nonmediastinal sites, raising questions about how these tumors should be classified. Here, we investigated whether these nonmediastinal lymphomas are indeed PMBLs or instead represent a distinct group within diffuse large B-cell lymphoma (DLBCL). From a cohort of 325 de novo DLBCL cases, we identified tumors from patients without evidence of anterior mediastinal involvement that expressed a PMBL expression signature (nm-PMBLsig+; n = 16; 5%). A majority of these tumors expressed MAL and CD23, proteins typically observed in bona fide PMBL (bf-PMBL). Evaluation of clinical features of nm-PMBLsig+ cases revealed close associations with DLBCL, and a majority displayed a germinal center B cell–like cell of origin (GCB). In contrast to patients with bf-PMBL, patients with nm-PMBLsig+ presented at an older age and did not show pleural disease, and bone/bone marrow involvement was observed in 3 cases. However, although clinically distinct from bf-PMBL, nm-PMBLsig+ tumors resembled bf-PMBL at the molecular level, with upregulation of immune response, JAK-STAT, and NF-κB signatures. Mutational analysis revealed frequent somatic gene mutations in SOCS1, IL4R, ITPKB, and STAT6, as well as CD83 and BIRC3, with the latter genes significantly more frequently affected than in GCB DLBCL or bf-PMBL. Our data establish nm-PMBLsig+ lymphomas as a group within DLBCL with distinct phenotypic and genetic features. These findings may have implications for gene expression– and mutation-based subtyping of aggressive B-cell lymphomas and related targeted therapies.
Introduction
Aggressive B-cell lymphomas account for >40% of newly diagnosed B-cell non-Hodgkin lymphoma (NHL) cases worldwide. Among these, diffuse large B-cell lymphoma (DLBCL) is by far the most common. It is a biologically and clinically heterogeneous disease that shows a wide range of clinical outcomes after standard treatments, highlighting the need to accurately define molecular subgroups that have prognostic significance and harbor potentially targetable biology.1
In contrast, primary mediastinal large B-cell lymphoma (PMBL) represents a relatively rare subtype, accounting for 2% to 3% of B-cell NHLs. It typically affects young adults, with a higher frequency in women, and is characterized by the presence of a bulky anterior mediastinal mass.2 Based on its anatomic location and immunophenotypic signature, PMBL has been postulated to be derived from thymic medullary B cells.3,4 Although its morphologic features are mostly reminiscent of DLBCL,5 gene expression (GE) profile analyses have shown that PMBL is more closely related to classic Hodgkin lymphoma (cHL).6,7 This relationship to cHL was further corroborated by recent studies that revealed frequent aberrations affecting immune response, JAK-STAT, and NF-κB signaling, representing molecular findings distinct from DLBCL but similar to cHL.8,9
PMBL has been recognized as a distinct lymphoma entity in the World Health Organization classification since 2001, but its diagnosis is, in most cases, still based on clinicopathologic consensus. In particular, the lack of histologic features that reliably distinguish PMBL from other types of B-cell NHL that may involve mediastinal lymph nodes can make a definitive diagnosis challenging.10 GE profiling has been shown to accurately differentiate PMBL from DLBCL with mediastinal involvement, but despite some translational efforts, diagnostic GE-based tests have not yet entered routine clinical practice.6,7,11,12
Yuan et al13 reported the identification of PMBL tumors observed at nonmediastinal sites. These tumors were initially diagnosed as DLBCL and showed no evidence of mediastinal involvement but expressed a PMBL-like signature. However, compared with PMBL tumors with anterior mediastinal involvement, they had significantly fewer aberrations involving CIITA or PDL1/PDL2. Patients with PMBL tumors without apparent mediastinal involvement were older, and there was more equal sex distribution compared with those with mediastinal PMBL. However, these differences did not reach statistical significance, likely reflecting a small sample size. Therefore, whether "nonmediastinal PMBLs" (nm-PMBLsig+) are indeed PMBLs or alternatively represent a distinct subtype of DLBCL remains an open question.
Methods
Patient cohorts
To enhance our understanding of the subset of DLBCL that exhibits a PMBL GE signature, we analyzed the mutational and clinical characteristics of 325 de novo tumors with DLBCL morphology, treated with rituximab plus cyclophosphamide, doxorubicin, vincristine, and prednisone (R-CHOP), selected from the BC Cancer (BCC) population-based registry. All DLBCL cases had MYC, BCL2, and BCL6 fluorescence in situ hybridization, RNA sequencing (RNAseq), OncoSNP copy-number (CN) call, and targeted capture sequencing results available from previously published studies.14,15
Mutational frequencies and clinical characteristics of nm-PMBLsig+ tumors were compared with those of bona fide PMBLs (bf-PMBLs) that were part of a previously described PMBL cohort drawn from the BCC population-based registry and centrally reviewed by the pathologist panel of the Lymphoma/Leukemia Molecular Profiling Project.11
This work was reviewed and approved by the University of British Columbia–BC Cancer Research Ethics Board in accordance with the Declaration of Helsinki.
DLBCL90 NanoString assay
The DLBCL90 NanoString subtyping assay for aggressive B-cell lymphoma was applied to 325 DLBCL samples to identify tumors with the PMBL GE signature, assign cell-of-origin (COO) group, and determine double-hit signature (DHITsig) status.11,16 Linear predictor scores and empiric Bayesian probabilities17 were calculated for all samples using the DLBCL90 R package.
IHC staining
Immunohistochemical staining (IHC) was performed on tissue microarrays, comprising cores from the diagnostic biopsies of the cohort, using antibodies and thresholds described in supplemental Table 1, available on the Blood Web site.
WES
Whole-exome sequencing (WES) was performed on 15 fresh frozen nm-PMBLsig+ tumors and 7 matched normal samples. Genomic libraries were prepared using the SureSelectXT Target Enrichment System for Illumina Paired-End Multiplexed Sequencing Library protocol and 200 ng of input genomic DNA. Each indexed library was then hybridized to the SureSelect Human All Exon V6+UTR capture library and sequenced on an Illumina NextSeq550 using 150-bp paired-end reads. Tumor and normal DNA samples were sequenced to an average depth of 117× (standard deviation, 30×). All reads were aligned to the human reference genome using bwa-mem (version 0.7.5a)18 with optical and polymerase chain reaction duplicates removed using the Picard tool (http://broadinstitute.github.io/picard/).
WES analysis
Somatic single-nucleotide variant (SNV)/indel variants were identified using the intersection of calls predicted by VarScan (version 2.3.6),19 Strelka (version 1.0.13),20 and MuTect (version 1.1.4).21 All variants were annotated using SnpEff (version 4.2)22 and filtered for effects predicted to have an impact at the protein level (nonsynonymous, stop gained, and splice site). Variants were filtered for a minimum of 3 variant reads, 10% variant allele frequency, and global minor allele frequency <1%. Unpaired tumors (n = 8) were matched against a pooled normal that was constructed from normal samples for paired tumors (n = 7). Variants from unpaired tumors were further filtered to remove potential germline single-nucleotide polymorphisms if the variant was present in dbSNP (version 137)23 but not COSMIC (version 68).24
Mutation calls from an external DLBCL cohort
Mutational frequencies in nm-PMBLsig+ tumors assessed by WES were compared with mutational frequencies observed in DLBCLs from Schmitz et al,25 who excluded cases with a PMBL signature from their study cohort. Germinal center B cell–like (GCB) DLBCLs were selected using the gene expression subgroup annotation that was based on RNAseq data. Variant calls were obtained using MuTect221 and postfiltered to remove germline and low-confidence variants as described in the supplemental Data.
GE analysis
RNAseq library construction and STAR alignment procedures have been described previously.26 Gene-level read counts were quantified using HTSEQ-count (intersection strict, reverse mode).27 Raw read counts were normalized and scaled using trimmed mean of M-values normalization and log2-transformed counts-per-million scaling, respectively.28
CN analysis from WES data
CN alterations in nm-PMBLsig+ and bf-PMBL tumors were called from WES data using CNVkit (version 0.9.1)29 with default thresholds. To identify relevant regions of CN alterations, GISTIC (version 2.0)30 was applied to the curated segmentation results. The supplemental Data provides a detailed description.
IL-4 receptor expression, site-directed mutagenesis, and reporter gene assays in HEK293-STAT6 cells
IL4R mutations were created using the GENEART site-directed mutagenesis system (Thermo Fisher Scientific) according to the manufacturer’s instructions. Wild-type (WT) IL4R and mutants were cloned into pcDNA3.1, and empty pcDNA3.1 was used as a mock vector. The plasmids were purified using the Spin Miniprep Kit (Qiagen), and 1 μg of plasmid was transfected into HEK293 cells expressing STAT6 (HEK293 interleukin-4 [IL-4]/IL-13; Invivogen), seeded the day before at 0.25 × 106 cells per well (12 multiwell plates) using Lipofectamine2000 (Invitrogen). Twenty-four hours after transfection, cell-free supernatants were collected to assess the secreted embryonic alkaline phosphatase levels according to the manufacturer’s protocol, and cells were lysed for immunoblotting analysis.
Preparation of doxycycline-inducible cell lines, retroviral transduction, and cell culture
Retroviral transduction of DEV cells was performed as previously described.31 Briefly, DEV cells were first transduced with a feline endogenous virus (FEV) expressing the ecotropic retroviral receptor (DEV-FEV). DEV-FEV cells were secondarily infected with a retrovirus expressing the bacterial tetracycline repressor, after which they were infected with retroviral particles containing the inducible vector pRETRO-TO-PuroGFP (donated by Louis M. Staudt, National Cancer Institute, Bethesda, MD), in which the cytomegalovirus promoter is used to drive expression of cloned WT IL4R or mutants in the presence of 20 ng/mL of doxycycline (Sigma-Aldrich). Stable GFP+ cells were sorted using BD FACSAria Fusion. Transduced DEV cells were cultured in RPMI 1640–GlutaMAX medium (Gibco) supplemented with 20% fetal bovine serum in the presence of doxycycline (20 ng/mL).
Western blotting and flow cytometry
Western blotting and flow cytometry were performed as previously described.31 Briefly, membranes were probed with the following primary antibodies at 1:1000 dilution ratio unless stated otherwise: phosphorylated STAT6 (pSTAT6; 9364; Cell Signaling Technology), STAT6 (ab32108; Abcam), and IL4 receptor (IL4R; sc-28361; 1:500; Santa Cruz Biotechnology). Glyceraldehyde-3-phosphate dehydrogenase (MAB374; 1:5000; Millipore) antibody was used as internal control. Bands were visualized using the enhanced chemiluminiscence system (GE Healthcare) on a Chemidoc digital imager (Bio-Rad), and intensities of bands were quantified using Image Laboratory software (Bio-Rad). Flow cytometric analysis for surface expression of IL4R (CD124) was performed using an LSRFortessa (Becton-Dickinson Biosciences).31
Quantitative reverse transcription polymerase chain reaction
Total RNA was isolated using the RNeasy Kit (Qiagen) and treated with DNAse I (Promega). CCL17 (Hs00171074_m1) and CD23 (Hs01077044_m1) TaqMan GE Assay probes were used to detect messenger RNA levels as previously described.31
Statistical analysis
For the comparison of baseline clinical characteristics of patients between lymphoma subsets, data were tested by Wilcoxon rank sum (continuous data) or χ2 (Pearson χ2) test (categorical data). A Fisher’s exact test was used to compare frequencies of mutations and IHC scores between lymphoma subsets. All analyses were performed using R software (version 3.6.3; https://cran.r-project.org/src/base/R-3/) For in vitro experiments, comparisons between groups were performed using a 2-sample Student t test, (GraphPad Prism 8).
Results
Identification of tumors with DLBCL morphology expressing a PMBL signature
The recently developed DLBCL90 NanoString assay was designed to classify tumors based on COO32 and identify those with a PMBL11 or DHIT16 GE signature. We applied the DLBCL90 assay, following the 3-tiered classification system depicted in Figure 1A, to 325 diagnostic biopsy samples of patients with de novo DLBCL treated with R-CHOP.26,33 Nineteen tumors (5.8%) were found to have a PMBL GE signature (PMBLsig+) (Figure 1B). This assignment was supported by RNAseq analysis that showed strong expression of PMBL signature genes published by Rosenwald et al6 (supplemental Figure 1). The remaining tumors were classified as DHITsig+ DLBCL (n = 66 samples; 20.3% of total), GCB DLBCL (n = 110; 33.8%), unclassified DLBCL (n = 32; 9.8%), or activated B-cell (ABC) DLBCL (n = 98; 30.2%).
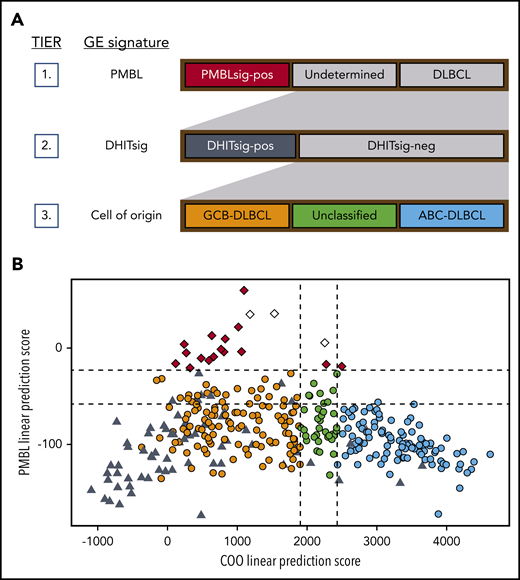
DLBCL90 assay: tiered classification system and scores. (A) Tiered classification of DLBCL tumors using the DLBCL90 NanoString assay. Tier 1: in the first layer of this 3-tiered classification system, PMBLsig+ samples are identified. Tier 2: DHITsig+ samples are then identified among the samples that do not have a PMBL GE signature. Tier 3: in the final tier, the remaining samples are categorized based on their COO. (B) DLBCL90 assay scores for 325 de novo DLBCL tumors from the BCC DLBCL cohort. PMBL linear prediction scores (LPSs) are plotted against COO LPSs for each sample. Low COO LPS result in an assignment to the GCB group, whereas high LPS result in an activated B-cell (ABC) assignment. High PMBL LPS results in an assignment to the PMBLsig+ group. Dotted lines represent the cutoffs between categories. Red, PMBLsig+; orange, GCB; green, unclassified; blue, ABC; black, DHITsig+; white, samples from patients found to have presented with bf-PMBL–like features and who were therefore excluded from further analysis.
DLBCL90 assay: tiered classification system and scores. (A) Tiered classification of DLBCL tumors using the DLBCL90 NanoString assay. Tier 1: in the first layer of this 3-tiered classification system, PMBLsig+ samples are identified. Tier 2: DHITsig+ samples are then identified among the samples that do not have a PMBL GE signature. Tier 3: in the final tier, the remaining samples are categorized based on their COO. (B) DLBCL90 assay scores for 325 de novo DLBCL tumors from the BCC DLBCL cohort. PMBL linear prediction scores (LPSs) are plotted against COO LPSs for each sample. Low COO LPS result in an assignment to the GCB group, whereas high LPS result in an activated B-cell (ABC) assignment. High PMBL LPS results in an assignment to the PMBLsig+ group. Dotted lines represent the cutoffs between categories. Red, PMBLsig+; orange, GCB; green, unclassified; blue, ABC; black, DHITsig+; white, samples from patients found to have presented with bf-PMBL–like features and who were therefore excluded from further analysis.
Clinical characteristics of nonmediastinal B-cell NHLs with a PMBL GE signature
The 19 tumors that expressed a PMBL GE signature were rereviewed by an expert panel of pathologists, radiologists, and oncologists. In 3 cases, the clinicopathologic features were deemed to be compatible with a diagnosis of PMBL; these cases were subsequently excluded from further analysis (Figure 1B). We then comprehensively characterized the clinicopathologic and genomic features of the remaining 16 tumors that did not show mediastinal involvement but expressed the PMBL GE signature (nm-PMBLsig+) and compared the findings with those of the remaining DLBCL cohort, as well as with a collection of bf-PMBLs published previously.8
From a clinical point of view, patients with nm-PMBLsig+ tumors were similar to those from the remaining DLBCL cohort but differed markedly from those with bf-PMBL (Table 1). Patients with nm-PMBLsig+ tumors were significantly older than those with bf-PMBL (median age at diagnosis, 66 vs 33 years; P < .001), and fewer presented with elevated serum lactate dehydrogenase levels or B symptoms (44% vs 78%; P = .006 and 19% vs 81%; P < .001, respectively). Pleural and/or pericardial effusions were rarely observed in patients with nm-PMBLsig+ tumors but were prevalent in patients with bf-PMBL (6% vs 30%; P = .045 and 0% vs 36%; P < .001, respectively). Involvement of bone marrow, which is highly unusual in bf-PMBL, was observed in 3 nm-PMBLsig+ cases (19% vs 0%; P < .001).
Baseline clinical characteristics of patients with nm-PMBLsig+ tumors, other DLBCLs, or bf-PMBL
| Characteristic | Other DLBCLs (n = 306) | nm-PMBLsig+ (n = 16) | bf-PMBL (n = 73) | ||
|---|---|---|---|---|---|
| n (% of total) | P* | n (% of total) | P† | n (% of total) | |
| Age, y | .42 | <.001 | |||
| Median | 64 | 66 | 33 | ||
| Range | 16-92 | 19-80 | 13-58 | ||
| Sex | .26 | .41 | |||
| Male | 196 (64) | 8 (50) | 29 (40) | ||
| Female | 110 (36) | 8 (50) | 44 (60) | ||
| ECOG PS | .54 | .45 | |||
| ≤1 | 205 (68) | 12 (75) | 47 (65) | ||
| >1 | 98 (32) | 4 (25) | 25 (35) | ||
| Unknown | 3 | — | 1 | ||
| Serum LDH level | .51 | .006 | |||
| Normal | 134 (48) | 9 (56) | 16 (22) | ||
| Elevated | 146 (52) | 7 (44) | 57 (78) | ||
| Unknown | 26 | — | — | ||
| Ann Arbor stage | .50 | .13 | |||
| I/II | 144 (48) | 9 (56) | 54 (75) | ||
| III/IV | 159 (52) | 7 (44) | 18 (25) | ||
| Unknown | 3 | — | 1 | ||
| B symptoms | .12 | <.001 | |||
| Absent | 187 (62) | 13 (81) | 14 (19) | ||
| Present | 115 (38) | 3 (19) | 58 (81) | ||
| Unknown | 4 | — | 1 | ||
| IPI | .78 | .50 | |||
| 0-2 | 192 (65) | 11 (69) | 51 (75) | ||
| >2 | 102 (35) | 5 (31) | 17 (25) | ||
| Unknown | 12 | — | 5 | ||
| Bulky disease (≥10 cm) | .08 | <.001 | |||
| No | 219 (74) | 15 (94) | 39 (46) | ||
| Yes | 77 (26) | 1 (6) | 28 (54) | ||
| Unknown | 10 | — | 1 | ||
| COO (Lymph2Cx) | .012 | .40 | |||
| GCB | 170 (56) | 14 (88) | 57 (78) | ||
| Non-GCB | 136 (44) | 2 (13) | 16 (22) | ||
| No. of identified extranodal sites | .38 | .007 | |||
| 0 or 1 | 261 (86) | 15 (94) | 28 (51) | ||
| ≥2 | 42 (14) | 1 (6) | 27 (49) | ||
| Unknown | 3 | — | 18 | ||
| Involvement of extranodal sites of interest | |||||
| Bone marrow | .43 | <.001 | |||
| Not identified | 269 (88) | 13 (81) | 73 (100) | ||
| Identified | 37 (12) | 3 (19) | 0 (0) | ||
| Pericardial effusion | .82 | .004 | |||
| Not identified | 305 (100) | 16 (100) | 47 (64) | ||
| Identified | 1 (0) | 0 (0) | 26 (36) | ||
| Pleural effusion | .52 | .045 | |||
| Not identified | 296 (97) | 15 (94) | 51 (70) | ||
| Identified | 10 (3) | 1 (6) | 22 (30) | ||
| Lung | .08 | .33 | |||
| Not identified | 295 (96) | 14 (88) | 56 (77) | ||
| Identified | 11 (4) | 2 (12) | 17 (23) | ||
| Characteristic | Other DLBCLs (n = 306) | nm-PMBLsig+ (n = 16) | bf-PMBL (n = 73) | ||
|---|---|---|---|---|---|
| n (% of total) | P* | n (% of total) | P† | n (% of total) | |
| Age, y | .42 | <.001 | |||
| Median | 64 | 66 | 33 | ||
| Range | 16-92 | 19-80 | 13-58 | ||
| Sex | .26 | .41 | |||
| Male | 196 (64) | 8 (50) | 29 (40) | ||
| Female | 110 (36) | 8 (50) | 44 (60) | ||
| ECOG PS | .54 | .45 | |||
| ≤1 | 205 (68) | 12 (75) | 47 (65) | ||
| >1 | 98 (32) | 4 (25) | 25 (35) | ||
| Unknown | 3 | — | 1 | ||
| Serum LDH level | .51 | .006 | |||
| Normal | 134 (48) | 9 (56) | 16 (22) | ||
| Elevated | 146 (52) | 7 (44) | 57 (78) | ||
| Unknown | 26 | — | — | ||
| Ann Arbor stage | .50 | .13 | |||
| I/II | 144 (48) | 9 (56) | 54 (75) | ||
| III/IV | 159 (52) | 7 (44) | 18 (25) | ||
| Unknown | 3 | — | 1 | ||
| B symptoms | .12 | <.001 | |||
| Absent | 187 (62) | 13 (81) | 14 (19) | ||
| Present | 115 (38) | 3 (19) | 58 (81) | ||
| Unknown | 4 | — | 1 | ||
| IPI | .78 | .50 | |||
| 0-2 | 192 (65) | 11 (69) | 51 (75) | ||
| >2 | 102 (35) | 5 (31) | 17 (25) | ||
| Unknown | 12 | — | 5 | ||
| Bulky disease (≥10 cm) | .08 | <.001 | |||
| No | 219 (74) | 15 (94) | 39 (46) | ||
| Yes | 77 (26) | 1 (6) | 28 (54) | ||
| Unknown | 10 | — | 1 | ||
| COO (Lymph2Cx) | .012 | .40 | |||
| GCB | 170 (56) | 14 (88) | 57 (78) | ||
| Non-GCB | 136 (44) | 2 (13) | 16 (22) | ||
| No. of identified extranodal sites | .38 | .007 | |||
| 0 or 1 | 261 (86) | 15 (94) | 28 (51) | ||
| ≥2 | 42 (14) | 1 (6) | 27 (49) | ||
| Unknown | 3 | — | 18 | ||
| Involvement of extranodal sites of interest | |||||
| Bone marrow | .43 | <.001 | |||
| Not identified | 269 (88) | 13 (81) | 73 (100) | ||
| Identified | 37 (12) | 3 (19) | 0 (0) | ||
| Pericardial effusion | .82 | .004 | |||
| Not identified | 305 (100) | 16 (100) | 47 (64) | ||
| Identified | 1 (0) | 0 (0) | 26 (36) | ||
| Pleural effusion | .52 | .045 | |||
| Not identified | 296 (97) | 15 (94) | 51 (70) | ||
| Identified | 10 (3) | 1 (6) | 22 (30) | ||
| Lung | .08 | .33 | |||
| Not identified | 295 (96) | 14 (88) | 56 (77) | ||
| Identified | 11 (4) | 2 (12) | 17 (23) | ||
Bold P values indicate significance.
ECOG PS, Eastern Cooperative Oncology Group performance status; IPI, International Prognostic Index; LDH, lactate dehydrogenase.
Compared with other DLBCLs using the Wilcoxon rank sum test for continuous data (age) and χ2 (Pearson χ2 test) for categorical data.
Compared with bf-PMBL using the Wilcoxon rank sum test for continuous data (age) and χ2 (Pearson χ2 test) for categorical data.
Translocations affecting MYC, BCL2, and BCL6 were found in 4 (25%), 5 (33%), and 2 (13%) PMBLsig+ tumors, respectively (Figures 2 and 3A). Three PMBLsig+ tumors were found to be HGBL-DH/TH. The results presented in the main article text reflect analyses including both DLBCL not otherwise specified (DLBCL, NOS) and HGBL-DH/TH, whereas analyses in the data supplement are restricted to DLBCL, NOS. All PMBLsig+ tumors were Epstein-Barr virus negative (Figure 2) and did not reveal structural rearrangements affecting CIITA, as assessed by a breakapart fluorescence in situ hybridization assay (S.B.-N.; data not shown). The vast majority of nm-PMBLsig+ tumors had a GCB GE signature, compared with approximately half of the remaining DLBCLs (88% vs 56%; P = .01; Table 1; Figure 1B).
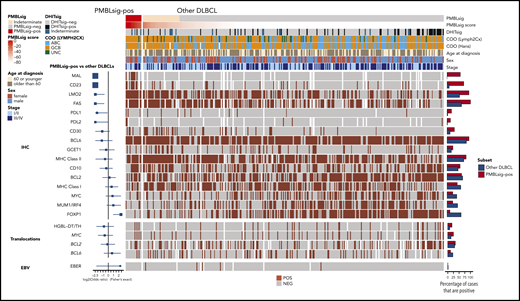
Immunophenotypic characteristics and translocation status of PMBLsig+ tumors and other tumors with DLBCL morphology. Forest plots at the left show enrichment or depletion of positive tumors among the PMBLsig+ group compared with other DLBCLs for each marker/translocation. Bar plots at the right represent the fraction of cases positive for each marker/translocation. ABC, activated B-cell–like; EBV, Epstein-Barr virus; HGBL-DH/TH, high-grade B-cell lymphoma with MYC and BCL2 and/or BCL6 rearrangement; MHC, major histocompatibility complex; UNC, unclassified.
Immunophenotypic characteristics and translocation status of PMBLsig+ tumors and other tumors with DLBCL morphology. Forest plots at the left show enrichment or depletion of positive tumors among the PMBLsig+ group compared with other DLBCLs for each marker/translocation. Bar plots at the right represent the fraction of cases positive for each marker/translocation. ABC, activated B-cell–like; EBV, Epstein-Barr virus; HGBL-DH/TH, high-grade B-cell lymphoma with MYC and BCL2 and/or BCL6 rearrangement; MHC, major histocompatibility complex; UNC, unclassified.
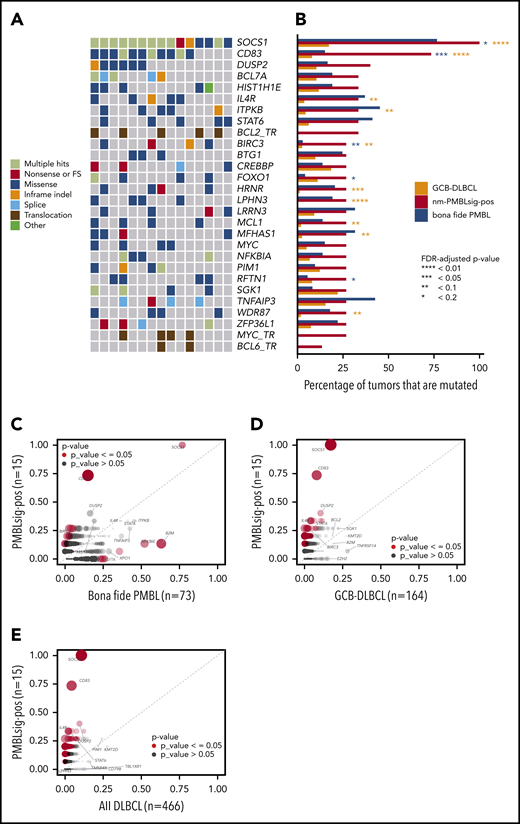
Mutational landscape of nm-PMBLsig+ tumors. (A) Oncoplot showing the somatic mutational landscape of nm-PMBLsig+ tumors. Genes mutated in at least 4 nm-PMBLsig+ tumors (≥27%) and translocations affecting MYC, BCL2, and BCL6 are shown. Colors represent various mutation types, as indicated. (B) Mutational frequency of selected genes in nm-PMBLsig+ tumors, GCB DLBCL, and bf-PMBL. Mutational frequencies of genes depicted in panel A are shown for nm-PMBLsig+ tumors (BCC DLBCL cohort), GCB DLBCL (Schmitz/Staudt cohort), and bf-PMBL (BCC PMBL cohort). Purple and orange asterisks represent significance of enrichment of mutations in nm-PMBLsig+ tumors vs bf-PMBL and nm-PMBLsig+ tumors vs GCB DLBCL, respectively (Fisher’s exact test). MYC, BCL2, and BCL6 translocation status was not assessed or has not been reported for the bf-PMBL and DLBCL samples from the Schmitz cohort. (C-E) Mutational enrichment plots showing mutational frequencies per gene in nm-PMBLsig+ tumors vs bf-PMBL (C), nm-PMBLsig+ tumors vs GCB DLBCL (D), and nm-PMBLsig+ vs all DLBCLs (E). Size and opacity of the data points are proportional to the significance of enrichment or depletion of the number of mutations affecting a given gene in nm-PMBLsig+ samples compared with bf-PMBL (C), GCB DLBCL (D), or all DLBCL (E) (Fisher’s exact test). FDR, false discovery rate; FS, frameshift.
Mutational landscape of nm-PMBLsig+ tumors. (A) Oncoplot showing the somatic mutational landscape of nm-PMBLsig+ tumors. Genes mutated in at least 4 nm-PMBLsig+ tumors (≥27%) and translocations affecting MYC, BCL2, and BCL6 are shown. Colors represent various mutation types, as indicated. (B) Mutational frequency of selected genes in nm-PMBLsig+ tumors, GCB DLBCL, and bf-PMBL. Mutational frequencies of genes depicted in panel A are shown for nm-PMBLsig+ tumors (BCC DLBCL cohort), GCB DLBCL (Schmitz/Staudt cohort), and bf-PMBL (BCC PMBL cohort). Purple and orange asterisks represent significance of enrichment of mutations in nm-PMBLsig+ tumors vs bf-PMBL and nm-PMBLsig+ tumors vs GCB DLBCL, respectively (Fisher’s exact test). MYC, BCL2, and BCL6 translocation status was not assessed or has not been reported for the bf-PMBL and DLBCL samples from the Schmitz cohort. (C-E) Mutational enrichment plots showing mutational frequencies per gene in nm-PMBLsig+ tumors vs bf-PMBL (C), nm-PMBLsig+ tumors vs GCB DLBCL (D), and nm-PMBLsig+ vs all DLBCLs (E). Size and opacity of the data points are proportional to the significance of enrichment or depletion of the number of mutations affecting a given gene in nm-PMBLsig+ samples compared with bf-PMBL (C), GCB DLBCL (D), or all DLBCL (E) (Fisher’s exact test). FDR, false discovery rate; FS, frameshift.
nm-PMBLsig+ tumors and bf-PMBL share a common immunophenotype
All PMBLsig+ tumors showed similar morphologic features on hematoxylin and eosin stain, with abundant eosinophilic cytoplasm, resembling the typical morphology of PMBL tumor cells. A majority of nm-PMBLsig+ tumors stained positive for PMBL markers CD23 (71%) and MAL (57%), whereas detectable expression of these proteins was rare or absent within the rest of the DLBCL cohort (9% and 0%, respectively; P < .001).34,35 Four nm-PMBLsig+ tumors (25%) stained positive for programmed death ligand 1 (PDL1) and/or PDL2, compared with 12 other DLBCLs (4%; P < .001). None of the PMBLsig+ tumors showed loss of FAS staining, compared with 35% of the other DLBCLs (P = .02). The GCB COO of the PMBLsig+ tumors was confirmed by IHC staining for CD10, BCL6, and MUM, followed by application of the Hans algorithm36 (Figure 2; supplemental Tables 2 and 3). Analysis of the cohort without HGBL-DH/TH tumors revealed a similar picture, showing positive staining of CD23, MAL, PDL1/2, and FAS to be prominently enriched among PMBLsig+ tumors (supplemental Figure 2; supplemental Table 4).
Mutational analysis of nm-PMBLsig+ tumors confirms molecular commonalities with bf-PMBL but suggests different modes of immune evasion
We performed WES on the 15 nm-PMBLsig+ tumors for which DNA was available. We identified, on average, 276 (range, 26-440) somatic nonsynonymous mutations with a variant allele frequency of at least 5% (supplemental Table 5; supplemental Figure 3). We then compared the mutational landscape of these nm-PMBLsig+ samples with the mutational patterns observed in 73 bf-PMBLs.8
Notably, all nm-PMBLsig+ tumors showed ≥1 mutation affecting genes encoding proteins involved in JAK-STAT signaling, including SOCS1, DUSP2, IL4R, and STAT6, akin to those found in bf-PMBL8,9 (Figures 3A-B and 4A).
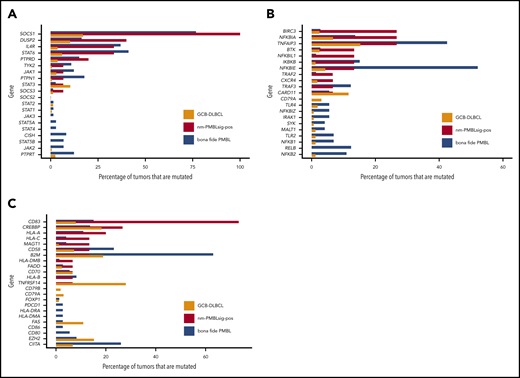
Mutational frequencies affecting genes involved in pathways of interest in GCB DLBCL, nm-PMBLsig+ tumors, and bf-PMBL. (A) JAK-STAT mutations are frequent in both nm-PMBLsig+ tumors and bf-PMBL. Frequencies of mutations affecting genes involved in JAK-STAT signaling are shown for GCB DLBCL, nm-PMBLsig+ tumors, and bf-PMBL. (B) NF-κB signaling mutations are frequent in both nm-PMBLsig+ tumors and bf-PMBL. Frequencies of mutations affecting genes involved in NF-κB signaling are shown for GCB DLBCL, nm-PMBLsig+ tumors, and PMBL. (C) Immune response mutations are common in all 3 subtypes. Frequencies of mutations affecting genes involved in immune response are shown for GCB DLBCL, nm-PMBLsig+ tumors, and bf-PMBL.
Mutational frequencies affecting genes involved in pathways of interest in GCB DLBCL, nm-PMBLsig+ tumors, and bf-PMBL. (A) JAK-STAT mutations are frequent in both nm-PMBLsig+ tumors and bf-PMBL. Frequencies of mutations affecting genes involved in JAK-STAT signaling are shown for GCB DLBCL, nm-PMBLsig+ tumors, and bf-PMBL. (B) NF-κB signaling mutations are frequent in both nm-PMBLsig+ tumors and bf-PMBL. Frequencies of mutations affecting genes involved in NF-κB signaling are shown for GCB DLBCL, nm-PMBLsig+ tumors, and PMBL. (C) Immune response mutations are common in all 3 subtypes. Frequencies of mutations affecting genes involved in immune response are shown for GCB DLBCL, nm-PMBLsig+ tumors, and bf-PMBL.
Mutations affecting BIRC3, which encodes an E3 ubiquitin ligase involved in NF-κB signaling,37 were highly enriched in nm-PMBLsig+ tumors compared with bf-PMBL (Figure 3A-C; supplemental Figure 4A). The BIRC3 p.Leu548X frameshift mutation, identified in 1 nm-PMBLsig+ tumor, is similar to the mutations that are frequent in mantle cell lymphoma and that invariably truncate the BIRC3 RING domain, resulting in loss of its ubiquitinating capacity (supplemental Figure 7).38-40 In 2 samples, we identified mutations affecting the BIR3 domain: an in-frame 9-bp deletion and a missense mutation categorized as highly likely to be damaging to the protein. The final BIRC3 mutation, a p.Gln515His missense mutation, affects the CARD domain, which is required for BIRC3 autoregulation.41 The recurrent aberrations affecting BIRC3 and NFKBIE identified in nm-PMBLsig+ tumors and bf-PMBL,8,42 respectively, suggest that although perturbation of NF-κB signaling is common in both entities, this disruption is most often established through distinct mechanisms (Figures 3A-C and 4B). In a similar fashion, mutations affecting immune response were prevalent in both nm-PMBLsig+ tumors (CD83) and bf-PMBL (B2M and CIITA), but the differences in the specific genes involved suggest distinct mechanisms of immune evasion (Figures 3A-C and 4C).43,44
IL4R mutation patterns and function differ significantly between nm-PMBLsig+ tumors and GCB DLBCL
Several of the frequently mutated genes in nm-PMBLsig+ tumors, including those affecting JAK-STAT genes and CD83, were found to be mutated in far lower fractions or were absent in GCB DLBCL (Figures 3B,D and 4A-C; supplemental Figure 4B).
In addition, analysis of the targeted sequencing results from the BCC DLBCL cohort15,26 revealed that IL4R mutations were more frequent in nm-PMBLsig+ tumors compared with GCB DLBCL (supplemental Figure 5), and the IL4R mutational patterns were different. While the previously reported transmembrane hotspot mutation p.I242N31 was present in both nm-PMBLsig+ tumors and GCB DLBCL, nm-PMBLsig+ samples showed an enrichment of mutations affecting the extracellular domain and less frequent cytoplasmic mutations compared with GCB DLBCL (Figure 5A).
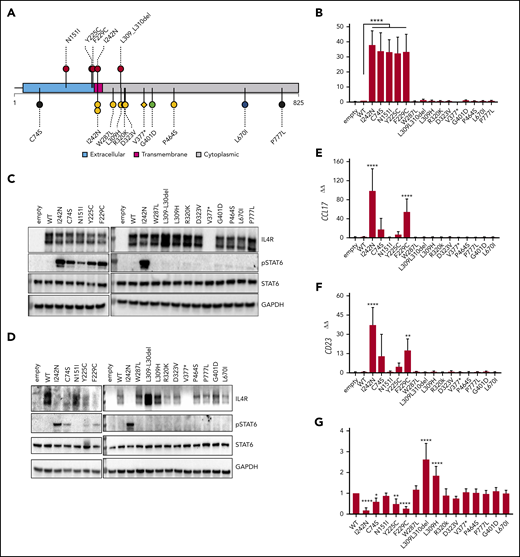
IL4R mutations in nm-PMBLsig+ tumors induce JAK-STAT activation. (A) Mutational pattern of IL4R (UniProtKB-P24394) in nm-PMBLsig+ tumors (top) and other DLBCL tumors (bottom) from the BCC cohort specified according to receptor domains (extracellular, transmembrane, or cytoplasmic). Colors and shapes of lollipops represent the DLBCL90 NanoString assay tiered classification of samples (see Figure 1) and type of mutation (circle, missense or in-frame indel; diamond, truncating), respectively. The plot was created using R package TrackViewer (version 1.24.0). (B-G) Ectopic expression of WT IL4R and mutants in HEK Blue IL-4/IL-13 (Invivogen) (B-C) and DEV cells (D-G). (B) Quantification of pSTAT6-dependent expression of secreted embryonic alkaline phosphatase (SEAP) in supernatant. (C-D) Immunoblot for pSTAT6, STAT6, IL4R, and glyceraldehyde-3-phosphate dehydrogenase (GAPDH). (E-F) CCL17 and CD23 messenger RNA expression measured by quantitative polymerase chain reaction. (G) IL4R surface expression measured by flow cytometry. Data are means ± standard deviation of 5, 4, 7, and 7 experiments in panels A, E, F, and G, respectively; significance was evaluated using a 1-sample Student t test. *P < .05, **P < .01, ****P < .0001.
IL4R mutations in nm-PMBLsig+ tumors induce JAK-STAT activation. (A) Mutational pattern of IL4R (UniProtKB-P24394) in nm-PMBLsig+ tumors (top) and other DLBCL tumors (bottom) from the BCC cohort specified according to receptor domains (extracellular, transmembrane, or cytoplasmic). Colors and shapes of lollipops represent the DLBCL90 NanoString assay tiered classification of samples (see Figure 1) and type of mutation (circle, missense or in-frame indel; diamond, truncating), respectively. The plot was created using R package TrackViewer (version 1.24.0). (B-G) Ectopic expression of WT IL4R and mutants in HEK Blue IL-4/IL-13 (Invivogen) (B-C) and DEV cells (D-G). (B) Quantification of pSTAT6-dependent expression of secreted embryonic alkaline phosphatase (SEAP) in supernatant. (C-D) Immunoblot for pSTAT6, STAT6, IL4R, and glyceraldehyde-3-phosphate dehydrogenase (GAPDH). (E-F) CCL17 and CD23 messenger RNA expression measured by quantitative polymerase chain reaction. (G) IL4R surface expression measured by flow cytometry. Data are means ± standard deviation of 5, 4, 7, and 7 experiments in panels A, E, F, and G, respectively; significance was evaluated using a 1-sample Student t test. *P < .05, **P < .01, ****P < .0001.
In vitro experiments showed that mutations affecting the extracellular domain of IL4R (p.N151I, p.Y225, and p.F229C) had the ability to constitutively activate the downstream JAK-STAT pathway, similarly to p.I242N.31 In contrast, mutations affecting the cytoplasmic domain of IL4R did not lead to JAK-STAT pathway activation. Upon ectopic expression of IL4R with extracellular domain mutations in HEK IL-4/IL-13 cells, the levels of STAT6-induced secreted embryonic alkaline phosphatase secreted into the cell culture supernatant (Figure 5B) and pSTAT6 (Figure 5C) were increased, independent of IL-4 ligand stimulation, but not upon expression of cytoplasmic IL4R mutations. Consistently, constitutive pSTAT6 activation was confirmed in a B-cell lymphoma–derived cell line (DEV) expressing these extracellular IL4R mutations (Figure 5D). In addition, high expression of CCL17 and CD23 (Figure 5E-F) was associated with JAK-STAT activation. In concordance, CCL17 transcript levels were high in clinical samples with mutations affecting the transmembrane or extracellular domains of IL4R but low in those with cytoplasmic IL4R mutations (supplemental Figure 6). The observation that most nm-PMBLsig+ tumors that had WT IL4R also showed high CCL17 transcript levels suggests alternative mechanisms for JAK-STAT activation in these tumors, presumably through mutation of STAT6, DUSP2, and/or SOCS1 (Figure 3A).12 Similar to findings reported by Viganò et al, we confirmed an inverse correlation between JAK-STAT activation (observed as pSTAT6, CD23, and CCL17 expression) and expression of IL4R protein on the plasma membrane (Figure 5G).31
Several gene mutations in nm-PMBLsig+ tumors show a pattern of aSHM
Remarkably, all nm-PMBLsig+ tumors harbored ≥1 mutation in SOCS1. SOCS1 is a known target for AID-mediated aberrant somatic hypermutation (aSHM) in lymphoma45,46 and is recurrently mutated in both bf-PMBL8 and cHL,47,48 although at lower frequencies. Several other known aSHM targets, including CD83, BCL7A, MYC, SGK1, and PIM1, were also frequently mutated in nm-PMBLsig+ tumors, and the mutational patterns in these genes were in concordance with aSHM (supplemental Figure 7). Moreover, transcript levels positively correlated with mutational status for these genes, consistent with the concept that SHM preferentially occurs in actively transcribed genes (supplemental Figure 8). While confirming the post-GC origin of nm-PMBLsig+ tumors, further investigation will be required to distinguish aSHM-induced driver mutations from passenger mutations.49
Recurrent CN changes in nm-PMBLsig+ tumors
Gain of 9p24, which is frequently observed in bf-PMBL (Figure 6), was identified in a significantly higher fraction of nm-PMBLsig+ tumors compared with GCB DLBCL (33% vs 10%; P = .03). 9p24 amplification was associated with increased transcript levels of JAK2, CD274, and PDCD1LG2 in nm-PMBLsig+ tumors (supplemental Figure 9).
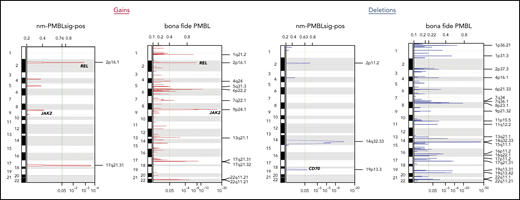
Recurrent CN aberrations in nm-PMBLsig+ tumors and bf-PMBL inferred from WES data. CNs were inferred from WES data using CNVkit; recurrent aberrations were then identified using the GISTIC algorithm (see “Methods”).
Recurrent CN aberrations in nm-PMBLsig+ tumors and bf-PMBL inferred from WES data. CNs were inferred from WES data using CNVkit; recurrent aberrations were then identified using the GISTIC algorithm (see “Methods”).
Besides JAK2, several other genes encoding proteins involved in JAK-STAT signaling showed increased CNs specifically in nm-PMBLsig+ samples (supplemental Figure 10). Beyond CN gains of 9p, we observed additional CN changes that add to the shared mutational patterns with bf-PMBL described at the SNV level, most prominently the amplification of the protooncogene c-REL located at 2p16.1 (Figure 6). In addition, we identified recurrent CN aberrations that seemed to be specific to nm-PMBLsig+ tumors. These included loss of the immune regulatory gene CD70 at 19p13.3, which was more frequently observed in nm-PMBLsig+ tumors than in GCB DLBCL (33% vs 10%; P = .02; supplemental Figure 11) and not detected as a recurring event in bf-PMBL (Figure 6). Four nm-PMBLsig+ tumors (25%) showed loss of 2q, with a minimally deleted region at 2q11.2 that included 5 genes, including REV1. REV1 encodes a TLS DNA polymerase, the knockdown of which has been shown to sensitize lymphomas to the crosslinking agent cisplatin.50 REV1 was deleted in only 1 GCB DLBCL (0.9%), and REV1 transcript levels were decreased in nm-PMBLsig+ tumors compared with GCB DLBCL (supplemental Figure 12).
Loss and concomitant decrease of transcript levels of RB1 (13q14.2) and PTEN (10q23.31) were found in 16% and 10% of GCB DLBCL samples, respectively, but were not observed in nm-PMBLsig+ samples (P = .1 and .3, respectively; supplemental Figures 13 and 14).
Discussion
Here, we have comprehensively characterized the mutational features and clinicopathologic characteristics of patients with de novo DLBCL presenting without evidence of anterior mediastinal involvement but expressing the PMBL GE signature (nm-PMBLsig+). Although previous reports have suggested that these tumors represent nonmediastinal PMBLs,13 our data indicate that they are clinically distinct from bf-PMBL and have molecular features that distinguish them from other DLBCLs. Identification of these cases from within our large population-based cohort suggests that ∼5% of de novo DLBCL, and therefore, ∼2% of all B-cell NHLs are nm-PMBLsig+. For perspective, these figures suggest an incidence rate similar to or greater than that of bf-PMBL.51
Although the clinical characteristics of nm-PMBLsig+ cases were similar to those of the other DLBCLs within our cohort, they differed considerably with respect to their immunophenotype and mutational landscape. The majority of nm-PMBLsig+ tumors expressed MAL and CD23, proteins typically expressed in bf-PMBL but rarely observed in DLBCL, and all were positive for LMO2 and FAS.
Similar to bf-PMBL,8,9 these tumors showed frequent aberrations affecting JAK-STAT signaling, including somatic point mutations in SOCS1, STAT6, IL4R, and DUSP2, and CN gains involving 9p24.1 that resulted in increased JAK2, CD274, and PDCD1LG2 transcript levels. Moreover, we found that mutations affecting IL4R were more frequent in nm-PMBLsig+ tumors than in GCB DLBCL and that the pattern of these mutations was also distinct. IL4R mutations identified in nm-PMBLsig+ samples predominantly affected the extracellular domain, whereas a majority of IL4R mutations in GCB DLBCL affected the cytoplasmic domain. We found that the extracellular domain mutations had the ability to activate JAK-STAT signaling in in vitro models, providing evidence that these mutations contribute to lymphomagenesis in nm-PMBLsig+ tumors. All investigated cytoplasmic IL4R mutations lacked this ability, suggesting that a majority of IL4R mutations identified in GCB DLBCL represent passenger mutations or contribute to lymphomagenesis through different mechanisms. The observation that cytoplasmic mutations that disrupt the dileucine motif L309-L310 caused an increase in the IL4R protein level (Figure 5C-D) and resulted in sustained IL4R surface receptor expression (Figure 5G) is interesting in this respect. In other receptors, this dileucine motif has been reported to function in clathrin-mediated endocytosis.52-54 It remains to be investigated whether this is also the case for IL4R and whether L309-L310 mutations contribute to lymphomagenesis by affecting endocytosis of IL4R.
Frequent disruption of immune response was an additional feature that nm-PMBLsig+ tumors shared with bf-PMBL.8,9 However, other than for the JAK-STAT pathway, the affected genes were different between nm-PMBLsig+ tumors and bf-PMBL, suggesting distinct mechanisms of disruption.
Interestingly, we identified somatic mutations affecting the E3 ubiquitin ligase gene BIRC3 in 4 of 15 nm-PMBLsig+ tumors. BIRC3 mutations and translocations are frequent in several hematologic malignancies,38,55-57 but mutations in DLBCL or bf-PMBL are rare. BIRC3 has, however, been reported to be involved in recurrently amplified regions in ABC DLBCL.58
Our findings are corroborated by 2 recent studies that created new DLBCL classification frameworks by applying computational algorithms to genetic features identified in DLBCL samples. Chapuy et al59 identified 5 distinct DLBCL subsets using SNVs, translocations, and CN aberrations. A subset of the Chapuy et al cluster 4 DLBCLs showed a mutational landscape similar to that of the nm-PMBLsig+ tumors, with frequent mutations affecting CD83, HIST1H1E, and SGK1. However, SOCS1 mutations were not reported in this study, because they did not pass MutsigCV filtering thresholds.
Lacy et al60 used targeted sequencing data to classify DLBCL tumors and identified 6 distinct DLBCL subsets, including a SOCS1/SGK1 group that harbored mutations also found in the nm-PMBLsig+ tumors from our DLBCL cohort, with recurrent mutations affecting SOCS1, SGK1, CD83, NFKBIA, HIST1H1E, and STAT3. This SOCS1/SGK1 group also included a majority of bf-PMBLs (12 of 20) as well as 98 (11.7%) of 839 DLBCL-NOS tumors. As observed for the nm-PMBCLsig+ tumors from our BCC DLBCL cohort, SOCS1/SGK1 DLBCLs did not show pathologic features of PMBL, were not enriched for mediastinal involvement, and were predominantly of GCB origin. However, BIRC3 mutations were not identified by Lacy et al as a defining feature of SOCS1/SGK1 DLBCL, and therefore, further research is required to assess the role this E3 ligase plays in the pathogenesis of such tumors.
The SOCS1/SGK1 group comprised a higher fraction of the investigated DLBCL cohort than our nm-PMBLsig+ subset (11.7% and 5.0%, respectively). Because DLBCL90 and Lymph3Cx assay results are currently unavailable for the Lacy et al60 DLBCL cohort, it remains an open question if the observed difference can be explained by between-cohort variation or whether the SOCS1/SGK1 group comprises a phenotypically mixed set of tumors that includes cases negative for the PMBL GE signature.
In addition to somatic mutations, our CN analysis revealed aberrations enriched in nm-PMBLsig+ tumors that could provide potentially novel therapeutic avenues for this subset of DLBCL. For example, patients with nm-PMBLsig+ tumors that show 9p24 gain might benefit from immunotherapy targeting PD-1/PD-L1.61
The shared GE pattern, which is the foundation of our study, likely represents a summation of heterogeneous somatic mutations that converge on common pathways and underpin hallmarks shared with bf-PMBL. The strong correlation between somatic mutations and GE phenotypes underscores the robustness of our findings, analogous to the original DLBCL COO classification.62
In summary, we have characterized DLBCLs with a PMBL GE signature, showing that they resemble bf-PMBL at the molecular level, with JAK-STAT signaling and immune response being frequently affected in both lymphoma entities. Our data suggest that dysregulation of the latter pathway is established through distinct evolutionary modes that are reflected in differential mutation patterns and anatomic and clinical presentations. With potential implications for classification and diagnostic procedures, this subset of DLBCL that expresses the PMBL signature can readily be identified using the DLBCL90 NanoString assay, emphasizing the value of GE assays in DLBCL subtyping.
The data reported in this article have been deposited in the European Genome-phenome Archive (accession number EGAS00001005057).
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Acknowledgments
This study was supported by Program Project Grant funding from the Terry Fox Research Institute (grant 1061), Large Scale Applied Research Project funding from Genome Canada (grant 13124; C.S.), Genome British Columbia (grant 271LYM; C.S.), Canadian Institutes of Health Research (CIHR; grant GP1-155873; C.S.) and the British Columbia Cancer Foundation, and a Foundation Grant from CIHR. C.S. is supported by the Michael Smith Foundation for Health Research, Career Investigator Award. D.W.S. is supported by a Michael Smith Foundation for Health Research, Health Professional Investigator Award. The Centre for Lymphoid Cancer acknowledges the support from the BC Cancer Foundation. E.V. was supported by a fellowship from the Michael Smith Foundation for Health Research.
Authorship
Contribution: G.D. and E.V. designed and performed the research, analyzed and interpreted data, and wrote the manuscript; D.E., C. Sarkozy, A.J., K.T., L.K.H., G.W.S., J.W.C., S.P.S., P.F., A.M., R.D.M., K.J.S., and E.D.H. analyzed and interpreted data; S.S.H. and C.R. analyzed exome sequencing data; E.C., S.B.-N., and B.W.W. performed experiments and interpreted data; R.D.G. provided study material; C. Steidl and D.W.S. supervised, designed the study, and wrote the manuscript; and all authors reviewed the manuscript.
Conflict-of-interest disclosure: C. Steidl has performed consultancy for Seattle Genetics, Curis, Inc, Roche, AbbVie, AstraZeneca, Juno Therapeutics, and Bayer and received research funding from Bristol-Myers Squibb, Epizyme, and Trillium Therapeutics, Inc. C. Steidl, D.W.S., and A.M. are coinventors on a patent (“Method for determining lymphoma type”) using NanoString technology. D.W.S. has performed consultancy for AbbVie, AstraZeneca, Celgene, and Janssen and received research funding from Janssen and NanoString Technologies. K.J.S. reports honoraria from Seattle Genetics, Bristol-Myers Squibb, Merck, AbbVie, AstraZeneca, and Gilead; consultancy for Servier; and institutional funds from Roche. J.W.C. has received honoraria from Seattle Genetics and Takeda Pharmaceuticals. E.D.H. has performed consultancy for Cytomx, Seattle Genetics, and Mitenyi and received research funding from AbbVie and Eli Lilly. S.P.S. is a shareholder in Canexia Health Inc. The remaining authors declare no competing financial interests.
Correspondence: Christian Steidl, Centre for Lymphoid Cancer, BC Cancer, 675 West 10th Ave, Vancouver, BC V5Z 1L3, Canada; e-mail: csteidl@bccancer.bc.ca.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal