

Key Points
Regnase-1 deficiency enhances CAR–T-cell persistence and CAR-T–mediated antitumor immunity in murine and human xenograft B-ALL models.
Regnase-1 targets Tcf7 mRNA to inhibit formation of TPEX cells critical for CAR–T-cell recall responses and survival.

Abstract
Chimeric antigen receptor (CAR)–T-cell therapeutic efficacy is associated with long-term T-cell persistence and acquisition of memory. Memory-subset formation requires T-cell factor 1 (TCF-1), a master transcription factor for which few regulators have been identified. Here, we demonstrate using an immune-competent mouse model of B-cell acute lymphoblastic leukemia (ALL; B-ALL) that Regnase-1 deficiency promotes TCF-1 expression to enhance CAR–T-cell expansion and memory-like cell formation. This leads to improved CAR-T–mediated tumor clearance, sustained remissions, and protection against secondary tumor challenge. Phenotypic, transcriptional, and epigenetic profiling identified increased tumor-dependent programming of Regnase-1–deficient CAR-T cells into TCF-1+ precursor exhausted T cells (TPEX) characterized by upregulation of both memory and exhaustion markers. Regnase-1 directly targets Tcf7 messenger RNA (mRNA); its deficiency augments TCF-1 expression leading to the formation of TPEX that support long-term CAR–T-cell persistence and function. Regnase-1 deficiency also reduces exhaustion and enhances the activity of TCF-1− CAR-T cells. We further validate these findings in human CAR-T cells, where Regnase-1 deficiency mediates enhanced tumor clearance in a xenograft B-ALL model. This is associated with increased persistence and expansion of a TCF-1+ CAR–T-cell population. Our findings demonstrate the pivotal roles of TPEX, Regnase-1, and TCF-1 in mediating CAR–T-cell persistence and recall responses, and identify Regnase-1 as a modulator of human CAR–T-cell longevity and potency that may be manipulated for improved therapeutic efficacy.
Introduction
Adoptive immunotherapy with CD19-specific chimeric antigen receptor (CAR)-T cells is an effective treatment of B-cell acute lymphoblastic leukemia (ALL; B-ALL).1 Nevertheless, 30% to 60% of treated patients relapse, in part due to poor long-term CAR–T-cell survival and function.2-4 Increased numbers of memory-like cells correlate with CAR–T-cell persistence and improved patient outcomes.5-7 Conversely, chronic antigen stimulation and tonic signaling lead to CAR–T-cell exhaustion, characterized by inhibitory receptor expression, loss of function, and failure to control tumor growth.5,8,9 Expanding the memory-like CAR–T-cell pool prior to treatment can improve engraftment and in vivo persistence.4,10-12 Modulating genes that foster memory formation or decrease exhaustion, and reducing CAR–T-cell tonic signaling can further support CAR–T-cell persistence and potency.13-17
Memory-like CAR-T cells are linked to upregulation of the gene encoding T-cell factor 1 (TCF-1), Tcf7.14,16 Upstream negative regulators of TCF-1 expression are unknown; clarifying TCF-1 regulation in CAR–T-cell fate determination may pinpoint targets supporting development of CAR-T cells with enhanced persistence and activity. One such potential target is posttranscriptional messenger RNA (mRNA) decay, which is well characterized to regulate T-cell activation, but has a less-defined role in memory formation. The ribonuclease Regnase-1 promotes decay of target mRNAs through recognition of a 3′ untranslated region (UTR) stem-loop motif.18 Regnase-1 targets c-rel, ox40, and il-2 to suppress T-cell activation and effector function, and regulates antitumor T-cell responses by targeting Batf, a transcriptional regulator of effector T-cell (TEFF) differentiation.18-23 We hypothesized that Tcf7 is additionally a direct target of Regnase-1.
Although exhaustion can impair CAR–T-cell activity, studies of chronic viral antigen exposure have identified a class of CD8+ exhausted T cells (TEX) with memory-like features that support T-cell persistence and function.24-26 These precursor exhausted T cells (TPEX) share features of central memory T cells (TCM), but are transcriptionally and epigenetically distinct.27-31 TCF-1 is an essential transcription factor for the development and maintenance of both TPEX and TCM.26,32-34 The role of TPEX in CAR–T-cell responses is unknown.
Our findings here describe a pivotal role for Regnase-1 in regulating CAR–T-cell longevity and function through increased TPEX formation. Regnase-1 deficiency supports CAR–T-cell expansion and persistence, effective recall responses, and sustained remissions in immunocompetent mouse and human xenograft models. By directly targeting TCF-1, Regnase-1 limits CAR–T-cell TPEX development and antitumor responses and promotes T-cell exhaustion. Additionally, Regnase-1 deficiency diminishes exhaustion and increases effector function in TCF-1− CAR-T cells. Our data provide new insights into CAR–T-cell fate regulation and identify TPEX as a key lineage regulated by Regnase-1 and TCF-1 to support long-lasting CAR-T–mediated immunity.
Methods
Mice
C57BL/6, Thy1.1, and Rosa26-Cas9 knock-in mice were obtained from The Jackson Laboratory.35 NSG mice were bred at St. Jude Children’s Research Hospital (SJCRH). Human CD19 CAR-transgenic (CAR-Tg) mice were generated as described in supplemental Methods (available on the Blood Web site). All studies used 8- to 12-week-old male mice housed in an American Association for Accreditation of Laboratory Animal Care (AAALAC)-accredited facility. Experimental protocols were approved by the SJCRH Animal Care and Use Committee in accordance with National Institutes of Health (NIH) guidelines.
Cell lines
Murine luciferase-expressing Arf−/−BCR-ABL1+Ph+ progenitor B-ALL cells were provided by Charles Mullighan (SJCRH) and transduced with MSCV-hCD19-IRES-RFP to generate hCD19+ B-ALL clones.36 Human Raji lymphoma cells from ATCC (CCL-86) were transduced with luciferase.
Murine CAR–T-cell studies
Naive CD4+ or CD8+ T cells from control or CAR-Tg mice were purified by magnetic-activated cell sorting (MACS) (Miltenyi Biotec) and activated with 5 µg/mL anti-mouse CD3 (TONBO) and CD28 (TONBO), followed by culture with 20 U/mL recombinant human interleukin 2 (IL-2; rhIL-2) (PeproTech), 5 ng/mL hIL-7 (PeproTech), and 25 ng/mL hIL-15 (PeproTech) for 3 to 4 days. Naive CD8+ T cells from Cas9-expressing hCD19 CAR-Tg mice were activated and transduced with regnase-1, Tcf7, or nontargeting single guide RNA (sgRNA) vectors as previously described to generate wild-type (WT), knockout (KO), or double knockout (DKO) CAR-T cells.23
For survival analyses, mice were injected IV with hCD19+ B-ALL cells, followed 6 days later by control or CAR–T-cell treatment. Surviving mice received another dose of hCD19+ B-ALL cells 50 days later. For cotransfers, the indicated populations were mixed and administered to tumor-bearing or tumor-free mice. Organs were harvested at the indicated time points for analysis. Detailed procedures are in supplemental Methods.
Luciferase assay
Luciferase activities were measured as previously described using a murine Tcf7 3′ UTR mRNA construct (GeneCopoeia).23
Microarray
Cotransferred cells were sorted from spleens based on fluorescent reporter and congenic marker expression. RNA was isolated and used with the Mouse Clariom S Assay (Thermo Fisher). Differential expression analysis, gene-set enrichment analysis (GSEA), and weighted gene correlation network analysis (WGCNA) were performed as previously described and are detailed in supplemental Methods.23,37
Bisulfite-sequencing methylation profiling
Genomic DNA was isolated and a bisulfite-modified DNA-sequencing library was generated as previously described and detailed in supplemental Methods.38
Human CAR–T-cell studies
CD4+ and CD8+ T cells were purified from peripheral blood mononuclear cells (PBMCs) of healthy donors obtained under an SJCRH-approved protocol, transduced with CD19 CAR lentivirus, and electroporated with Regnase-1–targeting sgRNA (KO CAR) or nontargeting sgRNA (WT CAR). Detailed procedures are described in supplemental Methods.
Cells were stimulated in vitro with irradiated CD19+ Raji cells (1:2 effector-to-target ratio [E:T]) for 7 days, quantified, and restimulated sequentially for 4 cycles. Phenotypes were assessed 24 hours after stimulation. For human xenograft modeling, NSG mice received 2 × 106 luciferase-expressing Raji cells IV followed by day 7 treatment with 2 × 106 (1:1 CD8+:CD4+) CAR or control T cells. Tumor burden was monitored by bioluminescence imaging. Organs were harvested at indicated time points for analysis.
Statistics
Statistical tests were performed using GraphPad Prism 6 software. Statistical analysis and number of replicates are indicated in the figure legends.
Results
Transgenic anti-human CD19 CAR-T cells treat B-ALL in an immunocompetent model
To assess CAR-T function in an immunocompetent and syngeneic model allowing assessment of memory formation, we generated transgenic mice expressing a CAR with an scFv from the clinically relevant FMC63 anti-human CD19 antibody linked to mouse CD8α, 4-1BB, and CD3ζ domains (Figure 1A; supplemental Figure 1A).39,40 CAR expression was detected on all thymic subsets and peripheral T cells (supplemental Figure 1B-C).
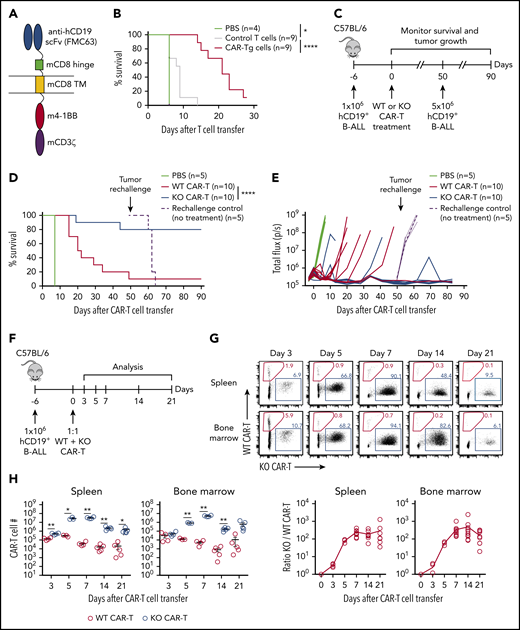
Targeting Regnase-1 leads to durable CAR–T cell-mediated protection in an immunocompetent leukemia model. (A) Schematic of hCD19 CAR-Tg construct. (B) Kaplan-Meier survival analysis of C57BL/6 mice bearing hCD19+ B-ALL tumors treated with phosphate-buffered saline (PBS), or with activated control or CAR-Tg cells (5 × 106 CD4+ and 5 × 106 CD8+). (C-E) Naive MACS-purified CD8+Cas9+ hCD19 CAR-Tg cells were activated and transduced with nontargeting (WT CAR-T) or Regnase-1–targeting (KO CAR-T) sgRNA. Mice bearing hCD19+ B-ALL tumors were treated with PBS or with WT or KO CAR-T cells. Surviving mice and control naive mice were rechallenged with hCD19+ B-ALL cells 50 days later. (C) Rechallenge experimental design. (D) Kaplan-Meier survival analysis. (E) Tumor growth monitored by bioluminescence imaging. (F-H) CD8+ WT and KO CAR-T cells were cotransferred 1:1 into tumor-bearing mice. Organs were harvested at the indicated time points for analysis. (F) Cotransfer experimental design. (G) Frequency and ratio of KO/WT CAR-T cells. (H) Absolute number of WT and KO CAR-T cells shown as mean plus or minus standard error of the mean (SEM) (n = 3-5 mice per group). Significance was determined by log-rank (Mantel-Cox) test (B,D) or paired Student t test (H). Data are representative of (D-E) or pooled from (B,G-H) 2 independent experiments. *P < .05; **P < .01; ***P < .001; ****P < .0001.
Targeting Regnase-1 leads to durable CAR–T cell-mediated protection in an immunocompetent leukemia model. (A) Schematic of hCD19 CAR-Tg construct. (B) Kaplan-Meier survival analysis of C57BL/6 mice bearing hCD19+ B-ALL tumors treated with phosphate-buffered saline (PBS), or with activated control or CAR-Tg cells (5 × 106 CD4+ and 5 × 106 CD8+). (C-E) Naive MACS-purified CD8+Cas9+ hCD19 CAR-Tg cells were activated and transduced with nontargeting (WT CAR-T) or Regnase-1–targeting (KO CAR-T) sgRNA. Mice bearing hCD19+ B-ALL tumors were treated with PBS or with WT or KO CAR-T cells. Surviving mice and control naive mice were rechallenged with hCD19+ B-ALL cells 50 days later. (C) Rechallenge experimental design. (D) Kaplan-Meier survival analysis. (E) Tumor growth monitored by bioluminescence imaging. (F-H) CD8+ WT and KO CAR-T cells were cotransferred 1:1 into tumor-bearing mice. Organs were harvested at the indicated time points for analysis. (F) Cotransfer experimental design. (G) Frequency and ratio of KO/WT CAR-T cells. (H) Absolute number of WT and KO CAR-T cells shown as mean plus or minus standard error of the mean (SEM) (n = 3-5 mice per group). Significance was determined by log-rank (Mantel-Cox) test (B,D) or paired Student t test (H). Data are representative of (D-E) or pooled from (B,G-H) 2 independent experiments. *P < .05; **P < .01; ***P < .001; ****P < .0001.
CAR-Tg T-cell function was assessed using mouse Arf−/−BCR-ABL1+ Philadelphia chromosome–positive (Ph+) B-cell progenitor ALL cells transduced with hCD19 (hCD19+ B-ALL).36 CAR-Tg cells lysed hCD19+ targets efficiently in vitro, whereas control T cells did not; both groups were ineffective against hCD19– B-ALL cells (supplemental Figure 1D). Treatment of mice bearing hCD19+ B-ALL tumors with CAR-Tg cells significantly extended survival and reduced tumor burden relative to control T-cell treatment (Figure 1B; supplemental Figure 1E). Thus, CAR-Tg cells exhibit antigen-specific cytotoxicity in an immunocompetent B-ALL model.
Regnase-1 deletion enhances CAR–T-cell persistence and supports durable antitumor responses
To evaluate the role of Regnase-1 on CAR–T-cell function, naive CD8+ T cells from Cas9+ hCD19 CAR-Tg mice were activated and transduced with nontargeting (WT CAR) or Regnase-1–targeting (KO CAR) sgRNA and transferred into mice bearing hCD19+ B-ALL tumors (Figure 1C). Both WT and KO CAR-T cells induced early tumor remission, but WT CAR-T–treated mice were more prone to relapse and death (Figure 1D-E). To determine whether Regnase-1–deficient CAR-T cells conferred long-lasting protection, surviving tumor-free mice were given a secondary tumor challenge at day 50 (Figure 1C). Secondary tumor engraftment was prevented in KO CAR-T–treated mice, with the mice surviving 40 days after rechallenge, whereas control mice without CAR-T cells rapidly succumbed (Figure 1D-E). Thus, Regnase-1 KO CAR–T-cell treatment reduced relapse incidence during the primary tumor response and conferred protection against secondary tumor challenge.
To compare WT and Regnase-1–deficient CAR-T cells in the same tumor environment, activated WT and KO cells were cotransferred 1:1 into tumor-bearing mice (Figure 1F; supplemental Figure 1F). KO CAR-T cells expanded at early time points, outnumbering WT cells by over 100-fold at day 7. This ratio was maintained through day 21 (Figure 1G-H). These results indicate that targeting Regnase-1 confers protection from primary and secondary tumors with markedly increased CAR–T-cell expansion and persistence.
Regnase-1 deletion promotes formation of memory-like CAR-T cells with recall capacity
We assessed the kinetics of WT and Regnase-1 KO CAR memory cell accumulation following 1:1 cotransfer into tumor-bearing mice. KO CAR-T cells had more CD44+CD62L− TEFF than WT cells at day 3 (Figure 2A). The Regnase-1 target basic leucine zipper ATF-like transcription factor (BATF) was increased in KO CAR-T cells, potentially fostering the early TEFF accumulation (supplemental Figure 2A).22,23 By days 14 to 21, CD44+CD62L+ memory-like cells became progressively dominant among KO cells, whereas CD44+CD62L− TEFF remained predominant in WT cells (Figure 2A).
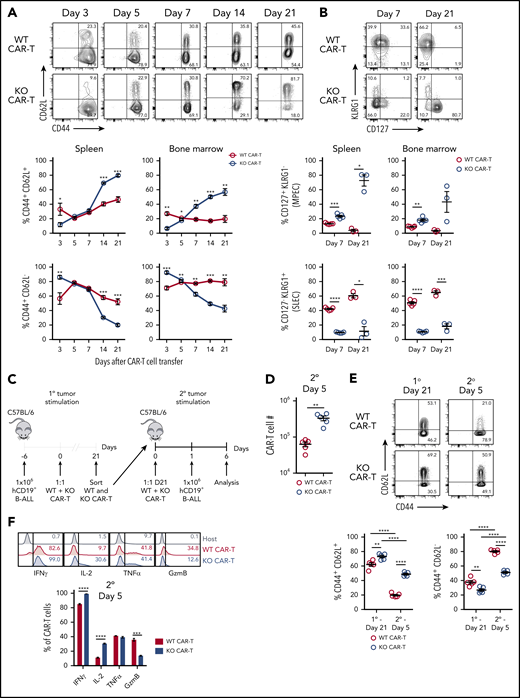
Regnase-1 deletion promotes formation of memory-like CAR-T cells with recall capacity. (A-B) CD8+ WT and KO CAR-T cells were cotransferred 1:1 into tumor-bearing mice. Organs were harvested at indicated time points. Representative plots are shown for spleen. (A) Frequencies of CD44+CD62L− and CD44+CD62L+ WT and KO CAR-T cells. (B) Frequencies of CD127+KLRG1− and CD127−KLRG1+ WT and KO CAR-T cells. (C-F) CD8+ WT and KO CAR-T cells were cotransferred 1:1 into tumor-bearing mice, sorted from spleens 21 days later, and cotransferred 1:1 again into naive recipients that received hCD19+ B-ALL cells the following day. Spleens were harvested 5 days later. (C) Experimental design schematic. (D) Absolute number of WT and KO CAR-T cells 5 days after secondary tumor stimulation. (E) Frequencies of CD44+CD62L− and CD44+CD62L+ WT and KO CAR-T cells 21 days after primary tumor stimulation (left) and 5 days after cotransfer and secondary tumor stimulation (right). (F) Frequencies of IFNγ+, IL-2+, TNFα+, and granzyme B+ (GzmB) WT and KO CAR-T cells from spleens 5 days after cotransfer and secondary tumor stimulation. Endogenous host CD8+ T cells are included as a gating control. Significance was determined by paired Student t test (A-B,D,F) or 2-way analysis of variance (ANOVA) with the Tukey posttest for multiple comparisons (E). Data are shown as mean plus or minus SEM and represent 2 independent experiments with 3 to 5 mice per group (A-B,D-F). *P < .05; **P < .01; ***P < .001; ****P < .0001.
Regnase-1 deletion promotes formation of memory-like CAR-T cells with recall capacity. (A-B) CD8+ WT and KO CAR-T cells were cotransferred 1:1 into tumor-bearing mice. Organs were harvested at indicated time points. Representative plots are shown for spleen. (A) Frequencies of CD44+CD62L− and CD44+CD62L+ WT and KO CAR-T cells. (B) Frequencies of CD127+KLRG1− and CD127−KLRG1+ WT and KO CAR-T cells. (C-F) CD8+ WT and KO CAR-T cells were cotransferred 1:1 into tumor-bearing mice, sorted from spleens 21 days later, and cotransferred 1:1 again into naive recipients that received hCD19+ B-ALL cells the following day. Spleens were harvested 5 days later. (C) Experimental design schematic. (D) Absolute number of WT and KO CAR-T cells 5 days after secondary tumor stimulation. (E) Frequencies of CD44+CD62L− and CD44+CD62L+ WT and KO CAR-T cells 21 days after primary tumor stimulation (left) and 5 days after cotransfer and secondary tumor stimulation (right). (F) Frequencies of IFNγ+, IL-2+, TNFα+, and granzyme B+ (GzmB) WT and KO CAR-T cells from spleens 5 days after cotransfer and secondary tumor stimulation. Endogenous host CD8+ T cells are included as a gating control. Significance was determined by paired Student t test (A-B,D,F) or 2-way analysis of variance (ANOVA) with the Tukey posttest for multiple comparisons (E). Data are shown as mean plus or minus SEM and represent 2 independent experiments with 3 to 5 mice per group (A-B,D-F). *P < .05; **P < .01; ***P < .001; ****P < .0001.
Consistently, KO CAR-T cells had a higher proportion of CD127+KLRG1− memory-precursor effector cells (MPECs) 7 days after transfer, whereas WT cells had more CD127−KLRG1+ short-lived effector cells (SLECs) (Figure 2B). These populations were further enriched at day 21, indicating preferential memory formation in KO CAR-T cells. This was associated with increased IL-2 and reduced tumor necrosis factor α (TNFα) and Ki67 in KO CAR-T cells (supplemental Figure 2B-C). Regnase-1 KO CAR-T cells maintained high interferon γ (IFNγ) expression, indicating retention of effector function (supplemental Figure 2C). Thus, Regnase-1 KO CAR-T cells progressively convert from an early predominantly TEFF phenotype into a largely memory-like population, with a crossover point at ∼7 days.
To evaluate recall responses, WT and KO CAR-T cells were isolated 21 days after cotransfer into tumor-bearing mice and cotransferred again 1:1 into naive recipients that then received tumors (Figure 2C). Five days later, both WT and KO CAR-T cells upregulated Ki67, but the KO cells outnumbered WT by approximately fivefold (Figure 2D; supplemental Figure 2D). KO CAR-T cells maintained a larger percentage of CD44+CD62L+ cells and increased IL-2 and reduced granzyme B production, further indicating preservation of a memory-like population during the recall response (Figure 2E-F). Thus, the increased expansion, persistence, and memory-like features associated with the primary antitumor response of Regnase-1 KO CAR-T cells are maintained upon secondary antigen exposure.
Regnase-1 KO CAR–T-cell reprograming is tumor dependent
To determine the antigen dependence of Regnase-1 on CAR–T-cell programming, WT and KO CAR-T cells were cotransferred 1:1 into tumor-bearing or tumor-free mice. More KO than WT cells were present 7 days after transfer regardless of tumor presence, but the KO-to-WT ratio was significantly higher in the presence of tumor (Figure 3A). At day 21, KO cells persisted in greater numbers with tumor, but both WT and KO CAR-T cells were depleted in tumor-free mice, demonstrating an antigen dependence for CAR-T persistence (Figure 3A). In tumor-free mice, most day 7 KO CAR-T cells were CD44+CD62L− TEFF, whereas WT CAR-T cells contained more naive CD44−CD62L+ and memory-like CD44+CD62L+ cells, consistent with inhibition of T-cell activation by Regnase-1 (Figure 3B; supplemental Figure 3A).19 Tumor stimulation increased the proportion of CD44+CD62L− TEFF cells among WT but not KO CAR-T cells. Consistently, KO cells included fewer CD127+KLRG1− MPECs than WT without tumor, and this trend was reversed upon tumor priming (supplemental Figure 3B).
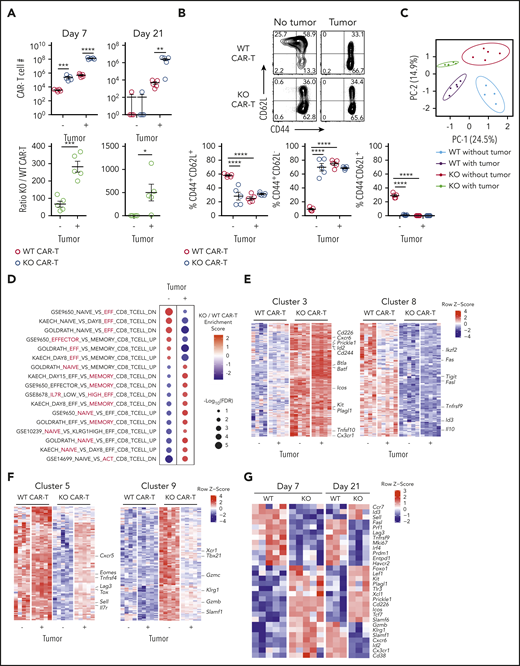
Regnase-1 KO CAR-T cells undergo tumor-dependent reprogramming from effector- to memory-like cells. CD8+ WT and KO CAR-T cells were cotransferred 1:1 into tumor-bearing or tumor-free mice. Spleens were harvested at the indicated time points. (A) Absolute number (top) and ratio of KO/WT CAR-T cells (bottom) 7 and 21 days after cotransfer. (B) Frequencies of CD44+CD62L−, CD44+CD62L+, and CD44+CD62L− WT and KO CAR-T cells 7 days after cotransfer. Data are shown as mean plus or minus SEM and represent 3 independent experiments with 5 mice per group. Significance was determined by the paired Student t test (A, top), unpaired Student t test (A, bottom), or 2-way ANOVA with the Tukey posttest for multiple comparison (B). (C-F) Microarray analysis of transcripts in CD8+ WT and KO CAR-T cells sorted 7 days after cotransfer from spleens of mice with or without tumors. (C) Principal component analysis (PCA) of all samples. (D) Gene-set enrichment analysis (GSEA) comparing differential gene expression between KO and WT CAR (KO/WT) with and without tumor to the C7 immunologic signatures gene-set collection.54 Red circles represent enrichment in KO CAR. Blue circles represent enrichment in WT CAR. The enriched subset in each gene set is highlighted in red. (E-F) WGCNA was performed on the top third of variable genes across 4 pairwise comparisons: KO vs WT without tumor, KO vs WT with tumor, KO with vs without tumor, and WT with vs without tumor; followed by filtering for differentially expressed (DE) genes at false discovery rate (FDR) < 0.05 and log2 fold change (FC) > 0.5 in at least 1 comparison. (E) Heatmaps of genes in clusters 3 and 8, and (F) clusters 5 and 9. (G) Expression of select genes in WT and KO CAR-T cells from tumor-bearing mice 7 and 21 days after cotransfer. Data represent biological replicates of 3 to 5 mice per group (C-G). *P < .05; **P < .01; ***P < .001; ****P < .0001. DN, downregulated gene set; UP, upregulated gene set.
Regnase-1 KO CAR-T cells undergo tumor-dependent reprogramming from effector- to memory-like cells. CD8+ WT and KO CAR-T cells were cotransferred 1:1 into tumor-bearing or tumor-free mice. Spleens were harvested at the indicated time points. (A) Absolute number (top) and ratio of KO/WT CAR-T cells (bottom) 7 and 21 days after cotransfer. (B) Frequencies of CD44+CD62L−, CD44+CD62L+, and CD44+CD62L− WT and KO CAR-T cells 7 days after cotransfer. Data are shown as mean plus or minus SEM and represent 3 independent experiments with 5 mice per group. Significance was determined by the paired Student t test (A, top), unpaired Student t test (A, bottom), or 2-way ANOVA with the Tukey posttest for multiple comparison (B). (C-F) Microarray analysis of transcripts in CD8+ WT and KO CAR-T cells sorted 7 days after cotransfer from spleens of mice with or without tumors. (C) Principal component analysis (PCA) of all samples. (D) Gene-set enrichment analysis (GSEA) comparing differential gene expression between KO and WT CAR (KO/WT) with and without tumor to the C7 immunologic signatures gene-set collection.54 Red circles represent enrichment in KO CAR. Blue circles represent enrichment in WT CAR. The enriched subset in each gene set is highlighted in red. (E-F) WGCNA was performed on the top third of variable genes across 4 pairwise comparisons: KO vs WT without tumor, KO vs WT with tumor, KO with vs without tumor, and WT with vs without tumor; followed by filtering for differentially expressed (DE) genes at false discovery rate (FDR) < 0.05 and log2 fold change (FC) > 0.5 in at least 1 comparison. (E) Heatmaps of genes in clusters 3 and 8, and (F) clusters 5 and 9. (G) Expression of select genes in WT and KO CAR-T cells from tumor-bearing mice 7 and 21 days after cotransfer. Data represent biological replicates of 3 to 5 mice per group (C-G). *P < .05; **P < .01; ***P < .001; ****P < .0001. DN, downregulated gene set; UP, upregulated gene set.
We also analyzed effects of Regnase-1 deletion in T cells lacking CAR. Equal numbers of CD8+ WT CAR-Tg, KO CAR-Tg, WT B6, and KO B6 T cells were cotransferred into tumor-bearing mice. No significant difference between the number of WT and KO B6 T cells was detected, whereas KO CAR-T cells were increased compared with other T cells types (supplemental Figure 3C). Together, these results indicate that the dramatic expansion of T cells observed in the presence of tumor requires both CAR presence and Regnase-1 deletion.
We next performed transcriptional profiling of WT and KO CAR-T cells isolated 7 days after cotransfer into tumor-bearing or tumor-free mice. Each group clustered independently, indicating distinct cell-intrinsic and tumor-dependent effects of Regnase-1 deletion (Figure 3C). KO CAR-T cells were enriched for effector gene sets in tumor-free mice and for naive- and memory-associated gene sets in tumor-bearing mice, relative to WT cells (Figure 3D). WGCNA across 4 pairwise comparisons (KO or WT with or without tumor) grouped 1856 highly variable genes into 10 clusters based on differential expression patterns (supplemental Figure 3D; supplemental Table 1). Tumor-independent gene upregulation in KO CAR-T cells relative to WT is shown in cluster 3 and downregulation in cluster 8, and indicates upregulation of effector-associated (Cd226, Id2, Icos, Tnfsf10, Cx3cr1) and survival-associated genes (Batf, Kit, Prickle1), and downregulation of genes associated with T-cell exhaustion (Ikzf2, Tigit, Tnfrsf9, Il10) and activation-induced cell death (Fas, Fasl) (Figure 3E).
Tumor-dependent gene-expression changes in KO vs WT CAR-T cells are in clusters 5 (upregulated) and 9 (downregulated) (Figure 3F). Cluster 5 is highly enriched for memory- and TPEX-associated genes (Eomes, Sell, Il7r, Cxcr5, Tox), whereas cluster 9 shows enrichment for effector-associated genes (Xcr1, Gzmb, Tbx21, and Klrg1) (Figure 3F; supplemental Figure 3E). Increased relative expression of memory-associated transcription factors (Tcf7, Foxo1, Lef1, Slamf6) in KO relative to WT CAR-T cells from tumor-bearing mice was evident by day 7 (Figure 3G). Transcriptional profiling of WT and KO CAR-T cells 21 days after cotransfer to tumor-bearing mice further indicated higher memory-associated (Ccr7, Id3, Sell) and lower effector-associated (Gzmb, Klrg1, Cxcr6, Id2, Cx3cr1) gene expression in KO CAR-T cells relative to WT (Figure 3G). These results support Regnase-1 KO CAR-T cells undergoing a tumor-dependent shift from an effector- to a memory-like phenotype, with reprogramming initiated early during the antitumor response and further developed at late time points.
Targeting Regnase-1 enhances formation of TCF-1+ TPEX CAR-T cells
Both WT and KO CAR-T cells from tumor-bearing mice upregulated Tox, Pdcd1, and Lag3 relative to cells from tumor-free mice, suggesting that tumor priming fosters exhaustion. However, KO CAR-T cells from tumor-bearing mice also upregulated memory-associated genes (Il7r, Sell, Tcf7, Id3, Lef1, Foxo1) while downregulating effector-associated (Tbx21, Gzmb, Irf4, Prdm1) and exhaustion-associated (Entpd1, Havcr2) genes (Figure 4A). This transcriptional program resembles that of memory-like TCF-1+ TPEX that control chronic infections and can maintain tumor immunity.26,31,41,42 Regnase-1 KO CAR-T cells had a higher proportion of TCF-1+ cells with higher TCF-1 expression levels than WT CAR-T cells after cotransfer into tumor-bearing mice (Figure 4B; supplemental Figure 4A). This enrichment was tumor dependent (supplemental Figure 4B). Consistent with an exhausted state, both WT and KO TCF-1+ CAR-T cells expressed thymocyte selection associated high mobility group box protein (TOX) and programmed cell death protein 1 (PD-1) (Figure 4C; supplemental Figure 4C).
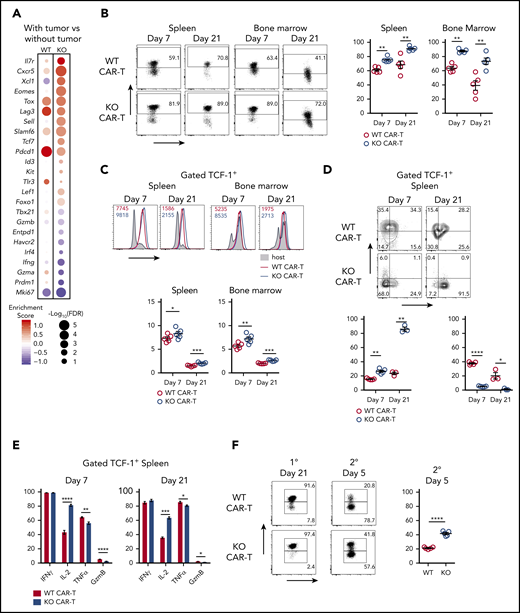
Regnase-1 limits formation of TPEXCAR-T cells and reduces their memory phenotype. (A) Comparison of TPEX-related gene expression in the presence vs absence of tumor in either WT (left) or KO (right) CAR-T cells. Cutoff FDR < 0.05 or log2FC > 0.5. Data represent biological replicates of 4 to 5 mice per group. (B-E) CD8+ WT and KO CAR-T cells were cotransferred 1:1 into tumor-bearing mice. Organs were harvested 7 and 21 days later. (B) Frequencies of TCF-1+ WT and KO CAR-T cells. (C) Mean fluorescence intensity (MFI) of TOX in TCF-1+ WT and KO CAR-T cells. (D) Frequencies of TCF-1+CD127+KLRG1− and TCF-1+CD127−KLRG1+ WT and KO CAR-T cells. (E) Frequencies of TCF-1+IFNγ+, TCF-1+IL-2+, TCF-1+TNFα+, or TCF-1+granzyme B+ WT and KO CAR-T cells. (F) CD8+ WT and KO CAR-T cells were cotransferred 1:1 into tumor-bearing mice and sorted from spleens 21 days later. Sorted cells (>90% TCF-1+) were cotransferred 1:1 into naive recipients that received hCD19+ B-ALL cells the following day. Spleens were harvested 5 days later. Frequencies of TCF-1+ WT and KO CAR-T cells 21 days after primary tumor stimulation (left) and 5 days after secondary cotransfer and tumor stimulation (right). Data are shown as mean plus or minus SEM and represent 2 independent experiments (B-F; n = 3-5 mice per group). Significance was determined by the paired Student t test (B-F). *P < .05; **P < .01; ***P < .001; ****P < .0001.
Regnase-1 limits formation of TPEXCAR-T cells and reduces their memory phenotype. (A) Comparison of TPEX-related gene expression in the presence vs absence of tumor in either WT (left) or KO (right) CAR-T cells. Cutoff FDR < 0.05 or log2FC > 0.5. Data represent biological replicates of 4 to 5 mice per group. (B-E) CD8+ WT and KO CAR-T cells were cotransferred 1:1 into tumor-bearing mice. Organs were harvested 7 and 21 days later. (B) Frequencies of TCF-1+ WT and KO CAR-T cells. (C) Mean fluorescence intensity (MFI) of TOX in TCF-1+ WT and KO CAR-T cells. (D) Frequencies of TCF-1+CD127+KLRG1− and TCF-1+CD127−KLRG1+ WT and KO CAR-T cells. (E) Frequencies of TCF-1+IFNγ+, TCF-1+IL-2+, TCF-1+TNFα+, or TCF-1+granzyme B+ WT and KO CAR-T cells. (F) CD8+ WT and KO CAR-T cells were cotransferred 1:1 into tumor-bearing mice and sorted from spleens 21 days later. Sorted cells (>90% TCF-1+) were cotransferred 1:1 into naive recipients that received hCD19+ B-ALL cells the following day. Spleens were harvested 5 days later. Frequencies of TCF-1+ WT and KO CAR-T cells 21 days after primary tumor stimulation (left) and 5 days after secondary cotransfer and tumor stimulation (right). Data are shown as mean plus or minus SEM and represent 2 independent experiments (B-F; n = 3-5 mice per group). Significance was determined by the paired Student t test (B-F). *P < .05; **P < .01; ***P < .001; ****P < .0001.
TCF-1+ Regnase-1 KO CAR-T cells also included more CD127+KLRG1− MPECs than WT cells, indicating a shift toward long-lived memory (Figure 4D; supplemental Figure 4D). This was paralleled with a higher frequency of CD62L positivity among TCF-1+ KO CAR-T, which also expressed higher levels of CD62L (supplemental Figure 4E-F), reduced Ki67, TNFα, and granzyme B expression, and increased IL-2 expression (Figure 4E; supplemental Figure 4G-H). Thus, Regnase-1–deficient CAR-T cells have an expanded pool of tumor-induced TCF-1+ TPEX cells with more memory-associated features than WT TCF-1+ cells.
To examine recall responses within the TCF-1+ subset, WT and KO CAR-T cells were isolated 21 days after cotransfer into tumor-bearing mice. WT and KO CAR-T (>90% TCF-1+) were then cotransferred 1:1 into naive mice that received tumors. Following restimulation, both WT and KO CAR-T cells showed reduced proportions of TCF-1+ cells, suggesting that the TCF-1+ population gave rise to TCF-1− cells. However, KO CAR-T cells retained a larger proportion of TCF-1+ cells than WT (Figure 4F). The TCF-1− cells retained TOX expression, consistent with their origin from transferred TCF-1+TOX+ TPEX cells (supplemental Figure 4I).32,43,44 These results indicate that Regnase-1 deficiency fosters the development of TCF-1+ TPEX CAR-T cells that respond to secondary tumor challenge.
Regnase-1 represses memory-associated epigenetic programs
To further resolve differentiation status, whole-genome DNA methylation profiles of Regnase-1 KO and WT CAR-T cells were compared with established T-cell subset epigenetic profiles.45 CAR-T cells isolated from tumor-bearing mice at day 21 were >95% Ly108+, a surrogate surface marker for TCF-1 (supplemental Figure 5A).32 Nearly 5000 differentially methylated regions (DMRs) between KO and WT cells were identified (supplemental Figure 5B). Methylation profiles of both cell types were most like TEX, with fewer DMRs than in comparison with other subsets (Figure 5A).
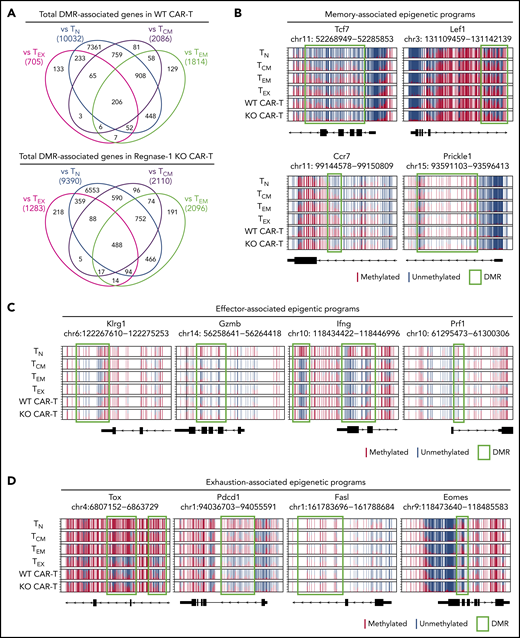
Regnase-1 KO CAR-T cells acquire memory-, effector-, and exhaustion-associated epigenetic programs. CD8+ WT and KO CAR-T cells were cotransferred 1:1 into tumor-bearing mice. CD8+Ly108+ cells were sorted from spleens 21 days later for whole-genome DNA methylation profiling. (A) Number of DMRs in Ly108+ WT and KO CAR–T-cell genomes relative to TN, TCM, effector memory T cell (TEM), and TEX genomes. The number of demethylated and methylated regions were calculated based on ≥30% and ≤30% methylation difference between the 2 populations, respectively. (B) Normalized CpG methylation plots at sites surrounding and within DMRs of memory-associated genes, (C) effector-associated genes, and (D) exhaustion-associated genes obtained by whole genome bisulfite sequencing (WGBS) analysis. Red and blue lines depict methylated and unmethylated CpG sites, respectively. DMRs are indicated by green boxes. Data represent 10 biological replicates pooled for triplicate analysis.
Regnase-1 KO CAR-T cells acquire memory-, effector-, and exhaustion-associated epigenetic programs. CD8+ WT and KO CAR-T cells were cotransferred 1:1 into tumor-bearing mice. CD8+Ly108+ cells were sorted from spleens 21 days later for whole-genome DNA methylation profiling. (A) Number of DMRs in Ly108+ WT and KO CAR–T-cell genomes relative to TN, TCM, effector memory T cell (TEM), and TEX genomes. The number of demethylated and methylated regions were calculated based on ≥30% and ≤30% methylation difference between the 2 populations, respectively. (B) Normalized CpG methylation plots at sites surrounding and within DMRs of memory-associated genes, (C) effector-associated genes, and (D) exhaustion-associated genes obtained by whole genome bisulfite sequencing (WGBS) analysis. Red and blue lines depict methylated and unmethylated CpG sites, respectively. DMRs are indicated by green boxes. Data represent 10 biological replicates pooled for triplicate analysis.
We next compared the methylation profiles at DMRs associated with CD8+ subset-specific memory, effector, and exhaustion programs. Regnase-1 KO CAR-T cells retained unmethylated programs resembling those found in naive T cells (TN) and TCM at the Tcf7, Lef1, Ccr7, and Prickle1 loci, indicating a transcriptionally permissive status of memory-associated genes, whereas WT cells were more methylated at these loci (Figure 5B). DMRs at effector-associated loci (Klrg1, Gzmb, Ifng, and Prf1) were generally unmethylated in both groups, indicating maintenance of effector functions (Figure 5C). The Fasl locus in KO cells was highly methylated, as in stem-like cells, whereas WT cells were similar to TEX (Figure 5D). DMRs of exhaustion-associated loci (Tox, Pdcd1, Eomes) in both WT and KO CAR-T cells were highly unmethylated and resemble the TEX profile (Figure 5D). These results substantiate our characterization of WT and KO Ly108+ (TCF-1+) CAR-T cells as TPEX and implicate Regnase-1 in the epigenetic repression of memory-associated programs.
Improved persistence and TPEX formation in Regnase-1 KO CAR-T cells is TCF-1 dependent
We next asked whether Regnase-1 directly targets Tcf7 mRNA using a 3′ UTR luciferase reporter. Regnase-1 inhibited luciferase activity in a dose-dependent manner, but the D141N Regnase-1 mutant lacking RNase activity did not (Figure 6A). Therefore, Tcf7 mRNA is a Regnase-1 target. To distinguish TCF-1–dependent and –independent effects of Regnase-1, activated CD8+Cas9+ CAR-Tg cells were transduced with sgRNAs targeting either Regnase-1 or both Regnase-1 and TCF-1. Approximately 30% of Regnase-1/TCF-1 DKO CAR-T cells expressed TCF-1, indicating incomplete biallelic disruption (supplemental Figure 6A). The KO and DKO cells were cotransferred 5:1 into tumor-bearing mice and assessed 7, 14, and 21 days later (supplemental Figure 6B). After normalization for input ratio, significantly more KO than DKO CAR-T cells were identified, and this proportion increased over time (Figure 6B; supplemental Figure 6C).
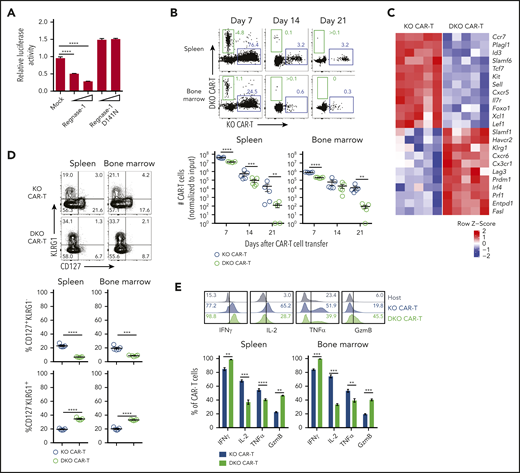
Improved persistence and expanded TPEX formation in Regnase-1 KO CAR-T cells is TCF-1 dependent. (A) Luciferase activity of HEK293T cells after transfection with Tcf7 mRNA 3′ UTR reporter, together with control (mock), Regnase-1 wild-type, or Regnase-1 D141N-expressing plasmid. (B-E) Naive MACS-purified CD8+Cas9+ hCD19 CAR-Tg cells were activated and transduced with sgRNA targeting Regnase-1 (KO CAR) or Regnase-1 and TCF-1 (DKO CAR). Cells were sorted based on fluorescent reporter expression and cotransferred 5:1 (KO:DKO) into tumor-bearing mice. Organs were harvested at the indicated time points. (B) Frequency and numbers of KO and DKO CAR-T cells (normalized to input ratio). (C) Relative expression of select genes from microarray analysis of KO and DKO CAR-T cells harvested from spleens of tumor-bearing mice 7 days after cotransfer (n = 5 mice per group). (D) Frequencies of CD127+KLRG1− and CD127−KLRG1+ KO and DKO CAR-T cells 7 days after cotransfer. (E) Representative histograms (spleen only) and frequencies of IFNγ+, IL-2+, TNFα+, and granzyme B+ KO and DKO CAR-T cells from the spleens and bone marrow of tumor-bearing mice 7 days after cotransfer. Endogenous host CD8+ T cells are shown as a gating control. Significance was determined by 1-way ANOVA with Tukey posttest for multiple comparisons (A) or paired Student t test (B,D-E). Data are shown as mean plus or minus SEM (A-B,D-E) and represent 3 (A) or 2 independent experiments with 5 mice per group (B,D-E). *P < .05; **P < .01; ***P < .001; ****P < .0001.
Improved persistence and expanded TPEX formation in Regnase-1 KO CAR-T cells is TCF-1 dependent. (A) Luciferase activity of HEK293T cells after transfection with Tcf7 mRNA 3′ UTR reporter, together with control (mock), Regnase-1 wild-type, or Regnase-1 D141N-expressing plasmid. (B-E) Naive MACS-purified CD8+Cas9+ hCD19 CAR-Tg cells were activated and transduced with sgRNA targeting Regnase-1 (KO CAR) or Regnase-1 and TCF-1 (DKO CAR). Cells were sorted based on fluorescent reporter expression and cotransferred 5:1 (KO:DKO) into tumor-bearing mice. Organs were harvested at the indicated time points. (B) Frequency and numbers of KO and DKO CAR-T cells (normalized to input ratio). (C) Relative expression of select genes from microarray analysis of KO and DKO CAR-T cells harvested from spleens of tumor-bearing mice 7 days after cotransfer (n = 5 mice per group). (D) Frequencies of CD127+KLRG1− and CD127−KLRG1+ KO and DKO CAR-T cells 7 days after cotransfer. (E) Representative histograms (spleen only) and frequencies of IFNγ+, IL-2+, TNFα+, and granzyme B+ KO and DKO CAR-T cells from the spleens and bone marrow of tumor-bearing mice 7 days after cotransfer. Endogenous host CD8+ T cells are shown as a gating control. Significance was determined by 1-way ANOVA with Tukey posttest for multiple comparisons (A) or paired Student t test (B,D-E). Data are shown as mean plus or minus SEM (A-B,D-E) and represent 3 (A) or 2 independent experiments with 5 mice per group (B,D-E). *P < .05; **P < .01; ***P < .001; ****P < .0001.
Transcriptional profiling of KO and DKO CAR-T cells isolated 7 days after cotransfer revealed downregulation of TPEX and memory-associated genes (Tcf7, Sell, Plagl1, Il7r, Id3, Kit, Ccr7, Slamf6) in DKO relative to KO cells, and upregulation of effector-associated (Tbx21, Prdm1, Klrg1, Prf1, Ccr6) and exhaustion-associated genes (Havcr2, Entpd1) (Figure 6C). Correspondingly, KO cells were enriched for memory- and naive-associated gene sets whereas DKO CAR-T cells were enriched for effector gene sets (supplemental Figure 6D). DKO CAR-T cells had more CD127–KLRG1+ cells and fewer CD127+KLRG1− and CD44+CD62L+ cells than KO CAR-T cells (Figure 6D; supplemental Figure 6E). DKO CAR-T cells also expressed more Ki67 and granzyme B, and reduced IL-2 (Figure 6E; supplemental Figure 6F). These results indicate that TCF-1 is required for the improved persistence and memory-like phenotype associated with increased TPEX abundance in Regnase-1 KO CAR-T cells.
Regnase-1 deletion prevents TCF-1− CAR–T-cell exhaustion
Although TCF-1+ CAR-T cells were enriched in tumor-bearing mice, a significant proportion remained TCF-1− at day 7 (Figure 4B). Among these, TCF-1− Regnase-1 KO CAR-T cells expressed less TOX than WT controls (supplemental Figure 7A). TOX expression was low in both WT and KO cells without tumor, and only increased in TCF-1− WT cells with tumor, indicating that Regnase-1 supports TOX expression in TCF-1− cells (supplemental Figure 7B). TCF-1− KO cells also expressed lower levels of inhibitory receptors PD-1, LAG3, and TIGIT and the transcription factor EOMES (supplemental Figure 7C-D), indicating that Regnase-1 deficiency reduces exhaustion in TCF-1− cells following tumor stimulation.46
More TCF-1− KO cells expressed IL-2, IFNγ, and granzyme B relative to WT cells, further indicating reduced exhaustion and increased effector function (supplemental Figure 7E). Despite this, TCF-1− KO CAR-T cells had fewer CD127−KLRG1+ cells than WT (supplemental Figure 7F). Thus, Regnase-1 deficiency leads to a less-exhausted TCF-1− CAR–T-cell population with an enhanced effector phenotype. These findings indicate that Regnase-1 deficiency enhances CAR-T–mediated antitumor immunity both by supporting the formation and function of TCF-1+ TPEX and by decreasing exhaustion in TCF-1− cells.
Regnase-1 deletion supports human CAR–T-cell expansion and function
To examine the effects of Regnase-1 deficiency on human CD19 CAR-T cells, we identified human Regnase-1–targeting sgRNAs with high editing efficiency (Figure 7A-B). Both CD8+ and CD4+ Regnase-1–deficient human CAR-T cells exhibited increased expansion after in vitro stimulation with CD19+ Raji cells, persisting through 4 rounds of restimulation (Figure 7C). Regnase-1 KO CAR-T cells maintained higher CD25 and CCR7 expression, and increased proportions of IL-2+, TNFα+, IFNγ+, and granzyme B+ cells than WT CAR-T cells (Figure 7D-F).
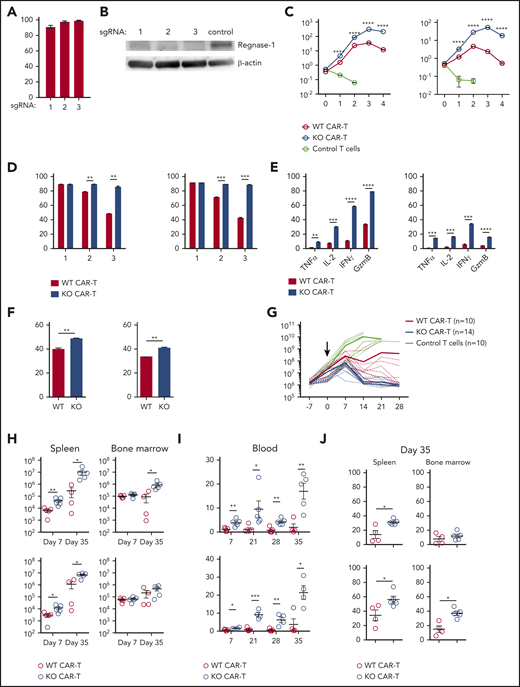
Regnase-1 deletion improves human CAR-T-cell expansion and function. (A) Editing efficacy of candidate Regnase-1 targeting sgRNAs in human CD19 CAR-T cells as determined by targeted deep sequencing. (B) Western blot analysis of Regnase-1 in hCD19 CAR-T cells 24 hours after ribonucleoprotein (RNP) transfection. (C-F) Human WT and Regnase-1 KO CAR-T cells were cocultured with irradiated Raji cells at a 1:2 E:T ratio. CAR-T cells were harvested and counted 7 days later, followed by restimulation with irradiated Raji cells. Phenotypic analysis was performed 24 hours after stimulation. Data are shown as mean plus or minus SEM and represent 3 independent experiments with CAR-T cells from 3 donors targeted with sgRNA-1. Similar results were obtained with sgRNA-3. (C) Number of CD8+ and CD4+ WT and Regnase-1 KO CAR-T cells or untransduced donor T cells after each round of stimulation. (D) Proportion of CD25+CD8+ and CD4+ CAR-T cells after each stimulation. (E) Proportion of TNFα+, IL-2+, IFNγ+, and granzyme B+CD8+ and CD4+ CAR-T cells after 3 rounds of stimulation. (F) Proportion of CCR7+CD8+ and CD4+ CAR-T cells after 3 rounds of stimulation. (G-J) NSG mice bearing Raji tumors were treated with untransduced human T cells, or with activated CD8+ and CD4+ (1:1) Regnase-1 KO or WT CAR-T cells. (G) Tumor growth monitored by bioluminescence imaging. Solid lines represent means and dotted lines represent individual mice. Data were pooled from 2 independent experiments. (H) CD8+ and CD4+ CAR-T-cell number in spleen and bone marrow 7 and 35 days after transfer. (I) Percentage of CD8+ and CD4+ CAR-T cells in blood at indicated time points. (J) Percentage of TCF-1+ CD8+ and CD4+ CAR-T cells in spleen and bone marrow 35 days after transfer. Significance was determined by multiple Student t test with Holm-Sidak correction for multiple comparisons (C-E), unpaired Student t test (F), or Mann-Whitney test (H-J). Data are shown as mean plus or minus SEM and represent 2 independent experiments with 4 to 5 mice per group (H-J). *P < .05; **P < .01; ***P < .001; ****P < .0001.
Regnase-1 deletion improves human CAR-T-cell expansion and function. (A) Editing efficacy of candidate Regnase-1 targeting sgRNAs in human CD19 CAR-T cells as determined by targeted deep sequencing. (B) Western blot analysis of Regnase-1 in hCD19 CAR-T cells 24 hours after ribonucleoprotein (RNP) transfection. (C-F) Human WT and Regnase-1 KO CAR-T cells were cocultured with irradiated Raji cells at a 1:2 E:T ratio. CAR-T cells were harvested and counted 7 days later, followed by restimulation with irradiated Raji cells. Phenotypic analysis was performed 24 hours after stimulation. Data are shown as mean plus or minus SEM and represent 3 independent experiments with CAR-T cells from 3 donors targeted with sgRNA-1. Similar results were obtained with sgRNA-3. (C) Number of CD8+ and CD4+ WT and Regnase-1 KO CAR-T cells or untransduced donor T cells after each round of stimulation. (D) Proportion of CD25+CD8+ and CD4+ CAR-T cells after each stimulation. (E) Proportion of TNFα+, IL-2+, IFNγ+, and granzyme B+CD8+ and CD4+ CAR-T cells after 3 rounds of stimulation. (F) Proportion of CCR7+CD8+ and CD4+ CAR-T cells after 3 rounds of stimulation. (G-J) NSG mice bearing Raji tumors were treated with untransduced human T cells, or with activated CD8+ and CD4+ (1:1) Regnase-1 KO or WT CAR-T cells. (G) Tumor growth monitored by bioluminescence imaging. Solid lines represent means and dotted lines represent individual mice. Data were pooled from 2 independent experiments. (H) CD8+ and CD4+ CAR-T-cell number in spleen and bone marrow 7 and 35 days after transfer. (I) Percentage of CD8+ and CD4+ CAR-T cells in blood at indicated time points. (J) Percentage of TCF-1+ CD8+ and CD4+ CAR-T cells in spleen and bone marrow 35 days after transfer. Significance was determined by multiple Student t test with Holm-Sidak correction for multiple comparisons (C-E), unpaired Student t test (F), or Mann-Whitney test (H-J). Data are shown as mean plus or minus SEM and represent 2 independent experiments with 4 to 5 mice per group (H-J). *P < .05; **P < .01; ***P < .001; ****P < .0001.
We next examined the effects of Regnase-1 KO CAR-T cells on tumor growth in vivo in a human xenograft B-ALL model. Tumor-bearing NSG mice treated with human KO CAR-T cells had reduced tumor burden relative to mice treated with WT CAR-T cells (Figure 7G). KO CAR-T cells were present in higher numbers than WT cells in the spleen 7 and 35 days after transfer and in the blood up to 35 days after transfer (Figure 7H-I). Human Regnase-1 KO CAR-T cells also had more CD8+TCF-1+ and CD4+TCF-1+ cells relative to WT CAR-T cells 35 days after transfer (Figure 7J). These results are consistent with findings obtained in the murine model and support a role for Regnase-1 as an important suppressor of human CAR-T-cell expansion, persistence, and tumoricidal activity.
Discussion
TCF-1 promotes long-term survival and homeostatic proliferation of T cells and is central to the development of TCM and TPEX linages.26,32-34 TCF-1 expression correlates with the potential for self-renewal, and TCF-1+ populations are associated with improved CAR–T-cell persistence and antitumor immunity.5,44,47 Negative regulators of TCF-1 that can be targeted to improve memory formation in adoptive cellular therapies have not been described.48 Here, we identify Regnase-1 as a key antigen-dependent regulator of CAR–T-cell fate, directly targeting TCF-1 to suppress long-term CAR–T-mediated antitumor immunity. Deletion of Regnase-1 promotes TCF-1–dependent TPEX development, dramatically enhancing CAR–T-cell potency and sustaining antitumor immunity.
TCF-1+ TPEX preserve TEX cell function in settings of persistent antigen stimulation.25,42,44 Both WT and Regnase-1 KO TCF-1+ CAR-T cells demonstrate TEX epigenetic signatures while also expressing gene profiles required for TPEX maintenance.26,28,29 TCF-1+ TPEX generate exhausted TCF-1− progeny.24,32,43 Consistently, WT and KO TCF-1+ CAR-T cells give rise to TCF-1−TOX+ cells during recall responses. Our findings, therefore, support a role for TCF-1+ TPEX in long-lived CAR-T–mediated antitumor responses. The transcriptional and epigenetic features of TCF-1+ TPEX are evident by day 7, supporting reports that TPEX development is established early with persistent antigen exposure.26,49
Regnase-1 deletion dramatically expands the TPEX pool and preserves memory-associated DNA methylation patterning. The Regnase-1 KO transcriptional program is also more consistent with reported TPEX populations, with higher Tcf7, Cxcr5, and Eomes expression and reduced Ki67, Havcr2, and Tbx21 expression, relative to WT.26,30-32,50,51 Transcriptional and phenotypic analyses of Regnase-1 KO CAR-T cells are consistent with the earliest TCF-1high TEX progenitors as defined previously with a 4-stage developmental hierarchy of the TEX lineage.52 Thus, Regnase-1 KO CAR-T cells appear to be composed of less differentiated TCF-1+ TPEX that are more effective in sustaining long-term CAR–T-cell function.
Regnase-1 further demonstrates divergent effects on TCF-1+ and TCF-1− subsets. Although Regnase-1 deficiency increases TOX expression in TCF-1+ TPEX cells, it decreases tumor-induced TOX expression in TCF-1− cells, preserving effector activity. This may be explained by Regnase-1 targeting of BATF, which impairs CD8+ TEFF accumulation and function.22,23 Regnase-1 deficiency therefore appears to enhance T-cell–mediated antitumor immunity by modulating 2 transcription factors critical for T-cell fate determination, BATF promoting TEFF accumulation and function, and TCF-1 fostering TPEX formation.
Our findings substantiate a role for Regnase-1 as a key modulator of human CAR–T-cell antitumor function and provide support for the clinical application of Regnase-1–deficient CAR-T cells. Improved in vivo expansion and persistence of Regnase-1–deficient human CAR-T cells accompanied by an expanded TCF-1+ population suggest that the mechanisms defined in our immunocompetent murine model translate to the human system, though further validation is important. TCF-1+ TPEX are responsible for the proliferative burst after anti–PD-1 therapy.32,53 Our data suggest that targeting Regnase-1 would not only improve CAR–T-cell function, but further enhance CAR–T-cell responses in conjunction with anti–PD-1 treatment. We also show increased activation and persistence of human CD4+ Regnase-1 KO CAR-T cells, consistent with reports of increased CD4+ T-cell activation in Regnase-1–deficient mice.19 CD4+ CAR-T cells potentiate CD8+ CAR–T-cell responses and the impact of their improved persistence and expansion due to Regnase-1 suppression will be important to evaluate. In summary, our findings provide new insight into the context-dependent control of CAR–T-cell memory and fate determination, and demonstrate how targeting Regnase-1 can improve the effectiveness of adoptive CAR-T immunotherapies.
The microarray data reported in this article have been deposited in the Gene Expression Omnibus database (accession number GSE154859) and will be made available to the public upon publication.
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Acknowledgments
The authors thank Rajshekhar Alli and Jacob Basham for thoughtful discussions, suggestions, and technical assistance; Charles Mullighan and his laboratory for providing B-ALL tumor cells; and the following St. Jude Children’s Research Hospital shared resources: Transgenic/Gene Knockout Shared Resource, Hartwell Center for Bioinformatics and Biotechnology, Flow Cytometry and Cell Sorting, Animal Resource Center, Center for In Vivo Imaging and Therapeutics, Center for Advanced Genome Engineering, and the Vector Development and Production Core. The authors also thank Richard Cross and the Department of Immunology Flow Cytometry Core.
This work was supported by funding from American Lebanese Syrian Associated Charities (ALSAC)/St. Jude Children’s Research Hospital (SJCRH), The Assisi Foundation of Memphis, and National Institutes of Health National Cancer Institute grants P30 CA021765 (H.C. and B.Y.), CA250533 and CA221290 (H.C.), and 1R01CA237311 (B.Y.).
The content of this article is solely the responsibility of the authors and does not necessarily represent the views of the National Institutes of Health.
Authorship
Contribution: W.Z. and T.L.G. designed studies; J.W. and L.L.J. assisted with study design; W.Z., J.W., J.M., and L.L. performed experiments; W.Z., J.W., L.L.J., and T.L.G. analyzed and interpreted results; C.C.Z. performed methylation data analysis and interpreted results; Y.D. and Y.-D.W. performed microarray data analysis; W.Z., L.L.J., C.C.Z., Y.D., and Y.-D.W. prepared figures; W.Z. and L.L.J. wrote the manuscript; W.Z., J.W., C.C.Z., L.L.J., and T.L.G. edited the manuscript; Y.F., B.Y., and H.C. provided resources and assisted in study design and interpretation of results; T.L.G. provided project supervision; and all authors reviewed the manuscript.
Conflict-of-interest disclosure: W.Z., J.W., H.C., and T.L.G. are authors of a patent application related to Regnase-1. The remaining authors declare no competing financial interests.
Correspondence: Terrence L. Geiger, St. Jude Children’s Research Hospital, 262 Danny Thomas Pl, Memphis, TN 38105; e-mail: terrence.geiger@stjude.org.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal