

Key Points
Genetic alterations in immune checkpoint–related genes are frequent in IVLBCL.
Plasma cfDNA is an alternative tumor DNA source of IVLBCL to detect genetic alterations by comprehensive analyses.

Abstract
Intravascular large B-cell lymphoma (IVLBCL) is a unique type of extranodal lymphoma characterized by selective growth of tumor cells in small vessels without lymphadenopathy. Greater understanding of the molecular pathogenesis of IVLBCL is hampered by the paucity of lymphoma cells in biopsy specimens, creating a limitation in obtaining sufficient tumor materials. To uncover the genetic landscape of IVLBCL, we performed whole-exome sequencing (WES) of 21 patients with IVLBCL using plasma-derived cell-free DNA (cfDNA) (n = 18), patient-derived xenograft tumors (n = 4), and tumor DNA from bone marrow (BM) mononuclear cells (n = 2). The concentration of cfDNA in IVLBCL was significantly higher than that in diffuse large B-cell lymphoma (DLBCL) (P < .0001) and healthy donors (P = .0053), allowing us to perform WES; most mutations detected in BM tumor DNA were successfully captured in cfDNA and xenograft. IVLBCL showed a high frequency of genetic lesions characteristic of activated B-cell–type DLBCL, with the former showing conspicuously higher frequencies (compared with nodal DLBCL) of mutations in MYD88 (57%), CD79B (67%), SETD1B (57%), and HLA-B (57%). We also found that 8 IVLBCL (38%) harbored rearrangements of programmed cell death 1 ligand 1 and 2 (PD-L1/PD-L2) involving the 3′ untranslated region; such rearrangements are implicated in immune evasion via PD-L1/PD-L2 overexpression. Our data demonstrate the utility of cfDNA and imply important roles for immune evasion in IVLBCL pathogenesis and PD-1/PD-L1/PD-L2 blockade in therapeutics for IVLBCL.
Introduction
Intravascular large B-cell lymphoma (IVLBCL) is a rare type of extranodal large B-cell lymphoma characterized by selective growth of lymphoma cells in the lumina of small vessels.1 IVLBCL presents with various clinical symptoms, including fever of unknown origin, malaise, and unexplained hypoxemia in the absence of marked lymphadenopathy, making accurate diagnosis difficult. Since being listed in the World Health Organization (WHO) classification,2 a significant increase in the incidence of IVLBCL has been reported in the United States,3,4 which suggests improved awareness of the disease. However, accurate and timely diagnosis is often difficult, requiring repetitive organ biopsies (including those of bone marrow [BM], skin, lung, etc) and precious time until a formal diagnosis can be rendered. Therefore, a diagnostic breakthrough is needed for IVLBCL. Recently, liquid biopsy has attracted attention as a powerful tool in the procedures used for diagnosis of various cancers.5,6 Notably, a report was published regarding the use of a target sequence in plasma for diagnosis of IVLBCL.7 Given the nature of IVLBCL, which involves the proliferation of lymphoma cells in small vessels, this type of cancer is presumed to generate large quantities of cell-free DNA (cfDNA) in plasma. However, the quantity and quality of cfDNA in plasma in IVLBCL are not fully understood.
In terms of clinical outcomes for IVLBCL, prognoses for patients with IVLBCL were very poor in the prerituximab era.8,9 The application of rituximab in patients with IVLBCL has provided improved clinical outcomes.10,11 The results of the first prospective trial in untreated patients with IVLBCL was reported very recently; treatment in this trial consisted of rituximab, cyclophosphamide, doxorubicin, vincristine, and prednisolone (R-CHOP) combined with central nervous system (CNS)-oriented therapy, including high-dose methotrexate (HDMTX) and intrathecal (IT) chemotherapies.12 The results of the trial were encouraging, but some patients developed relapse and exhibited refractory disease. Given that most of the IVLBCL cases are considered to be assigned to the activated B-cell (ABC) type,13 refractory patients may have corresponded to the MCD or C5 genetic subtypes, which have been associated with a poor prognostic phenotype.14,15 However, the prognosis of IVLBCL based on the genetic abnormalities is largely unknown. Herein, we examined the utility of liquid biopsy in patients with IVLBCL, with the intent of uncovering the genetic landscape and association between genetic abnormalities; our analysis used whole-exome sequencing (WES) and clinical outcomes.
Methods
Patients’ characteristics
Appropriate informed consent was obtained from all participants. The resulting study population included patients newly diagnosed with IVLBCL (n = 21) at Nagoya University Hospital and affiliated institutions in Japan. Diagnosis was assigned according to the WHO classification. All but 1 case for which no appropriate pathological materials were available, and were subjected to a centralized review to confirm the diagnosis of IVLBCL in 2 independent institutions (S.S., S.N., H.M., and K.O.). This study was done in accordance with the Declaration of Helsinki, and the domestic ethical guidelines issued by the Ministry of Health, Labor, and Welfare in Japan. The study protocol was approved by the institutional review boards at Nagoya University Hospital (approval number, 2014-0081) and Fujita Health University (approval number, HG17-032 and HG18-017), followed by those at all participating institutions. Patient characteristics are summarized in Table 1.
Patient characteristics
| No. | Age, y | Sex | Clinical stage | PS | LDH, >WLN | IPI risk | Presence of B symptom | COO | IHC | cfDNA, ng/p-mL | Prognosis | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| CD5 | CD10 | BCL6 | MUM1 | PD-L1 | PD-L2 | PFS, mo | OS, mo | Outcome | ||||||||||||
| 28-8 | E1J2J | SP142 | ||||||||||||||||||
| IVL_01 | 76 | F | IV | 4 | Y | H | + | Non-GCB | + | − | − | + | + | + | − | − | 1109.5 | 6 | 7 | DOD |
| IVL_02 | 80 | M | IV | 1 | Y | H | + | Non-GCB | − | − | + | + | + | + | + | − | 286.6 | 77 | 77 | AND |
| IVL_03 | 66 | M | IV | 1 | Y | H | + | Non-GCB | − | − | − | + | − | − | − | − | 1058.7 | 70 | 70 | AND |
| IVL_04 | 81 | M | IV | 1 | Y | H | + | Non-GCB | − | − | − | + | − | − | − | − | 631.9 | 68 | 68 | AND |
| IVL_05 | 61 | F | IV | 3 | Y | H | − | Non-GCB | − | − | + | + | − | − | − | + | 70.4 | 66 | 66 | AND |
| IVL_06 | 59 | M | IV | 2 | Y | H | − | Non-GCB | + | − | −/+ | + | n/a | n/a | n/a | n/a | n/a | 8 | 57 | AWD |
| IVL_07 | 72 | M | IV | 2 | Y | H | + | Non-GCB | + | − | − | + | + | + | − | − | n/a | 1 | 1 | DOD |
| IVL_08 | 77 | M | IV | 2 | Y | H | + | Non-GCB | − | − | + | + | − | − | − | − | n/a | 62 | 62 | DOO |
| IVL_09 | 59 | F | IV | 0 | Y | H-I | + | Non-GCB | + | − | +/− | + | + | n/a | + | − | 619.7 | 43 | 43 | AND |
| IVL_10 | 78 | F | IV | 1 | N | H-I | − | Non-GCB | − | − | − | + | + | n/a | − | − | 37.0 | 21 | 22 | DOD |
| IVL_11 | 66 | M | IV | 2 | Y | H | + | n/a | n/a | n/a | n/a | n/a | n/a | n/a | n/a | n/a | 631.5 | 48 | 48 | AND |
| IVL_12 | 77 | M | IV | 2 | Y | H | + | Non-GCB | + | − | + | + | + | + | + | − | 321.5 | 12 | 25 | DOD |
| IVL_13 | 84 | F | IV | 3 | Y | H | + | Non-GCB | n/a | − | + | + | − | − | − | − | 130.9 | 1 | 1 | DOD |
| IVL_14 | 68 | F | IV | 3 | Y | H | + | Non-GCB | n/a | − | − | + | + | + | − | − | 290.5 | 35 | 35 | AND |
| IVL_15 | 47 | M | IV | 2 | Y | H | + | n/a | n/a | n/a | n/a | n/a | + | n/a | − | − | 451.3 | 27 | 27 | AND |
| IVL_16 | 68 | M | IV | 1 | Y | H-I | + | Non-GCB | n/a | − | − | + | − | − | − | − | 353.3 | 47 | 47 | AND |
| IVL_18 | 80 | M | IV | 1 | Y | H | + | Non-GCB | n/a | − | − | − | − | − | − | − | 476.4 | 49 | 49 | AND |
| IVL_19 | 54 | M | IV | 1 | Y | H-I | + | Non-GCB | + | − | − | + | + | + | − | − | 111.0 | 66 | 66 | AND |
| IVL_20 | 62 | M | IV | 2 | Y | H | + | Non-GCB | + | − | − | + | + | n/a | − | − | 624.9 | 25 | 25 | AND |
| IVL_21 | 75 | F | IV | 2 | Y | H | + | Non-GCB | n/a | − | − | + | + | n/a | − | − | 57.7 | 31 | 31 | AND |
| IVL_22 | 71 | F | IV | 2 | Y | H | + | GCB | + | + | − | + | + | + or +/− | − | − | 3679.9 | 36 | 36 | AND |
| No. | Age, y | Sex | Clinical stage | PS | LDH, >WLN | IPI risk | Presence of B symptom | COO | IHC | cfDNA, ng/p-mL | Prognosis | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| CD5 | CD10 | BCL6 | MUM1 | PD-L1 | PD-L2 | PFS, mo | OS, mo | Outcome | ||||||||||||
| 28-8 | E1J2J | SP142 | ||||||||||||||||||
| IVL_01 | 76 | F | IV | 4 | Y | H | + | Non-GCB | + | − | − | + | + | + | − | − | 1109.5 | 6 | 7 | DOD |
| IVL_02 | 80 | M | IV | 1 | Y | H | + | Non-GCB | − | − | + | + | + | + | + | − | 286.6 | 77 | 77 | AND |
| IVL_03 | 66 | M | IV | 1 | Y | H | + | Non-GCB | − | − | − | + | − | − | − | − | 1058.7 | 70 | 70 | AND |
| IVL_04 | 81 | M | IV | 1 | Y | H | + | Non-GCB | − | − | − | + | − | − | − | − | 631.9 | 68 | 68 | AND |
| IVL_05 | 61 | F | IV | 3 | Y | H | − | Non-GCB | − | − | + | + | − | − | − | + | 70.4 | 66 | 66 | AND |
| IVL_06 | 59 | M | IV | 2 | Y | H | − | Non-GCB | + | − | −/+ | + | n/a | n/a | n/a | n/a | n/a | 8 | 57 | AWD |
| IVL_07 | 72 | M | IV | 2 | Y | H | + | Non-GCB | + | − | − | + | + | + | − | − | n/a | 1 | 1 | DOD |
| IVL_08 | 77 | M | IV | 2 | Y | H | + | Non-GCB | − | − | + | + | − | − | − | − | n/a | 62 | 62 | DOO |
| IVL_09 | 59 | F | IV | 0 | Y | H-I | + | Non-GCB | + | − | +/− | + | + | n/a | + | − | 619.7 | 43 | 43 | AND |
| IVL_10 | 78 | F | IV | 1 | N | H-I | − | Non-GCB | − | − | − | + | + | n/a | − | − | 37.0 | 21 | 22 | DOD |
| IVL_11 | 66 | M | IV | 2 | Y | H | + | n/a | n/a | n/a | n/a | n/a | n/a | n/a | n/a | n/a | 631.5 | 48 | 48 | AND |
| IVL_12 | 77 | M | IV | 2 | Y | H | + | Non-GCB | + | − | + | + | + | + | + | − | 321.5 | 12 | 25 | DOD |
| IVL_13 | 84 | F | IV | 3 | Y | H | + | Non-GCB | n/a | − | + | + | − | − | − | − | 130.9 | 1 | 1 | DOD |
| IVL_14 | 68 | F | IV | 3 | Y | H | + | Non-GCB | n/a | − | − | + | + | + | − | − | 290.5 | 35 | 35 | AND |
| IVL_15 | 47 | M | IV | 2 | Y | H | + | n/a | n/a | n/a | n/a | n/a | + | n/a | − | − | 451.3 | 27 | 27 | AND |
| IVL_16 | 68 | M | IV | 1 | Y | H-I | + | Non-GCB | n/a | − | − | + | − | − | − | − | 353.3 | 47 | 47 | AND |
| IVL_18 | 80 | M | IV | 1 | Y | H | + | Non-GCB | n/a | − | − | − | − | − | − | − | 476.4 | 49 | 49 | AND |
| IVL_19 | 54 | M | IV | 1 | Y | H-I | + | Non-GCB | + | − | − | + | + | + | − | − | 111.0 | 66 | 66 | AND |
| IVL_20 | 62 | M | IV | 2 | Y | H | + | Non-GCB | + | − | − | + | + | n/a | − | − | 624.9 | 25 | 25 | AND |
| IVL_21 | 75 | F | IV | 2 | Y | H | + | Non-GCB | n/a | − | − | + | + | n/a | − | − | 57.7 | 31 | 31 | AND |
| IVL_22 | 71 | F | IV | 2 | Y | H | + | GCB | + | + | − | + | + | + or +/− | − | − | 3679.9 | 36 | 36 | AND |
AND, alive with no evidence of disease; AWD, alive with disease; COO, cell of origin; CRP, C-reactive protein; DOD, dead of disease; DOO, dead of other diseases; GCB, germinal center B-cell type; H, high; H-I, high-intermediate; IHC, immunohistochemistry; IVL, intravascular large B-cell lymphoma; IPI, International Prognostic Index; LDH, lactate dehydrogenase; n/a, not available; ng/p-mL, nanogram per 1 mL of plasma; non-GCB, non germinal center B-cell type; PS, performance status; UPN, unique patient number; WBC, white blood cell count; WLN, within limit of normal; Y, yes.
Sample preparation
Plasma and serum from peripheral blood were collected from the enrolled patients. Plasma from 14 healthy volunteer donors also was harvested. Serial blood samples were collected from randomly selected patients during their clinical courses. Sample collection and processing to plasma and serum were performed as described previously.16,17 Collection of blood for plasma was conducted primarily with EDTA-Na2-containing blood collection tubes. Collection of blood for serum was performed using serum separator tubes. The primary blood samples were stored at 4°C immediately after blood collection from patients, and further processing of these samples was completed within 6 hours after blood collection. Processing consisted of centrifugation for 10 minutes at 430g for plasma or at 1710g for serum; the resulting plasma and serum specimens were portioned into aliquots and stored at −80°C pending analysis. Mononuclear cells (MNCs) from BM (BMMNCs) and peripheral blood (PB) (PBMNCs) were collected using Ficoll-Paque (GE Healthcare, Uppsala, Sweden), and the tumor cell content in BMMNCs and tumor biopsy samples was calculated by flow cytometry analysis by measuring CD19+ and CD20+ cells and evaluating immunoglobulin light chain restriction. Patient-derived xenograft (PDX) tumors were obtained as described previously.13 Genomic DNA from tumor cells (from patients and PDX models, BMMNCs, PBMNCs, and buccal mucosa) was extracted using the QIAamp DNA Blood Mini Kit (Qiagen, Valencia, CA). cfDNA from plasma and serum was extracted using the QIAamp Circulating Nucleic Acid Kit (Qiagen). The concentration of cfDNA was confirmed by agarose gel electrophoresis (supplemental Figure 1, available on the Blood Web site) and using a Qbit 4 fluorometer (Thermo Fisher Scientific, Waltham, MA) as described previously.18 In some cases, the concentration and quality of cfDNA were also analyzed using a BioAnalyzer (Agilent Technologies, Inc, Santa Clara, CA).
WES
WES was performed for samples derived from 21 patients with IVLBCL, including cfDNA samples (n = 18), PDX IVLBCL tumors (n = 4), and DNA obtained from BMMNCs containing substantial tumor cells (n = 2). DNA extracted from PBMNCs without tumor cells, BMMNCs from patients in complete remission (CR), and buccal mucosa was used as germline control DNA. WES was performed as previously reported.19 Whole-exome capture was accomplished by liquid-phase hybridization of sonicated genomic DNA using a bait complementary RNA library (SureSelect Human ALL Exon V5; Agilent Technologies) according to the manufacturer’s protocol. Massive parallel sequencing of the captured targets was performed using a HiSeq 2000/2500 sequencing system (Illumina, San Diego, CA) with the 126-bp read option, according to the manufacturer’s instructions. The mean sequencing depth of all samples was 164× (95-228×). Detailed WES procedures are described in supplemental Methods.
Targeted capture sequencing
To validate mutations in 223 genes (supplemental Table 1), including recurrently mutated genes identified by WES and known/putative driver genes, and to comprehensively detect structural variants (SVs) involving programmed cell death 1 ligand 1 and 2 (PD-L1/PD-L2), targeted sequencing was performed for 21 IVLBCL samples as well as 2 BM tumor samples. All exons of 223 genes and all exons, introns, and untranslated regions (UTRs) of the PD-L1 and PD-L2 genes and 983 single nuclear polymorphism (SNP) probes for copy-number analysis were captured using a custom-designed SureSelect kit. Sample preparation and sequencing were performed in a fashion similar to that used for WES. The mean sequencing depth of 21 IVLBCL samples was 433× (208-649×), and the mean depth of targeted exons was 383×. In total, 1083 of 1113 (97%) of the mutations identified by WES were confirmed by targeted capture sequencing with variant allele frequency (VAF) >0.02. In addition, 149 mutations were newly identified by targeted sequencing. Copy-number analysis was performed using CNACS (https://github.com/papaemmelab/toil_cnacs). For cycline-dependent kinase inhibitor 2A (CDKN2A) deletions, >3 consecutive probes showing <1.5 total copy numbers were considered to be deletions.
Definition of driver genes
Analysis with MutSigCV identified multiple genes as being significantly mutated. When genes known to be targeted by somatic hypermutation (SHM), namely HIST1H1C, PIM1, and TMSB4X, were excluded, the 9 remaining loci were considered driver genes. Known tumor-suppressor genes with recurrent loss-of-function mutations and genes with driver mutations reported in Catalogue of Somatic Mutations in Cancer (COSMIC) for ≥4 cases were also considered driver genes.
Immunohistochemistry
Formalin-fixed, paraffin-embedded tissues were evaluated by routine hematoxylin-and-eosin staining and immunohistochemical staining. Detailed immunohistochemistry (IHC) procedures for using anti–PD-L1 and –PD-L2 antibodies are indicated in supplemental Methods.
Statistical analysis
Statistical analyses with a 2-tailed, unpaired Student t test and nonlinear regression were performed with Prism software (version 5; GraphPad Software, Inc, San Diego, CA). The Mann-Whitney U test and the Fisher exact test were performed with R, version 3.4.2. P values <.05 were considered statistically significant. Details of the survival analysis were described in supplemental Methods.
Results
Concentration of PB cfDNA in IVLBCL significantly correlates with serum lactate dehydrogenase level and reflects disease status
To investigate the pathogenesis, we performed comprehensive detection of driver mutations in IVLBCL using WES of cfDNA derived from the PB of 18 patients with IVLBCL to test the hypothesis that IVLBCL cells produce a large amount of tumor-derived cfDNA for next-generation sequencing for detection of somatic events with high sensitivity and accuracy. All evaluable patients but 1 were diagnosed with the non–germinal center B-cell type using IHC (Table 1).1,20 For 3 other patients and 1 of the 18 patients with cfDNA available, we established PDX models, and genomic DNA was extracted from the tumors formed in the spleen. For 2 patients, tumor DNA was also obtained from BM infiltrated with IVLBCL. BM- or PDX-derived DNA was also analyzed with WES.
A sufficient amount of cfDNA was obtained from all 18 patients with a median recovery of 451.3 ng/mL plasma, which was 10 times higher than that obtained from patients with diffuse large B-cell lymphoma (DLBCL) (22.6 ng/mL plasma) with a lower variance across patients (P < .0001) (Figure 1A). cfDNA amounts from both IVLBCL and DLBCL were significantly higher than that from healthy donors (P = .0053 and P = .036, respectively). The cfDNA concentration in plasma was significantly correlated with that in serum, and the amount in plasma was higher than that in serum (supplemental Figure 2a). The recovery of cfDNA was significantly correlated with serum lactate dehydrogenase (LDH) levels (P < .0001), C-reactive protein (P < .0001) levels, and the status of BM invasion (P = .0432) (supplemental Figure 2b-d), but not with several other clinical parameters (supplemental Figures 2e-j and 3). The cfDNA concentration rapidly decreased just after starting the first chemotherapy (Figure 2B-D; supplemental Figure 3a-c), but was elevated in the refractory phase (supplemental Figure 3d-e), suggesting that cfDNA mainly originated from lymphoma cells.
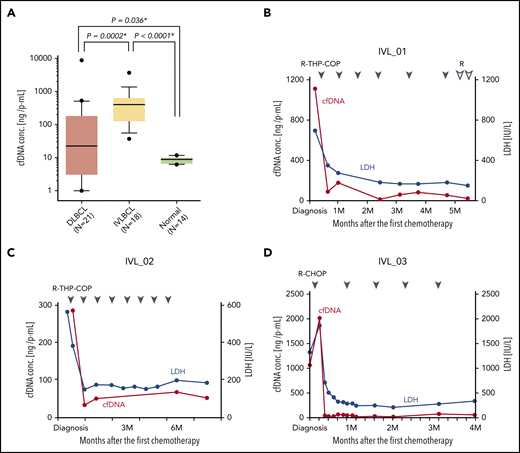
cfDNA concentration in IVLBCL. (A) Plasma (P) cfDNA concentration at diagnosis in patients with DLBCL, IVLBCL, and normal volunteer donors are compared. Note that the P-cfDNA concentration in IVLBCL is significantly higher than that in DLBCL and normal donors. Box-and-whisker plot with 10% to 90% is shown. Asterisks indicate significant differences. (B-D) Time-dependent analyses of the cfDNA concentration in an individual patient with IVLBCL during the clinical course is shown. After the first chemotherapy, the cfDNA concentration (red line) in each patient was markedly decreased as well as the serum LDH level (blue line). Blue and white triangles indicate the first day of each chemotherapy regimen. R, rituximab; THP, pirarubicin.
cfDNA concentration in IVLBCL. (A) Plasma (P) cfDNA concentration at diagnosis in patients with DLBCL, IVLBCL, and normal volunteer donors are compared. Note that the P-cfDNA concentration in IVLBCL is significantly higher than that in DLBCL and normal donors. Box-and-whisker plot with 10% to 90% is shown. Asterisks indicate significant differences. (B-D) Time-dependent analyses of the cfDNA concentration in an individual patient with IVLBCL during the clinical course is shown. After the first chemotherapy, the cfDNA concentration (red line) in each patient was markedly decreased as well as the serum LDH level (blue line). Blue and white triangles indicate the first day of each chemotherapy regimen. R, rituximab; THP, pirarubicin.
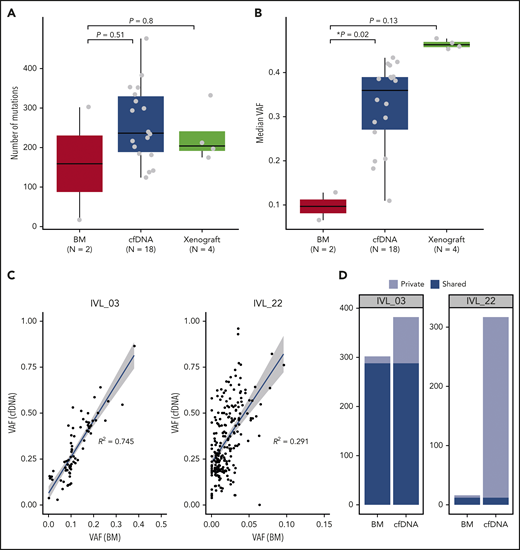
Comparison of mutations identified in BM, cfDNA, and xenograft samples. (A) Number of mutations identified in BM, cfDNA, and xenograft samples. Box-and-whisker plots show the boxes indicating median and interquartile range, and the whiskers denoting the range. The overlaid points are the observed mutation burden of individual samples. (B) Median VAFs of mutations identified in BM, cfDNA, and xenograft samples. Box-and-whisker plots show the boxes indicating median and interquartile range, and the whiskers denoting the range. The overlaid points are the observed mutation burden of individual samples. Asterisks indicate significant differences (Mann-Whitney U test). (C) VAFs of mutations detected in cfDNA and BM cells of IVL_03 and IVL_22 are plotted. VAFs are calculated using the allelic counts of amplicon sequencing (IVL_03) and WES (IVL_22). (D) Mutations identified in BM and cfDNA samples in IVL_03 and IVL_22 are shown. Unique mutations and shared mutations between the 2 samples are shown in different colors.
Comparison of mutations identified in BM, cfDNA, and xenograft samples. (A) Number of mutations identified in BM, cfDNA, and xenograft samples. Box-and-whisker plots show the boxes indicating median and interquartile range, and the whiskers denoting the range. The overlaid points are the observed mutation burden of individual samples. (B) Median VAFs of mutations identified in BM, cfDNA, and xenograft samples. Box-and-whisker plots show the boxes indicating median and interquartile range, and the whiskers denoting the range. The overlaid points are the observed mutation burden of individual samples. Asterisks indicate significant differences (Mann-Whitney U test). (C) VAFs of mutations detected in cfDNA and BM cells of IVL_03 and IVL_22 are plotted. VAFs are calculated using the allelic counts of amplicon sequencing (IVL_03) and WES (IVL_22). (D) Mutations identified in BM and cfDNA samples in IVL_03 and IVL_22 are shown. Unique mutations and shared mutations between the 2 samples are shown in different colors.
WES analyses in IVLBCL using cfDNA
WES was performed on 18 cfDNA samples, 4 PDX samples, and 2 BM samples from 21 patients with IVLBCL (supplemental Table 2; supplemental Figure 4a). Mutations were detected in all cfDNA and PDX samples with a median of 236.5 and 204 mutations per sample, respectively, which were higher than the number found in BM-derived DNA (159 per sample) (Figure 2A). Validation was performed for selected mutations using Sanger sequencing or amplicon-based deep sequencing, which confirmed high accuracy (92%) of mutation calling in cfDNA samples. Based on analysis using PMSignature,21 mutations were enriched for age-related C < T transitions at CpG sites (signature 1) and the classical activation-induced deaminase (AID) signature that mainly affected somatic hypermutations (SHMs) at WRCY motifs (signature 2) (supplemental Figure 4b).
VAFs, which reflect the presence of substantial intratumor heterogeneity, of detected mutations were generally highly variable even within the same patient. However, we detected mutations with very high (>40%) VAFs in most cfDNA samples, confirming the major contribution of tumor components to cfDNA (supplemental Figure 5). In 3 patients with IVLBCL, WES was performed for both cfDNA and BM- (n = 2) or PDX-derived DNA (n = 1). Mutations in cfDNA had substantially higher VAFs compared with those in BM-derived tumor DNA (Figure 2B), suggesting that tumor-derived DNA was significantly enriched in cfDNA. Most mutations detected in BM tumor DNA were successfully captured in cfDNA (Figure 2C), whereas many mutations were detected in cfDNA alone (Figure 2D), suggesting that cfDNA can be used to detect mutations with much higher sensitivity. In a similar way, most mutations (70 of 71) identified in BM tumor DNA were also detected in PDX-derived DNA (supplemental Figure 6a). Some mutations and copy-number changes were detected in BM DNA (Figure 2D) or PDX tumor DNA alone (supplemental Figure 6b-d), likely reflecting the regional heterogeneity of tumor populations or unique positive selection in the xenograft environment.
Identification of recurrently mutated genes in IVLBCL
Significantly mutated genes were interrogated using MutSigCV.22 Twelve genes were significantly mutated in IVLBCL, and they all overlapped with known mutational targets in nodal DLBCL and/or other extranodal lymphomas (supplemental Figure 7; supplemental Table 3).14,15,23-25 To identify mutations in these genes in small fractions of tumor populations, all 21 samples were further investigated using targeted-panel sequencing (supplemental Figure 8a-c), which was designed to capture known targets of mutations and translocations in lymphomas, comprising 223 genes (supplemental Table 1). Most mutations identified with WES were validated with targeted capture sequencing. Driver mutations (Figure 3A; supplemental Table 4) were significantly enriched in the B-cell receptor/NF-κB signaling pathways: CD79B (67%), MYD88 (57%), IRF4 (38%), ITPKB (14%), NFKBIE (14%), and TNFAIP3 (24%). Nine of the 21 samples (43%) had both MYD88 and CD79B mutations. IgH-BCL6 translocation was found in a patient. SHMs, suggestive of AID-related events, were frequently confirmed in known targets of AID (supplemental Methods), such as PIM1 (95%) and IGLL5 (90%) (Figure 3B; supplemental Table 5).26 Mutations also commonly affected genes related to B-cell development, including PRDM1 (43%) and TOX (33%). Other common mutational targets included histone/chromatin modification factors, such as SETD1B (57%), KMT2D (24%), and EP300 (14%). The landscape of genetic alterations in IVLBCL is similar to that in ABC-type DLBCL,23 consistent with our previous gene expression profiling and copy-number alterations (CNAs).13 Several mutations were seen in IVLBCL at significantly higher frequencies than in nodal ABC-type DLBCL,15,25 including MYD88, CD79B, TBL1XR1, and SETD1B, which were also frequently mutated in primary CNS lymphoma (PCNSL) (K.Y. and S.O., manuscript in preparation) (Figure 3C).
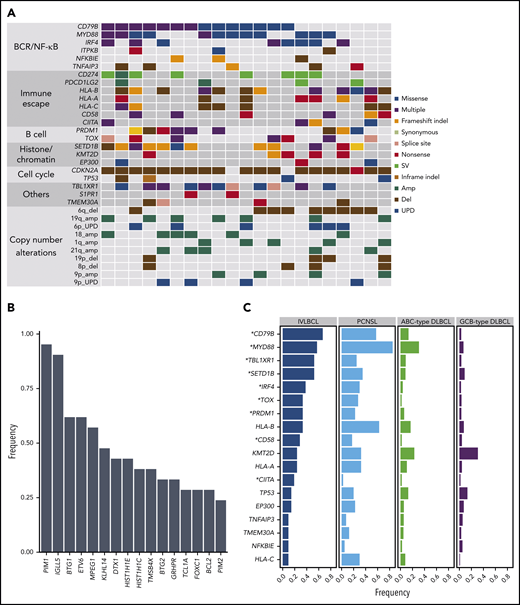
Landscape of genetic alterations in IVLBCL. (A) Driver mutations and CNAs detected in 21 patients with IVLBCL are shown. The vertical line indicates 1 patient. (B) Genes frequently affected by SHMs identified in IVLBCL. (C) Comparison of the frequency of driver mutations among IVLBCL, PCNSL, ABC-type DLBCL, and germinal center B-cell (GCB)-type DLBCL. Asterisks indicate genes that are significantly more frequently mutated in IVLBCL compared with ABC-type DLBCL (Fisher exact test).
Landscape of genetic alterations in IVLBCL. (A) Driver mutations and CNAs detected in 21 patients with IVLBCL are shown. The vertical line indicates 1 patient. (B) Genes frequently affected by SHMs identified in IVLBCL. (C) Comparison of the frequency of driver mutations among IVLBCL, PCNSL, ABC-type DLBCL, and germinal center B-cell (GCB)-type DLBCL. Asterisks indicate genes that are significantly more frequently mutated in IVLBCL compared with ABC-type DLBCL (Fisher exact test).
Many of these recurrent mutational targets were also affected by CNAs, which were sensitively detected with sequencing-based copy-number analysis. Among the most frequently observed was deletion of CDKN2A/2B (also called P16INK4A and P15INK4B, respectively), which was found in ∼86% of the cases (supplemental Figure 9a-b). Other common targets of CNAs included molecules involved in antigen presentation to T cells, including HLA-A/B/C (33%) and HLA class II (9%). Recurrent arm-level CNAs were also frequent, affecting chromosomal 6q deletion (43%), 19q amplification (32%), 6p uniparental disomy (29%), 18 amplification (29%), and 21q amplification (24%), and were confirmed in >20% of cases (Figure 3A; supplemental Figure 9a).
We attempted to classify IVLBCL cases into the recently described subtypes of DLBCL (supplemental Methods).14,27 As expected from the high frequency of MYD88 and CD79B mutations, 17 of 21 IVLBCL cases were classified into the MCD subtypes (n = 15) or as a composite case with features of MCD (supplemental Table 6), suggesting genetic similarity between the MCD-type DLBCL and IVLBCL.
SVs in PD-L1 (CD274) and PD-L2 (PDCD1LG2) genes in IVLBCL
SVs were also explored using WES data. We detected recurrent SVs involving PD-L1 and PD-L2 in 8 of 21 IVLBCL cases (supplemental Table 7). PD-L1–involving SVs were previously reported in 26.5% of adult T-cell leukemia/lymphoma18 and 6% to 19% of DLBCL.28,29 These SVs cause truncation of the 3′-UTR of these genes, leading to stabilization and markedly elevated expression of PD-L1 transcripts, and thus cancer immune evasion. PD-L2–involving SVs also cause overexpression of PD-L2 protein via a similar mechanism.30 Based on these findings, we further interrogated our 21 IVLBCL cases for SVs and other CNAs affecting PD-L1/PD-L2, using targeted-capture sequencing of cfDNA, in which the entire PD-L1 and PD-L2, including 5′ and 3′ noncoding regions, were captured to sensitively detect intronic breakpoints. PD-L1/PD-L2–involving SVs or copy-number gains were detected in 10 of the 21 cases (48%) (Figure 4A-B). Except for EEF1A1-PD-L2 translocation with a breakpoint in the 5′-UTR of PD-L2 in IVL_05 (Figure 4C) and IgK–PD-L1 translocation in the same patient, all SVs, including deletions, inversions, and translocations, resulted in the truncation of 3′-UTR sequences of the affected genes.
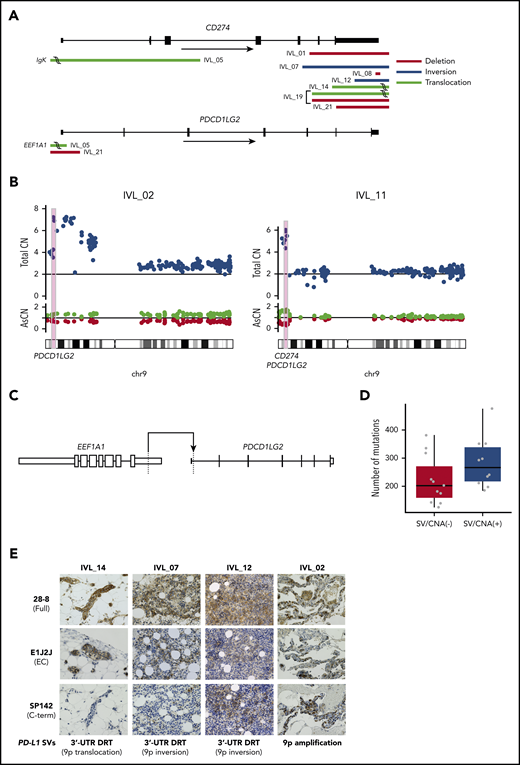
PD-L1 and PD-L2 genetic alterations and aberrant expression of PD-L1 protein in IVLBCL. (A) Distributions and types of SVs involving PD-L1 (CD274) and PD-L2 (PDCD1LG2) are depicted. (B) CNAs of PD-L1 and PD-L2 in 2 patients with IVLBCL, IVL_02 and IVL_11. (C) Novel translocation involving PDCD1LG2 identified in IVLBCL (IVL_05). (D) Number of somatic mutations in patients with and without SVs/CNAs in PD-L1/PD-L2. Box-and-whisker plots show the boxes indicating median and interquartile range, and the whiskers denoting the range. The overlaid points are the observed mutation burden of individual samples. (E) PD-L1 protein expression with IHC analyses using 3 different anti–PD-L1 antibodies: 28-8, E1J2J, and SP142. Skin biopsy samples (IVL_14), BM clot section samples (IVL_07, 12, and 18), and lung biopsy samples (IVL_02) were used for IHC analysis. Hematoxylin and eosin stain; original magnification ×400. AsCN, allele-specific copy number; CN, copy number; DRT, disruption; EC, extracellular domain.
PD-L1 and PD-L2 genetic alterations and aberrant expression of PD-L1 protein in IVLBCL. (A) Distributions and types of SVs involving PD-L1 (CD274) and PD-L2 (PDCD1LG2) are depicted. (B) CNAs of PD-L1 and PD-L2 in 2 patients with IVLBCL, IVL_02 and IVL_11. (C) Novel translocation involving PDCD1LG2 identified in IVLBCL (IVL_05). (D) Number of somatic mutations in patients with and without SVs/CNAs in PD-L1/PD-L2. Box-and-whisker plots show the boxes indicating median and interquartile range, and the whiskers denoting the range. The overlaid points are the observed mutation burden of individual samples. (E) PD-L1 protein expression with IHC analyses using 3 different anti–PD-L1 antibodies: 28-8, E1J2J, and SP142. Skin biopsy samples (IVL_14), BM clot section samples (IVL_07, 12, and 18), and lung biopsy samples (IVL_02) were used for IHC analysis. Hematoxylin and eosin stain; original magnification ×400. AsCN, allele-specific copy number; CN, copy number; DRT, disruption; EC, extracellular domain.
The numbers of somatic mutations in IVLBCL with SVs/CNAs involving PD-L1/PD-L2, although not significant (P = .10), were higher than those in cases without SVs/CNAs (Figure 4D), suggesting driver roles via immune evasion. Although we recently found strong enrichment of PD-L1/PD-L2–involving SVs in Epstein-Barr virus (EBV)-associated lymphomas,30 none of the 21 cases were positive for EBV sequences in WES.
PD-L1 and PD-L2 protein expression by IHC in IVLBCL
PD-L1 protein expression was investigated in 4 cases with PD-L1–involving amplifications or SVs using IHC. In IVL_12, the SV had a breakpoint within a 3′ noncoding exon and did not disrupt the authentic open reading frame. Accordingly, strong staining of tumor cells was observed with both N terminus– (E1J2J) and C terminus–directed (SP142) antibodies, and 1 recognizing full-length PD-L1 (clone 28-8). PD-L1 overexpression was confirmed with all 3 antibodies in 2 patients with 9p amplification (IVL_02) and 9p inversion (IVL_12). In contrast, in IVL_07 and IVL_14, the authentic reading frame was truncated within exon 6. Consistent with this, these cases showed strong staining with E1J2J and 28-8, but not with SP142, suggesting expression of a truncated protein (Figure 4E). These results confirmed a previous finding demonstrating a strong association between PD-L1 overexpression and PD-L1–involving genetic lesions.18 IHC analysis using anti–PD-L2 antibody (D7U8C) confirmed PD-L2 expression in 1 case (IVL_05) with EEF1A1-PD-L2 translocation (Figure 4A,C; Table 1; supplemental Table 7), but not in the remaining 2 cases (IVL_02 and IVL_21) having PD-L2 amplification and deletion, respectively. In IVL_02, PD-L2 gain occurred as a part of arm-level gain involving the whole 9p arm. The lack of PD-L2 staining, therefore, rather support that PD-L2 should not be the target of the 9p gain. In the remaining case (IVL_21), the SV had 2 breakpoints, 1 in PD-L1 and the other in PD-L2, causing a deletion affecting 3′-UTR of PD-L1 and the 5′ part of PD-L2. The deletion of 3′-UTR is a well-known cause of PD-L1 overexpression, whereas the deletion of the 5′ PD-L2 region likely resulted in the loss of PD-L2 promoter activity, explaining the lack of PD-L2 staining.
Survival based on genetic abnormalities in IVLBCL
Finally, we analyzed the survival of 19 patients who received immunochemotherapies (R-CHOP with HDMTX plus IT chemotherapies, n = 12; R-CHOP or R-CHOP-like with IT, n = 4; R-CHOP or R-CHOP-like, n = 2; and R-CHOP with IT followed by high-dose chemotherapy, n = 1) based on genetic abnormalities. Excluded from the survival analysis were 2 patients who were diagnosed postmortem; given their advanced age, these patients had received the best available supportive care but not chemotherapy. With a median follow-up duration of surviving patients of 49 months, median progression-free survival (PFS) and overall survival (OS) were not reached, and PFS and OS at 5 years were 79% and 84%, respectively (Table 1).
To test the prognostic value of genetic alterations, we investigated associations between genetic alterations and patients’ survival, focusing on 20 genes altered in ≥3 but ≤18 cases. We found possible association with OS for CIITA, which however, was no longer significant after correction for multiple testing (supplemental Table 8).
Discussion
In the present study, we demonstrated the genetic landscape of IVLBCL using cfDNA to perform WES analyses. In the present study, we investigated the genetic profile of IVLBCL using different DNA sources, including cfDNA, tumor-bearing BMMNCs, and PDX. On the evaluation in a small number of cases, in which the analysis was performed on multiple DNA sources, these different DNA sources provided almost identical genetic profiles. However, we still observed a subset of mutations that were only detected in cfDNA or xenograft samples of the same individuals, whereas other mutations were only seen in the primary tumor samples. These differences are thought to reflect intratumor heterogeneity, although we could not exclude a possibility that some lesions were acquired during the establishment of PDX in the mouse. However, the almost identical profile should argue for the use of PDX and cfDNA. In particular, given the integrative nature of cfDNA from multiple tumor components31 and high recovery and purity of cfDNA in IVLBCL, cfDNA might be a well-reasoned noninvasive alternative to BM or other biopsies.
We showed that IVLBCL harbored PD-L1/PD-L2 rearrangements at a high frequency, and SVs in PD-L1/PD-L2 likely correlate with overexpression of PD-L1/PD-L2 protein. These results suggested that immune evasion is deeply involved in IVLBCL pathogenesis. Considering the peculiar nature of IVLBCL, which involves the proliferation of tumor cells in blood vessels, elucidation of the molecular mechanisms whereby IVLBCL cells escape immune surveillance is expected to clarify the biology underlying this disease and to facilitate the development of novel treatment strategies.
In accordance with the ABC phenotype found in most cases, IVLBCL is characterized by a high prevalence of mutations in molecules involved in B-cell receptor/NF-κB signaling.7,32 The mutation frequencies in IVLBCL are higher than those reported in ABC-type DLBCL.23-25 The IVLBCL mutation landscape better resembles those of PCNSL,29,33,34 primary testicular lymphoma,29,35 DLBCL of the leg,36 and a small subset of ABC-type DLBCL termed MCD14 or C5.15 Molecular classification using the LymphGen algorithm27 indicated that 76% of IVLBCL cases in this cohort were classified as the MCD subtype (supplemental Table 6). The MCD- and C5-subtype lymphomas frequently involve the CNS, which may partly explain the frequent CNS invasion seen in IVLBCL. Although also reported in PCNSL (13%)29 and DLBCL,15,30 SVs involving PD-L1/PD-L2 are extremely common in IVLBCL, accounting for 38% of patients. Although the pattern of other driver mutations in IVLBCL was similar to those seen in PCNSLs and in MCD or C5 DLBCLs, the distinctively elevated frequency of PD-L1/PD-L2 rearrangements in IVLBCL suggested that IVLBCL constitutes a genetically unique subset of DLBCL. Combined with frequent deletions and mutations in HLA molecules, the high incidence of alterations in PD-L1 and PD-L2 suggests an important role for evasion from antitumor immunity in the pathogenesis of IVLBCL. This observation also suggests that IVLBCL positive for these PD-L1/PD-L2 lesions might be successfully treated by checkpoint blockade using anti–PD-1 or anti–PD-L1 antibodies.
In the present study, 12 of 21 patients (57%) received R-CHOP with CNS-oriented therapy including HDMTX and IT. As reported in the recent phase 2 trial, R-CHOP combined with CNS-oriented therapy is the currently recommended active treatment.12 Indeed, the present study’s PFS and OS at 5 years of 79% and 84%, respectively, are quite favorable. Although the significant association between genetic alterations and patients’ survival was not observed, the fact that patients harboring tumors with a MYD88 mutation and alterations and rearrangement of immune evasion–related genes displayed poorer outcomes might be meaningful. The use of treatment strategies blocking these molecules might be investigated in future clinical trials.
The present study’s demonstration of the utility of cfDNA is an important finding. Specifically, we showed that cfDNA concentrations were significantly higher in patients diagnosed with IVLBCL than in patients with DLBCL and healthy donors, and that cfDNA from patients with IVLBCL could be successfully used for WES (Figure 1a). These data strongly suggest that genetic analyses using cfDNA in IVLBCL may facilitate the detection and/or diagnosis of IVLBCL. Another important feature of cfDNA is that the cfDNA concentration in IVLBCL correlated strongly with disease status (Figure 1B-D; supplemental Figure 2b-d; supplemental Figure 3a-b), suggesting that liquid biopsy followed by comprehensive genetic analysis in IVLBCL may be a safer and less painful strategy than repeated biopsies for longitudinal monitoring of minimal residual disease. The next important step in the development of this strategy may be the confirmation of sensitivity. Further analyses and accumulation of patient numbers are required.
In summary, the present study demonstrated the utility of cfDNA in IVLBCL as a reliable source of uncovering details of genetic alterations; our analysis also provided information about the genetic landscape of IVLBCL. Furthermore, our findings also implied that mechanisms of immune evasion are deeply involved in IVLBCL pathogenesis and clinical outcomes. Although our data are based on a limited number of patients, the present study provides meaningful starting points for future analyses, considering the rarity of IVLBCL. Further investigations regarding this potential breakthrough method in diagnosis and novel treatment of intractable patients with IVLBCL are warranted.
The sequencing data reported in this article have been deposited at the European Genome-Phenome Archive (http://www.ebi.ac.uk/ega/) under accession ID EGAS00001003970.
Please e-mail the corresponding authors for sharing of the data.
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Acknowledgments
The authors thank Kunoyoshi Kitou (Nagoya University) and Kazutaka Nakashima (Kurume Univerisity) for IHC work.
This work was supported, in part, by Grants-in-Aid from the Program to Disseminate Tenure Tracking System; Ministry of Education, Culture, Sports, Science and Technology (MEXT), Japan; Japan Society for the Promotion of Science (JSPS) Grant-in-Aid for Young Scientists (B; 26860724 [K.S.]); MEXT, Japan (15K09473 [A.T.], 15H05912 [S.M.]); the National Cancer Center Research and Development Fund (26-A-4 [A.T.]); the Japan Agency for Medical Research and Development (AMED) (JP19cm0106501 [S.O.]); and JSPS (JP15H05909 [S.O.]).
Authorship
Contribution: A.T., K.Y., and S.O. designed the study; K.S., Y. Suzuki, C.I., M.Y., Y.T., S.K., Y.M., T.I., Y. Inagaki, A. Okamoto, and A.T. collected patient materials and clinical data; Y. Suzuki, K.S., and C.I. prepared the DNA samples; K.Y., M.S., Y.N., and Y. Suzuki performed sequencing experiments; K.Y., M.S., K. Kataoka, Y. Inoue, Y. Shiraishi, K.C., H.T., S.M., and Y. Shiozawa performed sequencing data analyses; S.S., K. Kohno, A. Okabe, and S.N. performed pathological diagnosis; S.S., S.N., H.M., and K.O. performed pathological central review; S.S., K. Kohno, and H.M. performed immunostaining of PD-L1/L2; K.S., Y.K., M.N., and Y.A. performed statistical analysis; K.Y., A.T., Y. Suzuki, and K.S. generated figures and tables; K.Y., A.T., K.S., and S.O. wrote the paper; A.T., S.O., and H.K. supervised the study; and all authors participated in discussions and interpretation of the data and results.
Conflict-of-interest disclosure: K.S. received research funding from Celgene, Otsuka Pharmaceutical, Merck Sharp & Dohme (MSD), Chugai Pharmaceutical, Kyowa Kirin (KK), and Daiichi Sankyo, and personal fees from AstraZeneca, Eizai, Celgene, Takeda Pharmaceutical, Janssen Pharmaceutical, Bristol Myers Squibb, Chugai Pharmaceutical, Kyowa Kirin, Nippon Shinyaku, and Daiichi Sankyo. K. Kataoka received research funding from Otsuka Pharmaceutical, Chugai Pharmaceutical, Takeda Pharmaceutical, and Bristol Myers Squibb; has ownership in Asahi Genomics; and has received honoraria from Celgene, Eisai, Astellas Pharma, Novartis, Chugai Pharmaceutical, AstraZeneca, Sumitomo Dainippon Pharma, Kyowa Kirin, Janssen Pharmaceutical, and MSD. S.K. received research funding from Chugai Pharmaceutical and Kyowa Kirin, and received honoraria from Chugai Pharmaceutical and Kyowa Kirin. Y. Inoue received lecture fees from Celgene Corporation, Takeda Pharmaceutical, Mundipharma KK, Bristol Myers Squibb, Eisai, Novartis Pharma KK, Kyowa Kirin, and Sumitomo Dainippon Pharma. M.N. received research funding from Boehringer Ingelheim. S.O. provided consultancy services to Chordia Therapeutics and KAN Research Institute; has ownership in Chordia Therapeutics, Asahi Genomics, and Rebirthel; received research funding from KAN Research Institute, Chordia Therapeutics, Sumitomo Dainippon Pharma, Otsuka Pharmaceutical, and Eisai; and served on advisory committees for Chordia Therapeutics and Asahi Genomics. A.T. received research funding from Chugai Pharmaceutical, Astellas Pharma, Eisai, Otsuka Pharmaceutical, Ono Pharmaceutical, Kyowa Kirin, Shionogi, Sumitomo Dainippon Pharma, Taiho Pharmaceutical, Takeda Pharmaceutical, Teijin, Nippon Shinyaku, Nihon Pharmaceutical, Pfizer Japan, Mochida Pharmaceutical, Yakult Honsha, and Perseus Proteomics, and lecture fees from Chugai Pharmaceutical, Kyowa Kirin, Eisai, Takeda Pharmaceutical, Astellas Pharma, Nippon Shinyaku, Janssen Pharmaceutical, Zenyaku Kogyo, AbbVie GK, Bristol Myers Squibb, and SymBio Pharmaceutical. H.K. received research funding from Chugai Pharmaceutical, Kyowa Kirin, Zenyaku Kogyo, FUJIFILM Corporation, Daiichi Sankyo, Astellas Pharma, Otsuka Pharmaceutical, Nippon Shinyaku, Eisai, Pfizer Japan, Takeda Pharmaceutical, Novartis Pharma KK, Sumitomo Dainippon Pharma, Sanofi KK, Perseus Proteomics, and Celgene Corporation; consulting fees from Astellas Pharma, Amgen Astellas BioPharma KK, and Daiichi Sankyo; and honoraria from Bristol Myers Squibb, Astellas Pharma, and Novartis Pharma KK. Y.A. received lecture fees from Astellas Pharma, Mochida Pharmaceutical, Meiji Seika Pharma, Chugai Pharmaceutical, Kyowa Kirin, and Janssen Pharmaceutical. The remaining authors declare no competing financial interests.
Correspondence: Akihiro Tomita, Department of Hematology, School of Medicine, Fujita Health University, 1-98 Dengakugakubo, Kutsukake-cho, Toyoake, Aichi 470-1192, Japan; e-mail: atomita@fujita-hu.ac.jp; and Seishi Ogawa, Department of Pathology and Tumor Biology, Graduate School of Medicine, Kyoto University, Kyoto, Japan, F-2-203 Yoshida-Konoe-cho, Sakyo-ku, Kyoto 606-8501, Japan; e-mail: sogawa-tky@umin.ac.jp.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal