

Key Points
Otub1 is a novel Dub of c-Maf and promotes its oncogenic transcriptional activity.
The Otub1/c-Maf axis is a potential therapeutic target for MM.

Abstract
The oncogenic transcription factor c-Maf has been proposed as an ideal therapeutic target for multiple myeloma (MM), but how to achieve it is still elusive. In the present study, we found the Otub1/c-Maf axis could be a potential target. Otub1, an OTU family deubiquitinase, was found to interact with c-Maf by mass spectrometry. Otub1 abrogates c-Maf K48-linked polyubiquitination, thus preventing its degradation and enhancing its transcriptional activity. Specifically, this deubiquitinating activity depends on its Lys71 and the N terminus but is independent of UBE2O, a known E2 of c-Maf. Otub1 promotes MM cell survival and MM tumor growth. In contrast, silence of Otub1 leads to c-Maf degradation and c-Maf-expressing MM cell apoptosis. Therefore, the Otub1/c-Maf axis could be a therapeutic target of MM. In order to explore this concept, we performed a c-Maf recognition element–driven luciferase-based screen against US Food and Drug Administration–approved drugs and natural products, from which the generic cardiac glycoside lanatoside C (LanC) is found to prevent c-Maf deubiquitination and induces its degradation by disrupting the interaction of Otub1 and c-Maf. Consequently, LanC inhibits c-Maf transcriptional activity, induces c-Maf-expressing MM cell apoptosis, and suppresses MM growth and prolongs overall survival of model mice, but without apparent toxicity. Therefore, the present study identifies Otub1 as a novel deubiquitinase of c-Maf and establishes that the Otub1/c-Maf axis is a potential therapeutic target for MM.
Introduction
Multiple myeloma (MM) is a malignancy of clonal plasma cells and evolves from asymptomatic monoclonal gammopathy of undetermined significance (MGUS) and smoldering MM.1 c-Maf, an oncogenic transcription factor, is correlated with high risk of poor outcomes of patients with MM.2 By upregulating the expression of several key genes, including cyclin D2 (CCND2),3 integrin β7 (ITGB7),3 and AMPK-related protein kinase 5 (ARK5),4 c-Maf promotes MM cell proliferation, progression, and survival.3,5,6 Moreover, c-Maf leads to MM resistance to proteasomal inhibitors.6 Therefore, c-Maf could be a potential target for MM treatment.6-8 However, how to target c-Maf is still elusive.
It is known that c-Maf can be processed via the ubiquitin (Ub)-proteasomal pathway6,8,9 mediated by the E2 enzyme UBE2O and the E3 ligase HERC4.8,10 Moreover, protein ubiquitination is a dynamic process in that the conjugated ubiquitin molecules can be hydrolyzed by deubiquitinases (Dubs). Recently, we found that the Dubs USP5 and USP7 can abolish c-Maf polyubiquitination and increases its stability.8,11 In the characterization of the c-Maf interactome by affinity purification-coupled tandem mass spectrometry (AP-MS),8 Otub1, a member of the OTU-domain-containing ubiquitin thioesterases, is found to a novel Dub of c-Maf. Moreover, as a proof of concept, we found that targeting the Otub1/c-Maf axis could be a novel strategy for MM treatment.
Materials and methods
Cells
MM cell lines were generously provided by Aaron D. Schimmer (Princess Margaret Cancer Center, Toronto, ON, Canada) and A.K.S. Primary bone marrow cells were donated by healthy donors (HDs) and patients with MM from the First Affiliated Hospital of Soochow University with written consent for research purposes and approved by the Review Board and Ethics Committee of Soochow University. All cells were cultured in Iscove's Modified Dulbecco's Medium supplemented with fetal bovine serum (ExCell Bio, Shanghai, China) and appropriate antibiotics (Sigma-Aldrich).
Chemicals, antibodies, and plasmids
MG132 and cycloheximide (CHX) were provided by MedChemExpress (Shanghai, China). Lanatoside C (LanC), the US Food and Drug Administration (FDA)-approved drug library, and the Natural Products Library were obtained from Targetmol (Wellesley Hills, MA). The Maf plasmids were obtained as described previously.8,11 The Otub1 mutants (K71R, D88A, C91S, and ΔN) were generated using a Site-Directed Mutagenesis Kit from Vazyme Biotech (Nanjing, China) as described previously.12 Antibody information is provided in supplemental Materials (available on the Blood Web site).
AP-MS
Detailed affinity purification–coupled mass spectometry (AP-MS) methodology was described previously.8 Otub1 was chosen because of its 6 unique peptides identified with a confidence >95% (Figure 1A).
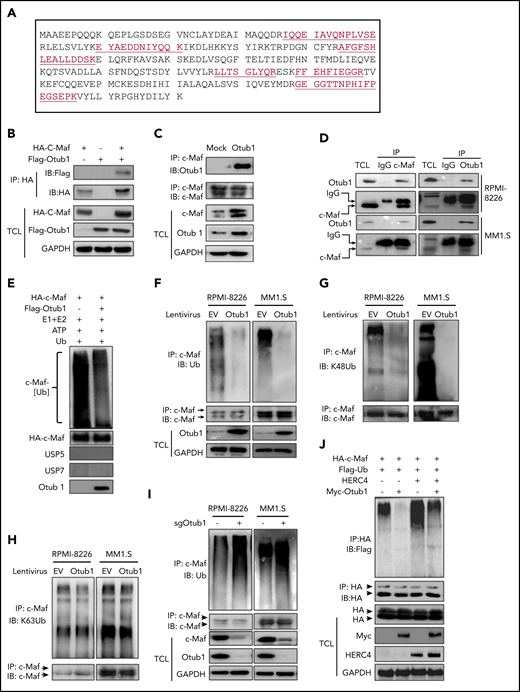
Otub1 interacts with c-Maf and inhibits its K48-linked polyubiquitination. (A) An AP-MS assay was performed against c-Maf. From the c-Maf immunoprecipitates, Otub1 was identified with 6 unique peptides as underlined. (B) c-Maf and Otub1 plasmids were cotransfected into HEK293T cells; 48 hours later, cell lysates were prepared for IP and IB assays. TCL, total cell lysate. (C) MM cell line RPMI-8226 cells were infected with lentiviral Otub1, followed by IP/IB assays as indicated. (D) Cell lysates from RPMI-8226 and MM1.S cells were subjected to IP/IB with a c-Maf or Otub1 antibody or immunoglobulin G (IgG), followed by IB assays. (E) Purified c-Maf and Otub1 were incubated with E1, E2, Ub, and adenosine triphosphate (ATP) in a microtube. The proteins were then subjected to IB assays. (F-H) RPMI-8226 and MM1.S cells were infected with lentiviral Otub1, followed by IP/IB assays to examine the ubiquitination type. EV, empty vector. (I) RPMI-8226 and MM1.S cells were infected with lentiviral sgOtub1 to knock out Otub1, followed by IP/IB assays to determine c-Maf ubiquitination levels. (J) c-Maf, HERC4, Otub1, and Ub plasmids were cotransfected to HEK293T cells, followed by IP/IB assays.
Otub1 interacts with c-Maf and inhibits its K48-linked polyubiquitination. (A) An AP-MS assay was performed against c-Maf. From the c-Maf immunoprecipitates, Otub1 was identified with 6 unique peptides as underlined. (B) c-Maf and Otub1 plasmids were cotransfected into HEK293T cells; 48 hours later, cell lysates were prepared for IP and IB assays. TCL, total cell lysate. (C) MM cell line RPMI-8226 cells were infected with lentiviral Otub1, followed by IP/IB assays as indicated. (D) Cell lysates from RPMI-8226 and MM1.S cells were subjected to IP/IB with a c-Maf or Otub1 antibody or immunoglobulin G (IgG), followed by IB assays. (E) Purified c-Maf and Otub1 were incubated with E1, E2, Ub, and adenosine triphosphate (ATP) in a microtube. The proteins were then subjected to IB assays. (F-H) RPMI-8226 and MM1.S cells were infected with lentiviral Otub1, followed by IP/IB assays to examine the ubiquitination type. EV, empty vector. (I) RPMI-8226 and MM1.S cells were infected with lentiviral sgOtub1 to knock out Otub1, followed by IP/IB assays to determine c-Maf ubiquitination levels. (J) c-Maf, HERC4, Otub1, and Ub plasmids were cotransfected to HEK293T cells, followed by IP/IB assays.
Otub1 lentivirus
The complete Otub1 complementary DNA (cDNA) was inserted into pLVX-AcGFP lentiviral vector (Clontech) to generate lentiviral particles as described previously.8
Colony-forming unit assay
Bone marrow cells from patients with MM or HDs were grown in LanC-containing MethoCult GF H4434 medium for 14 days. Colonies containing ≥20 cells were counted for statistical analysis. The protocol is described in detail in the supplemental Methods.
RT-PCR
qRT-PCR
The quantitative RT-PCR (qRT-PCR) primers for Otub1 were 5′-AACACGTTCATGGACCTGATTG-3′ (forward) and 5′-TGCTCTGGTCATTGAAGGAGG-3′ (reverse). The specific primers for c-Maf, glyceraldehyde-3-phosphate dehydrogenase, CCND2, ARK5, and ITGB7 and the qRT-PCR protocol were described previously.8 Data quantification was carried out by the 2−ΔΔCt method.
In-tube ubiquitination assay
Purified hemagglutinin (HA)-c-Maf and Flag-Otub1 proteins were added to the reaction mixture containing adenosine triphosphate and Myc-Ub, with or without E1 and E2 (Boston Biochem, Boston, MA), for deubiquitination according to the manufacturer’s instructions.
IB
After transfection with appropriate plasmids, cells were lysed in a lysis buffer followed by sodium dodecyl sulfate-polyacrylamide gel electrophoresis and immunoblotting (IB), as described previously.8
IP
Forty eight hours following transfection with appropriate plasmids, total cell lysates (TCLs) were prepared and subjected to immunoprecipitation (IP), as described previously.8
Apoptosis detection with flow cytometry
Treated MM cells were stained with Annexin V–fluorescein isothiocyanate (FITC)/propidium iodide (PI) (MultiSciences Biotech, Hangzhou, China). Flow cytometry was performed on a BD flow cytometer, as described previously.10
Luciferase-based screen
HEK293T cells cotransfected with the pMARE.Luci,10 c-Maf, and Otub1 plasmids were incubated with each compound (5 µM) from TargetMol collections of FDA-approved drugs and natural products. Luciferase activity was analyzed with the Bright-Glo substrate (Promega), as described previously.8 The screen is described in detail in the supplemental Materials.
Transcriptomic sequencing
RPMI-8226 cells were independently infected with lentiviral Otub1 or empty virus (n = 3) for 72 hours, and cells were collected for total RNA extraction using Trizol reagent (Invitrogen). After enrichment of messenger RNA (mRNA) by OligoT and random hexamer-primed cDNA synthesis, a second PCR was performed before HiSeq 2000 sequencing and analysis by Shanghai Personalbio Technology (Shanghai, China).
Data mining
Two MM data sets (Agnelli Myeloma 313 and Zhan Myeloma 314 ) from the Oncomine database (www.oncomine.org) were analyzed for Otub1 expression per instructions of the database. The clinical relevance of Otub1 in patients with MM was analyzed based on the GSE2658 data set from 559 patients with MM2 using the PROGgeneV2 platform (http://genomics.jefferson.edu/proggene/).15
Myeloma models in nude mice
Three types of MM models were established to examine the effects of Otub1 and LanC treatment. One is a subcutaneous xenograft model by inoculating RPMI-8226 and LP1 cells into BALB/c nude mice. The other 2 are disseminated models. One is generated by tail-vein injection of RPMI-8226 and KMS11 cells expressing green fluorescent protein (GFP)-Otub1 or GFP; the other one is generated by tail-vein injection of GFP-labeled RPMI-8226 and KMS11 cells followed by LanC treatment. All mice were obtained from the Slac Laboratory Animal Co. (Shanghai, China). These experiments were approved by the Review Board for Animal Welfare and Ethics of Soochow University. The protocols are described in detail in the supplemental Methods.
Statistics
Statistical differences between the control and experimental groups were analyzed by Student t test or analysis of variance. Survival time was calculated using the Kaplan-Meier method and compared by log-rank test, as described previously.16
Results
Otub1 interacts with c-Maf and reduces its K48-linked polyubiquitination
To identify ubiquitination-associated enzymes for c-Maf, we performed an affinity purification using a HA-c-Maf–specific antibody to pull down c-Maf–interacting proteins, followed by trypsin digestion and tandem mass spectrometry,8 from which UBA1,8 UBE2O,10 HERC4,8 some proteasome proteins, and the Dub Otub1 were identified (supplemental Figure 1; Figure 1A). To verify the c-Maf-Otub1 interaction, both plasmids were cotransfected into HEK293T cells. The reciprocal IP/IB assays demonstrated that Otub1 interacted with c-Maf in HEK293T cells (Figure 1B; supplemental Figure 2). This interaction was also confirmed in MM cells at both exogenous and endogenous contexts (Figure 1C-D). Because Otub1 is a putative Dub, we wondered whether Otub1 could decrease c-Maf ubiquitination. To this end, c-Maf deubiquitination was measured in a cell-free system. As shown in Figure 1E, in the presence of Otub1, but not the proven USP5 or USP7, c-Maf polyubiquitination was markedly reduced, which was confirmed in MM cells by lentiviral Otub1 infection (Figure 1F). Furthermore, Otub1 decreased c-Maf ubiquitination in a K48-linked, but not K63-linked, manner (Figure 1G-H). Moreover, when Otub1 was knocked out by its specific single guide RNA, c-Maf ubiquitination markedly increased (Figure 1I). It is known that HERC4 mediates c-Maf polyubiquitination as an E3,8 but to find out whether Otub1 prevents HERC4-mediated c-Maf ubiquitination, HEK293T cells were cotransfected with c-Maf, Ub, HERC4, and Otub1. The subsequent IP/IB assay showed that HERC4 increased c-Maf polyubiquitination, which was markedly reduced by coexpression of Otub1 (Figure 1J). Interestingly, Otub1 also deubiquitinated MafA, but not MafB, the close family member of c-Maf (supplemental Figure 3B). Taken together, the above results demonstrated that Otub1 interacts with c-Maf and decreases its polyubiquitination.
Otub1 stabilizes c-Maf protein
Because Otub1 can decrease c-Maf K48-linked polyubiquitination, a hallmark tag of protein degradation, we wondered whether Otub1 could stabilize c-Maf. To this end, c-Maf or its lysine-free (K0) mutant was cotransfected with Otub1 into HEK293T cells, and the subsequent IB assay showed that Otub1 increased protein levels of wild-type (WT), but not K0, c-Maf (Figure 2A), suggesting that polyubiquitination is required for Otub1 to stabilize c-Maf. Moreover, Otub1 increased c-Maf stability in a time- and concentration-dependent manner in both HEK293T (Figure 2B) and MM cells (Figure 2C). Moreover, the proteasomal inhibitor MG132 could abolish c-Maf degradation induced by siOtub1 (Figure 2D). This finding was further confirmed by the CHX chase assay. In the presence of CHX, an inhibitor of de novo protein synthesis, Otub1 markedly increased the half-life of c-Maf protein in both HEK293T (Figure 2E) and RPMI-8226 cells (Figure 2F). Notably, Otub1 also stabilized MafA, but not MafB, consistent with the effects on their polyubiquitination (supplemental Figure 3A-B). However, Otub1 failed to induce c-Maf expression in L363 and U266 cells that lack endogenous c-Maf (supplemental Figure 4). Taken together, the above results suggested that Otub1 stabilizes c-Maf by decreasing its polyubiquitination and preventing its degradation in proteasomes.
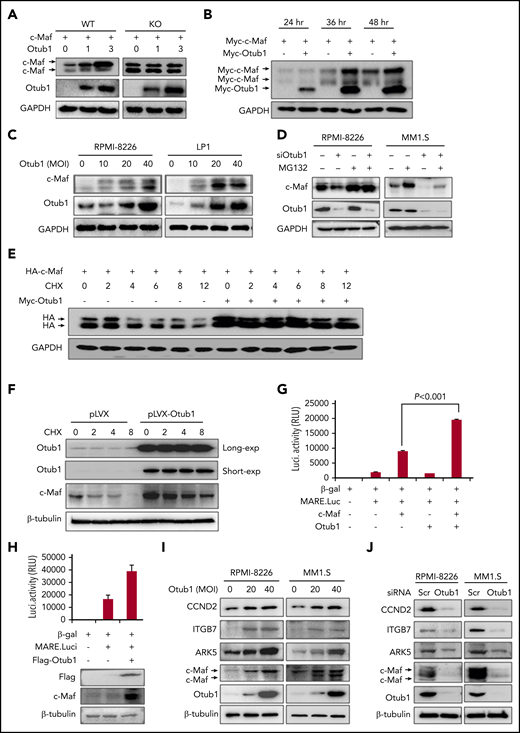
Otub1 stabilizes c-Maf and promotes its transcriptional activity. (A) Otub1 was cotransfected with c-Maf and its lysine-free variant (K0) in HEK293T cells; 48 hours later, cell lysates were prepared for IB assay as indicated. GAPDH, glyceraldehyde 3-phosphate dehydrogenase. (B) Otub1 plasmids were cotransfected with c-Maf into HEK293T cells for the indicated periods, followed by IB assays. (C) MM cell lines RPMI-8226 and LP1 were infected with lentiviral Otub1 for 72 hours, followed by an IB assay. MOI, multiplicity of infection. (D) MM cell lines were transfected with siOtub1 for 48 hours, followed by MG132 treatment (8 hours) before being collected for IB assays. (E) c-Maf was cotransfected with Otub1 for 24 hours, followed by CHX treatment for the indicated periods and IB assays. (F) RPMI-8226 cells were infected with lentiviral Otub1, followed by CHX treatment and IB assays for endogenous c-Maf stability. (G) Otub1, MARE.Luci, c-Maf, and β-galactosidase (β-gal) plasmids were cotransfected into HEK293T cells; 24 hours later, cell lysates were prepared for luciferase (Luci.) activity measurement and IB assays. RLU, relative light units. (H) MARE.Luci, Otub1, and β-gal were cotransfected into RPMI-8226 cells for 48 hours, followed by luciferase and IB assays. (I-J) MM cell lines RPMI-8226 and MM1.S were infected with lentiviral Otub1 (I) for 72 hours or were transfected with siOtub1 or scramble (Scr) for 48 hours (J), followed by IB assays to evaluate the expression levels of c-Maf-targeted genes.
Otub1 stabilizes c-Maf and promotes its transcriptional activity. (A) Otub1 was cotransfected with c-Maf and its lysine-free variant (K0) in HEK293T cells; 48 hours later, cell lysates were prepared for IB assay as indicated. GAPDH, glyceraldehyde 3-phosphate dehydrogenase. (B) Otub1 plasmids were cotransfected with c-Maf into HEK293T cells for the indicated periods, followed by IB assays. (C) MM cell lines RPMI-8226 and LP1 were infected with lentiviral Otub1 for 72 hours, followed by an IB assay. MOI, multiplicity of infection. (D) MM cell lines were transfected with siOtub1 for 48 hours, followed by MG132 treatment (8 hours) before being collected for IB assays. (E) c-Maf was cotransfected with Otub1 for 24 hours, followed by CHX treatment for the indicated periods and IB assays. (F) RPMI-8226 cells were infected with lentiviral Otub1, followed by CHX treatment and IB assays for endogenous c-Maf stability. (G) Otub1, MARE.Luci, c-Maf, and β-galactosidase (β-gal) plasmids were cotransfected into HEK293T cells; 24 hours later, cell lysates were prepared for luciferase (Luci.) activity measurement and IB assays. RLU, relative light units. (H) MARE.Luci, Otub1, and β-gal were cotransfected into RPMI-8226 cells for 48 hours, followed by luciferase and IB assays. (I-J) MM cell lines RPMI-8226 and MM1.S were infected with lentiviral Otub1 (I) for 72 hours or were transfected with siOtub1 or scramble (Scr) for 48 hours (J), followed by IB assays to evaluate the expression levels of c-Maf-targeted genes.
Otub1 promotes c-Maf transcriptional activity
The above results demonstrated that Otub1 deubiquitinates and stabilizes c-Maf; therefore, we wondered whether Otub1 upregulates c-Maf transcriptional activity. To this end, Otub1 was cotransfected with c-Maf and a Maf recognition element–driven luciferase reporter (pMARE.Luci)17 into HEK293T cells. The luciferase assay showed that Otub1 significantly increased luciferase activity (Figure 2G). This finding was also confirmed in RPMI-8226 cells (Figure 2H). Consistently, Otub1 markedly upregulated the expression of CCND2, ITGB7, and ARK5, 3 typical genes modulated by c-Maf (Figure 2I). Furthermore, when Otub1 was knocked down, these genes were markedly downregulated, along with c-Maf degradation (Figure 2I). Furthermore, Otub1-induced CCND2 expression was verified in RPMI-8226 cells by transcriptomic sequencing. Consistently, knockdown of Otub1 arrested the cell cycle at the G1 phase (supplemental Figure 5). Therefore, all of these findings demonstrated that Otub1 upregulates c-Maf transcriptional activity by reducing its polyubiquitination and increasing its stability.
Otub1 does not interact with UBE2O but decreases c-Maf ubiquitination via both canonical and noncanonical pathways
Otub1 functions as a canonical deubiquitinase by cleaving conjugated Ub chains18 or acts in a noncanonical manner by binding to certain E2 enzymes to prevent Ub chain transfer.19 To find which mechanism is important for Otub1 in c-Maf ubiquitination, we mutated K71, D88, and C91, which are critical for its protease activity, and deleted the N terminus, which is essential for Otub1 to interact with E2,12,18,19 as shown in Figure 3A. Following cotransfection of c-Maf and individual Otub1 variants, the IP/IB assays showed that the D88A and C91S variants dramatically decreased c-Maf ubiquitination to a level similar to WT Otub1, but the K71R and ΔN mutants did not (Figure 3B). Therefore, D88 and C91 are dispensable, and K71 and the N terminus are required for Otub1 to modulate c-Maf ubiquitination. To confirm these findings, c-Maf was cotransfected with K71R or C91S variants followed by a CHX chase assay. The IB assay demonstrated that both WT and C91S Otub1 stabilized c-Maf, while c-Maf was steadily downregulated in the presence of K71R (Figure 3C-D), suggesting the K71R substitution inactivated Otub1. To solidify this finding, MM cells were infected with lentiviral Otub1 mutants. The subsequent IP/IB assay confirmed that the K71R mutation abolished Otub1 activity and failed to deubiquitinate c-Maf (Figure 3E). Moreover, USP5 and USP7 are proven Dubs of c-Maf polyubiquitination,11,20 but cotransfection of Otub1 and USP5/7 led to more c-Maf accumulation (Figure 3F), suggesting that these Dubs are not redundant for c-Maf stability. Furthermore, Otub1 prevents substrate ubiquitination by binding to some E2s,19 while UBE2O is a hybrid E2/E3 for c-Maf ubiquitination and degradation.10 To find out whether Otub1 stabilizes c-Maf via UBE2O, Otub1 and/or its ΔN mutant were cotransfected with UBE2O. The IP/IB assays showed that Otub1 did not bind to UBE2O (Figure 3G) but interacted with UBE2D3 (Figure 3H), a proven E2 partner of Otub1. Therefore, Otub1 might display a unique action on c-Maf polyubiquitination and stability by both canonical and noncanonical processes.
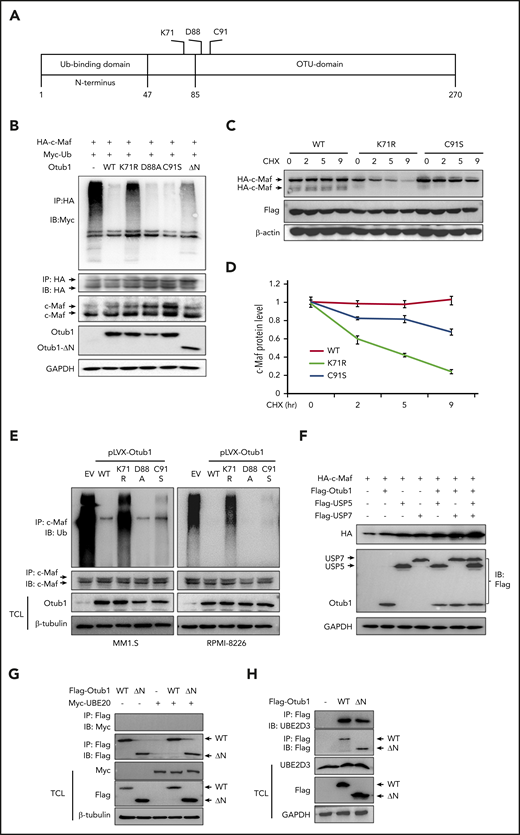
Otub1 deubiquitinates c-Maf in both canonical and noncanonical manners. (A) A skeptical illustration of Otub1 domains and key residues in its catalytic activity. (B) c-Maf, Ub, and Otub1 variants were cotransfected into HEK293T cells followed by IP/IB assays as indicated. (C) WT, K71R, or C91S Otub1 variants were cotransfected into HEK293T cells with c-Maf plasmids for 24 hours, followed by CHX treatment and IB assays. (D) The density of c-Maf bands from panel C was analyzed. (E) Lentivirus containing Otub1 and its mutants were infected into MM cell lines for 96 hours, followed by IP/IB assays to evaluate c-Maf polyubiquitination levels. EV, empty virus. (F) Otub1 and USP5 or USP7 plasmids were cotransfected into HEK293T cells for 24 hours, followed by IB assays. (G-H) WT or ΔN Otub1 cells were transfected into HEK293T cells alone or together with UBE2O (G) or UBE2D3 (H) for 24 hours, followed by IP/IB assays as indicated.
Otub1 deubiquitinates c-Maf in both canonical and noncanonical manners. (A) A skeptical illustration of Otub1 domains and key residues in its catalytic activity. (B) c-Maf, Ub, and Otub1 variants were cotransfected into HEK293T cells followed by IP/IB assays as indicated. (C) WT, K71R, or C91S Otub1 variants were cotransfected into HEK293T cells with c-Maf plasmids for 24 hours, followed by CHX treatment and IB assays. (D) The density of c-Maf bands from panel C was analyzed. (E) Lentivirus containing Otub1 and its mutants were infected into MM cell lines for 96 hours, followed by IP/IB assays to evaluate c-Maf polyubiquitination levels. EV, empty virus. (F) Otub1 and USP5 or USP7 plasmids were cotransfected into HEK293T cells for 24 hours, followed by IB assays. (G-H) WT or ΔN Otub1 cells were transfected into HEK293T cells alone or together with UBE2O (G) or UBE2D3 (H) for 24 hours, followed by IP/IB assays as indicated.
Otub1 promotes MM cell survival and correlates with poor prognosis of patients with MM
Since Otub1 increases c-Maf oncogenic transcriptional activity, we wondered whether Otub1 expression affects clinical outcomes of patients with MM. To this end, Otub1 expression profiles were evaluated in various stages of MM based on 2 independent data sets. The results showed that Otub1 was steadily upregulated from HDs to patients with MGUS to patients with SMM (Figure 4A) or from HDs to patients with MGUS to patients with MM to patients with PCL (Figure 4B). Otub1 expression was confirmed in primary bone marrow cells from HDs and patients with MM by qRT-PCR (Figure 4C) and regular RT-PCR (supplemental Figure 6). These findings indicated Otub1 might promote MM cell survival and MM tumor growth. To further determine the correlation between Otub1 and clinic outcomes of patients with MM, a large public data set2 was analyzed, which revealed that Otub1 specifically predicted inferior prognosis in the myloid and the proliferation subgroups of patients with MM (Figure 4D-E; the higher the Otub1 expression, the lower the overall survival of patients with MM). To verify this finding, MM cell lines stably infected with lentiviral GFP-Otub1 were injected into nude mice via the tail vein, followed by mice survival analysis. It showed that Otub1 strikingly increased the death rate of nude mice carrying RPMI-8226 that expressed a high level of c-Maf, but not nude mice carrying KMS11 with a low level of c-Maf (Figure 4F-G). Therefore, Otub1 predicts poor prognosis of patients with MM in association with c-Maf.

Otub1 is overexpressed in MM and confers poor prognosis to patients with MM. (A) Otub1 cDNA expression (exp.) was analyzed from HDs (n = 23), monoclonal gammopathy of undetermined significance (MGUS) (n = 44), and smoldering MM (SMM) (n = 12) based on the Zhan Myeloma 3 data set.13 (B) Otub1 cDNA was analyzed from HD (n = 5), MGUS (n = 11), MM (n = 133), and plasma cell leukemia (PCL) (n = 9) based on the Agnelli Myeloma 3 data set.14 (C) qRT-PCR analysis of Otub1 in bone marrow cells from HDs (n = 10) and patients with MM (n = 30). (D-E) Otub1 expression and clinical outcomes were evaluated based on the GSE2658 GEP data set2 on the PROGgeneV2 platform.15 Otub1 predicted an inferior prognosis for patients with MM in the myloid (MY) (D) and the proliferation (E) subgroups. (F-G) RPMI-8226 (F) with high c-Maf and KMS11 (G) with low c-Maf were stably infected with lentiviral GFP or GFP-Otub1. Cells were injected into nude mice via the tail vein. Mice survival was then monitored. (H) RPMI-8226 and MM1.S cells were stably infected with lentiviral Otub1, followed by cell viability assay with MTT. (I-J) MM cells were transfected with siOtub1 for 48 hours, followed by IB (I) and apoptotic assays on a flow cytometer (J). **P < .01; ***P < .001. NS, not significant; Scr, scramble.
Otub1 is overexpressed in MM and confers poor prognosis to patients with MM. (A) Otub1 cDNA expression (exp.) was analyzed from HDs (n = 23), monoclonal gammopathy of undetermined significance (MGUS) (n = 44), and smoldering MM (SMM) (n = 12) based on the Zhan Myeloma 3 data set.13 (B) Otub1 cDNA was analyzed from HD (n = 5), MGUS (n = 11), MM (n = 133), and plasma cell leukemia (PCL) (n = 9) based on the Agnelli Myeloma 3 data set.14 (C) qRT-PCR analysis of Otub1 in bone marrow cells from HDs (n = 10) and patients with MM (n = 30). (D-E) Otub1 expression and clinical outcomes were evaluated based on the GSE2658 GEP data set2 on the PROGgeneV2 platform.15 Otub1 predicted an inferior prognosis for patients with MM in the myloid (MY) (D) and the proliferation (E) subgroups. (F-G) RPMI-8226 (F) with high c-Maf and KMS11 (G) with low c-Maf were stably infected with lentiviral GFP or GFP-Otub1. Cells were injected into nude mice via the tail vein. Mice survival was then monitored. (H) RPMI-8226 and MM1.S cells were stably infected with lentiviral Otub1, followed by cell viability assay with MTT. (I-J) MM cells were transfected with siOtub1 for 48 hours, followed by IB (I) and apoptotic assays on a flow cytometer (J). **P < .01; ***P < .001. NS, not significant; Scr, scramble.
Next, we evaluated Otub1 on MM cell viability in vitro. RPMI-8226 and MM1.S were infected with lentiviral Otub1 for viability assay. The results showed ectopic Otub1 markedly increased cell viability of c-Maf-expressing RPMI-8226 and MM1.S (Figure 4H), but not c-Maf–deficient L363 and U266 (supplemental Figure 7A). To confirm the effects of Otub1 on MM cell survival, we knocked down Otub1 to check cell apoptosis. As expected, Otub1 knockdown downregulated c-Maf and CCND2 and induced marked cleavage of poly (adenosine 5′-diphosphate-ribose) polymerase (PARP), a hallmark of apoptosis, in c-Maf-expressing, but not in c-Maf–deficient, cells (Figure 4I). siOtub1-induced cell apoptosis was confirmed by flow cytometric analysis after Annexin V and propidium iodide staining (Figure 4J). Moreover, Otub1-promoted MM cell viability could be fully inhibited by c-Maf knockdown (supplemental Figure 7B). In contrast, siOtub1-induced MM cell apoptosis could be rescued by stabilized c-Maf (supplemental Figure 8A). Therefore, Otub1 promotes MM cell survival by deubiquitinating and stabilizing c-Maf.
LanC is an inhibitor of the Otub1/c-Maf axis
The above results demonstrated that the Otub1/c-Maf axis promotes MM survival and growth, while inhibition of Otub1/c-Maf leads to MM cell apoptosis; therefore, targeting the Otub1/c-Maf axis might be a potential strategy for MM treatment. To explore this concept, we established a drug screen system in HEK293T cells that expressed MARE.Luci, c-Maf, and Otub1. These cells were incubated with 5 μM of each compound from FDA-approved drugs and the natural compounds. Luciferase activity was measured after 24-hour incubation, and those compounds that reduced luciferase activity >75% were considered for further studies. Both screens returned that several natural compounds, including LanC, displayed great potency in suppressing MARE.Luci activity (Figure 5A-B). To examine the effects of LanC on c-Maf polyubiquitination, MM cells were incubated with LanC. The IP/IB assays showed that LanC drastically increased c-Maf polyubiquitination in both cell lines (Figure 5C). Moreover, LanC increased the K48-linked, but not K63-linked, polyubiquitination form on c-Maf (Figure 5D), consistent with effects of Otub1 on c-Maf ubiquitination (Figure 1G-H). Furthermore, when Otub1 was knocked down, LanC failed to increase c-Maf polyubiquitination (Figure 5E), suggesting that LanC suppressed c-Maf ubiquitination via Otub1. To demonstrate this hypothesis, Otub1-knockdown cells were transfected with c-Maf and MARE.Luci, followed by LanC treatment. The results showed that LanC inhibited c-Maf-driven luciferase activity in intact cells (Figure 5F). However, when Otub1 was knocked down, c-Maf stability and its transcriptional activity were less affected by LanC (Figure 5F). Moreover, LanC markedly decreased CCND2 and ARK5 expression at both the protein and mRNA levels in MM cells (Figure 5G-H) but had no effect on c-Maf mRNA (Figure 5H), suggesting that LanC suppressed c-Maf transcriptional activity by targeting its protein stability. Furthermore, LanC disrupted the interaction between c-Maf and Otub1 (Figure 5I), which is probably the mechanism by which LanC inhibits the Otub1/c-Maf axis, thus decreasing MM cell viability.

Identification of LanC as an inhibitor of the Otub1/c-Maf axis. FDA-approved drugs (A) and natural products (B) provided by TargetMol were subjected to screen inhibitors of the Otub1/c-Maf axis as described in “Materials and methods.” (C) RPMI-8226 and LP1 were incubated with LanC at 100 nM for 24 hours, followed by IP/IB assays. (D) RPMI-8226 cells were treated with LanC for 24 hours, followed by IP/IB assays. (E) HEK293T cells were transfected with an HA-c-Maf plasmid and siOtub1 for 48 hours, followed by LanC treatment (24 hours) before being subjected to IP/IB assays. (F) HEK293T cells were cotransfected with c-Maf, MARE.Luci, and siOtub1 for 24 hours, followed by LanC treatment (8 hours) and a luciferase assay. (G-H) MM cells were incubated with LanC overnight, followed by IB (G) or RT-PCR (H) assays for indicated proteins or genes. (I) HEK293T cells were cotransfected with c-Maf and Otub1 for 24 hours, followed by LanC treatment (8 hours) and IP/IB assays to examine the interaction between Otub1 and c-Maf. *P < .05; **P < .01; ***P < .001. NS, not significant.
Identification of LanC as an inhibitor of the Otub1/c-Maf axis. FDA-approved drugs (A) and natural products (B) provided by TargetMol were subjected to screen inhibitors of the Otub1/c-Maf axis as described in “Materials and methods.” (C) RPMI-8226 and LP1 were incubated with LanC at 100 nM for 24 hours, followed by IP/IB assays. (D) RPMI-8226 cells were treated with LanC for 24 hours, followed by IP/IB assays. (E) HEK293T cells were transfected with an HA-c-Maf plasmid and siOtub1 for 48 hours, followed by LanC treatment (24 hours) before being subjected to IP/IB assays. (F) HEK293T cells were cotransfected with c-Maf, MARE.Luci, and siOtub1 for 24 hours, followed by LanC treatment (8 hours) and a luciferase assay. (G-H) MM cells were incubated with LanC overnight, followed by IB (G) or RT-PCR (H) assays for indicated proteins or genes. (I) HEK293T cells were cotransfected with c-Maf and Otub1 for 24 hours, followed by LanC treatment (8 hours) and IP/IB assays to examine the interaction between Otub1 and c-Maf. *P < .05; **P < .01; ***P < .001. NS, not significant.
Targeting the Otub1/c-Maf axis with LanC leads to MM cell apoptosis
Next, we examined MM cell apoptosis induced by LanC. Compared with MM1.S, LP1, and RPMI-8226 cells, which express c-Maf, KMS11 and U266 cells expressed little or no c-Maf (Figure 6A), consistent with a previous study.6 After treatment with LanC, cells were subjected to IB assays that showed that LanC induced PARP cleavage in MM1.S, LP1, and RPMI-8226 cells, but not in U266 and KMS11 cells (Figure 6A). Notably, when Otub1 was knocked down, LanC activity was partly abolished, as evidenced by PARP cleavage (Figure 6B). The Annexin V staining and flow cytometric assays confirmed this finding that LanC induced marked apoptosis in MM1.S, RPMI-8226, and LP1 cells, but not in KMS11 or U266 cells (Figure 6C). Moreover, when WT c-Maf was introduced to Otub1-knockdown cells, cell apoptosis was strikingly reduced (Figure 6D). When a lysine-free c-Maf mutant (K0) was introduced, either siOtub1 or LanC almost completely lost its ability to induce MM cell apoptosis (supplemental Figure 8), further suggesting that LanC acts on the Otub1/c-Maf axis. We also examined the effects of LanC on primary MM cells using a colony-forming unit assay, which revealed that LanC significantly suppressed colony formation in bone marrow cells from patients with MM, but not HDs (Figure 6E; supplemental Figure 9). Interestingly, fewer colonies were found in MM species with high c-Maf than in those with low-c-Maf, further suggesting c-Maf is critical for the LanC-induced decrease in colony formation (Figure 6F). Moreover, enforced expression of c-Maf significantly abolished PARP cleavage induced by LanC in typical MM cells (Figure 6G-H), consistent with apoptosis analyses (Figure 6D; supplemental Figure 8). Notably, although LanC induced striking PARP and caspase-3 cleavage, it did not show marked effects on proapoptotic proteins (such as Bax and Bim), with the exception of Rb, and had no effects on prosurvival proteins (such as Bcl-xL and Bcl-2), with the exception of p-mTOR and c-IAP1 (supplemental Figure 10). Furthermore, consistent with the effects of WT-Otub1, the K71R and ΔN mutants partly ablated PARP cleavage induced by LanC (supplemental Figure 11). Therefore, all of these results show that LanC induces MM cell apoptosis by targeting the Otub1/c-Maf axis.

Inhibition of the Otub1/c-Maf axis by LanC induces MM cell apoptosis. (A) MM cells were incubated with LanC overnight, followed by IB assays. (B) RPMI-8226 cells were transfected with siOtub1 for 48 hours, followed by LanC treatment and IB assays. (C) MM cells were incubated with LanC overnight, followed by Annexin V/PI staining and flow cytometric analysis. (D) MM cells were transfected with siOtub1 alone or with c-Maf for 48 hours, followed by Annexin V-FITC staining and flow cytometric analysis (n = 3). (E) Bone marrow mononuclear cells from patients with MM (n = 23) or HDs (n = 5) were mixed in LanC (10 nM)–containing MethoCult medium and cultured in 3.5-cm dishes for 14 days before colony counts. (E) Based on the c-Maf measurement of qRT-PCR results, 23 patients with MM from panel D were divided into 3 groups (c-Maflow, c-Mafmid, and c-Mafhi); colonies were then statistically analyzed. (F) RPMI-8226 and MM1.S cells were transfected with c-Maf, followed by LanC treatment of 24 hours and IB assays. (G) The Cle-PARP/Pro-PARP ratios from panel F were quantified. *P < .05; **P < .01. NS, not significant.
Inhibition of the Otub1/c-Maf axis by LanC induces MM cell apoptosis. (A) MM cells were incubated with LanC overnight, followed by IB assays. (B) RPMI-8226 cells were transfected with siOtub1 for 48 hours, followed by LanC treatment and IB assays. (C) MM cells were incubated with LanC overnight, followed by Annexin V/PI staining and flow cytometric analysis. (D) MM cells were transfected with siOtub1 alone or with c-Maf for 48 hours, followed by Annexin V-FITC staining and flow cytometric analysis (n = 3). (E) Bone marrow mononuclear cells from patients with MM (n = 23) or HDs (n = 5) were mixed in LanC (10 nM)–containing MethoCult medium and cultured in 3.5-cm dishes for 14 days before colony counts. (E) Based on the c-Maf measurement of qRT-PCR results, 23 patients with MM from panel D were divided into 3 groups (c-Maflow, c-Mafmid, and c-Mafhi); colonies were then statistically analyzed. (F) RPMI-8226 and MM1.S cells were transfected with c-Maf, followed by LanC treatment of 24 hours and IB assays. (G) The Cle-PARP/Pro-PARP ratios from panel F were quantified. *P < .05; **P < .01. NS, not significant.
Targeting the Otub1/c-Maf axis with LanC displays potent antimyeloma activity in vivo
The above results demonstrated that LanC induces MM cell apoptosis via the Otub1/c-Maf axis in vitro. To verify this effect in vivo, 2 MM models were examined in mice. One was a subcutaneous model established by subcutaneous injection of LP1 and RPMI-8226 cells into severe combined immunodeficiency nude mice. When tumors were palpable, each model was randomly divided into 3 groups (n = 5 per group) followed by administration of vehicle or LanC and monitoring of tumor volumes and mice body weights on a daily basis. The results showed that LanC inhibited MM tumor growth in a dose-dependent manner (Figure 7A-D). LanC at 6 mg/kg almost completely suppressed tumor growth (Figure 7A-D) but did not show marked effects on mice body weights (supplemental Figure 12A-B) or blood chemical analysis, including urea, creatine, total protein, aspartate aminotransferase and alanine aminotransferase (supplemental Figure 12C-D). These results thus suggested inhibition of the Otub1/c-Maf axis by LanC suppressed myeloma xenograft growth but did not show overt toxicity. To further confirm the benefit of LanC in the treatment of MM, a disseminated MM model was established by injection of GFP-labeled KMS11 or RPMI-8226 into nude mice via the tail veins. Compared with the control group, LanC strikingly improved the survival periods of mice carrying RPMI-8226, but not KMS11 (Figure 7E). Analyses of GFP-MM cells from bone marrow showed that LanC significantly reduced GFP-RPMI-8226, but not GFP-KMS11, cells (Figure 7F). This finding was consistent with the in vitro assay in which LanC failed to induce KMS11 cell apoptosis (Figure 6A,C). The tumor tissues dissected at the end of the subcutaneous model were subjected to IB assays. As shown in Figure 7G, LanC downregulated c-Maf, CCND2, and ITGB7 and induced PARP cleavage in myeloma tissues. Therefore, all results collectively demonstrated that targeting the Otub1/c-Maf axis with LanC exerts potent anti-MM activity in vivo.

Inhibition of the Otub1/c-Maf axis by LanC impairs the growth of myeloma xenografts in immunodeficient mice and improves survival of immunodeficient mice. Severe combined immunodeficiency mice were injected subcutaneously with the MM cell line LP1 or RPMI-8226. When tumors were palpable, each model was randomly divided into 3 groups (n = 5 for each group), and vehicle or 3 or 6 mg/kg of LanC was administered intraperitoneally for 15 continuous days. (A-B) Tumor growth curves for the LP1 (A) or the RPMI-8226 model (B). (C-D) Dissected tumors from the LP1 (C) or RPMI-8226 model (D). (E) RPMI-8226 and KMS11 cells were injected into the tail vein of nude mice followed by treatment with LanC (6 mg/kg) on a 5-day-on 2-day-off protocol. Mice survival was monitored. (F) Bone marrow cells from mice treated with LanC or vehicle (as shown in panel E) were collected for flow cytometric analysis for GFP-expressing MM cells. (G) Total proteins were extracted from tumor tissues of the subcutaneous models, followed by IB assay against indicated antigens. **P < .01; ***P < .001. NS, not significant.
Inhibition of the Otub1/c-Maf axis by LanC impairs the growth of myeloma xenografts in immunodeficient mice and improves survival of immunodeficient mice. Severe combined immunodeficiency mice were injected subcutaneously with the MM cell line LP1 or RPMI-8226. When tumors were palpable, each model was randomly divided into 3 groups (n = 5 for each group), and vehicle or 3 or 6 mg/kg of LanC was administered intraperitoneally for 15 continuous days. (A-B) Tumor growth curves for the LP1 (A) or the RPMI-8226 model (B). (C-D) Dissected tumors from the LP1 (C) or RPMI-8226 model (D). (E) RPMI-8226 and KMS11 cells were injected into the tail vein of nude mice followed by treatment with LanC (6 mg/kg) on a 5-day-on 2-day-off protocol. Mice survival was monitored. (F) Bone marrow cells from mice treated with LanC or vehicle (as shown in panel E) were collected for flow cytometric analysis for GFP-expressing MM cells. (G) Total proteins were extracted from tumor tissues of the subcutaneous models, followed by IB assay against indicated antigens. **P < .01; ***P < .001. NS, not significant.
Discussion
As a Dub, Otub1 has been shown to inhibit monoubiquitination,21 K48-linked ubiquitination,18 and K63-linked ubiquitination.19 By preventing the K48-linked polyubiquitination, Otub1 stabilizes several important oncoproteins, including Snail,18 Deptor,22 cIAP1,23 and FoxM1.24 The present study found that c-Maf is a novel substrate of Otub1. Identified by the AP-MS assay, Otub1 interacts with c-Maf and decreases its polyubiquitination levels and therefore increases its protein stability. However, different from previous studies, we found that Otub1 might exert unique mechanisms on c-Maf deubiquitination. It is well known that Otub1 could decrease substrate ubiquitination via the canonical manner to cleave conjugated Ub chains from specific substrates by its protease activity12,18 or the noncanonical one to prevent Ub transfer from E2 to E3 by binding to E2 ubiquitin-conjugating enzymes, such as UBC13 and other E2s.19 For example, Otub1 cleaves ubiquitin chains from Snail while binding to UBE2N to prevent K63-linked Ub chain synthesis.19 Our present study demonstrated that both mechanisms might exist in Otub1-mediated c-Maf deubiquitination. It is known that K71, D88, and C91 are essential in maintaining Otub1 catalytic activity and the N terminus is essential for its binding to E2 enzymes; however, we found that the D88A and C91S Otub1 variants show deubiquitination activity similar to WT, but K71R and ΔN mutants have largely lost their activity, suggesting that D88 and C91 are dispensable but K71 and the N terminus are essential for Otub1-mediated c-Maf deubiquitination. Because the N terminus is required for the non-canonical deubiquitinating activity of Otub1, but Otub1 fails to interact with UBE2O, the known E2 for c-Maf ubiquitination,10 therefore, the E2 partnering with Otub1 to mediate c-Maf ubiquitination needs to be further identified. Thus, different from previous reports,18,21-24 the present study demonstrated that both the N terminus and the catalytic domain are required for Otub1 to deubiquitinate c-Maf, suggesting Otub1 probably modulates c-Maf polyubiquitination via both canonical and noncanonical mechanisms.
The present study also demonstrated that the Otub1/c-Maf axis is a promising therapeutic target against MM. Otub1 is dysregulated in MM and is correlated with poor prognosis of patients with MM. Moreover, inhibition of Otub1 leads to MM cell apoptosis; in contrast, its ectopic expression promotes MM cell survival. This action of Otub1 probably contributes to its deubiquitination on c-Maf and promotion of c-Maf oncogenic transcription, because Otub1 upregulates CCND2, ITGB7, and AKR5, modulated by c-Maf and critical for MM proliferation, adherence, and survival. Furthermore, as a proof of concept, we identified several off-patent drugs from FDA-approved drugs and natural products. The representative one is LanC, which targets the Otub1/c-Maf axis and displays anti-MM activity both in vitro and in vivo. LanC is a generic cardiac glycoside that could be used for congestive heart failure and cardiac arrythmia.25 Recent studies show that LanC induces apoptosis of several types of cancer cells, including gastric,26 liver,27 and colorectal cancers,28 and glioblastomas.29 The present study is the first report identifying LanC as an inhibitor of the Otub1/c-Maf axis. Consistent with the Otub1 action on c-Maf ubiquitination, stability and MM cell survival, LanC inhibits Otub1 deubiquitination activity therefore leading to c-Maf polyubiquitination and degradation. Moreover, LanC-induced MM cell apoptosis depends on the expression of both Otub1 and c-Maf. When Otub1 is knocked down or c-Maf is deficient, LanC loses this activity; in contrast, Otub1 and c-Maf can rescue LanC-induced MM cell death. In addition to c-Maf, some other Otub1 substrates, such as Deptor22 and c-IAP1,23 probably play a key role in LanC-induced MM cell death, because LanC also downregulates c-IAP1 and p-mTOR in LanC-sensitive, but not resistant, MM cells (supplemental Figure 10); however, the detailed mechanisms should be further investigated.
In summary, the present study identifies Otub1 as a novel Dub of c-Maf that promotes MM tumor survival and leads to a poor prognosis in patients with MM. Moreover, targeting the Otub1/c-Maf axis using compounds such as LanC could be a novel and promising strategy for myeloma treatment.
All data are ready to share with researchers of interest. Please contact X.M. (xinliangmao@gzhmu.edu.cn).
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Acknowledgments
This work was partly supported by the National Natural Science Foundation of China (grants 81970194, 81770154, 81320108023, 81800205, 81770215, and 81730003). This study was also partly supported by the Guangzhou Municipal Science and Technology Project (grant 202002030059), the National Science and Technology Major Project (2017ZX09304021), National Key R&D Program of China (2019YFC0840604), the Key R&D Program of Jiangsu Province (BE2019798), the Priority Academic Program Development of Jiangsu Higher Education Institutions (PAPD), and the Guangzhou Science Technology and Innovation Commission Technology Research Projects (201805010005).
Authorship
Contribution: X.M. designed the study; Y.X., J.T., J.C., X.C., Z.Z., and B.C. conducted experiments; M.X., X.T., and D.W. provided key agents and analyzed clinical data; X.M., Y.X., J.T., A.K.S., and M.F.M. analyzed data; and X.M. and Y.X. wrote the manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Xinliang Mao, Guangzhou Institute of Cardiovascular Diseases, Guangdong Key Laboratory of Vascular Diseases, State Key Laboratory of Respiratory Diseases, The Second Affiliated Hospital–Guangdong Key Laboratory of Protein Modification and Degradation, School of Basic Medical Sciences, Guangzhou Medical University, Guangzhou 510260, People’s Republic of China; e-mail: xinliangmao@gzhmu.edu.cn; and Depei Wu, Department of Hematology, The First Affiliated Hospital of Soochow University, Suzhou, Jiangsu 215100, People’s Republic of China; e-mail: wudepei@suda.edu.cn.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal