

Key Points
EBV-associated PCNSL typically has absent MYD88, CD79B, and PIM1 mutations, is rarely ABC cell of origin, and has intact HLA class I/II.
The tumor microenvironment in EBV-associated PCNSL adapts to tolerate expression of an immunogenic virus.

Abstract
Primary central nervous system lymphoma (PCNSL) is confined to the brain, eyes, and cerebrospinal fluid without evidence of systemic spread. Rarely, PCNSL occurs in the context of immunosuppression (eg, posttransplant lymphoproliferative disorders or HIV [AIDS-related PCNSL]). These cases are poorly characterized, have dismal outcome, and are typically Epstein-Barr virus (EBV)-associated (ie, tissue-positive). We used targeted sequencing and digital multiplex gene expression to compare the genetic landscape and tumor microenvironment (TME) of 91 PCNSL tissues all with diffuse large B-cell lymphoma histology. Forty-seven were EBV tissue-negative: 45 EBV− HIV− PCNSL and 2 EBV− HIV+ PCNSL; and 44 were EBV tissue-positive: 23 EBV+ HIV+ PCNSL and 21 EBV+ HIV− PCNSL. As with prior studies, EBV− HIV− PCNSL had frequent MYD88, CD79B, and PIM1 mutations, and enrichment for the activated B-cell (ABC) cell-of-origin subtype. In contrast, these mutations were absent in all EBV tissue-positive cases and ABC frequency was low. Furthermore, copy number loss in HLA class I/II and antigen-presenting/processing genes were rarely observed, indicating retained antigen presentation. To counter this, EBV+ HIV− PCNSL had a tolerogenic TME with elevated macrophage and immune-checkpoint gene expression, whereas AIDS-related PCNSL had low CD4 gene counts. EBV-associated PCNSL in the immunosuppressed is immunobiologically distinct from EBV− HIV− PCNSL, and, despite expressing an immunogenic virus, retains the ability to present EBV antigens. Results provide a framework for targeted treatment.
Introduction
Primary central nervous system lymphoma (PCNSL) is a rare form of extranodal non-Hodgkin lymphoma (NHL) that is confined to the brain, eyes, and cerebrospinal fluid without evidence of systemic spread. It most commonly has diffuse large B-cell lymphoma (DLBCL) histology, has a median age of 65 years, and accounts for ∼1% of all cases of NHL.1,2 In recent years, understanding of the molecular pathogenesis of PCNSL has increased. Recurrent mutations have been identified in the B‐cell receptor (BCR) signaling axis and its downstream target: nuclear factor κ-light-chain enhancer of activated B cells (NF-κB). These principally involve MYD88, CD79B, and less often CARD11 and TNFAIP3.3-7 Phylogenetic analysis indicates MYD88 mutation is an early clonal event.8 Among other high-frequency mutations is the cell-cycle/adhesion gene PIM1.9,10
Rarely, PCNSL occurs with immunosuppression (eg, posttransplant lymphoproliferative disorder [PTLD] or HIV [AIDS-related PCNSL]).11 Although no accurate figures exist, it has been estimated that overall PCNSL after immunosuppression accounts for <10% of PCNSL cases (<0.1% of NHL).12,13 These patients are typically younger than nonimmunosuppressed PCNSL, have dismal outcomes,13,14 and because (with a few worthy exceptions)15,16 clinical trials for PCNSL typically exclude patients with PTLD and HIV, the optimal management is unknown and there are no consensus guidelines. Because of its rarity, characterization of the immunobiological features of PCNSL after immunosuppression remains minimal, with the largest published series restricted to microRNA profiling of 9 cases.17 It is known however that the malignant B cells are typically tissue-positive for the lymphotropic Epstein-Barr virus (EBV), termed EBV-associated PCNSL.13
Here, we present, to our knowledge, the first large-scale comparative data on the genetic and gene expression landscape of PCNSL subtypes, stratified by EBV tissue and HIV status. PCNSL tissues were subdivided into: EBV tissue-negative: EBV− HIV− PCNSL and EBV− HIV+ PCNSL; and EBV tissue-positive: EBV+ HIV+ PCNSL and EBV+ HIV− PCNSL. The findings provide a rationale for targeted therapy.
Methods
Samples
There were 91 samples in total (90 formalin-fixed paraffin-embedded [FFPE] patient samples and 1 cell line). FFPE samples were obtained retrospectively from Australia (60 samples), Germany (7 samples), and the United States (23 samples). Selection criteria were PCNSL cases with DLBCL histology in immunosuppressed and immunocompetent settings. For non-HIV cases, samples were obtained from hospitals that were tertiary referral centers for both lymphoma and organ transplantation. This ensured that PCNSL cases were enriched for PTLD. HIV cases (with the exception of samples from 3 Australian patients) were from US HIV biobanks. The consort diagram (Figure 1) provides details. Median age for EBV− HIV− PCNSL was 64 (range, 28-82) years, 61% male; for AIDS-related PCNSL 37 (range, 31-60) years, 100% male; and for EBV+ HIV− PCNSL 55 (range, 30-71) years, 45% male. Clinical details are provided in the supplemental Data on the Blood Web site. In line with previous reports, EBV+ HIV− PCNSL resulting from PTLD occurred late following transplant (mean >9 years) as distinct from systemic EBV+ PTLD.13 All tumors were classified as PCNSL with DLBCL histology according to the World Health Organization classification.1 Systemic staging was performed and only cases of isolated CNS lymphoma were included. EBV-tissue status was determined as published.17,18 There were: 44 EBV− HIV− PCNSL, 2 EBV− HIV+ PCNSL, 23 EBV+ HIV+ PCNSL, and 21 EBV+ HIV− PCNSL (18 with PTLD, 3 without PTLD/immunosuppression) FFPE samples. These were macrodissected to enrich for tumor content. Testing was based on quality and quantity of FFPE-extracted DNA/RNA, with all samples suitable for either sequencing and/or gene expression. The 44 EBV− HIV− PCNSL samples were supplemented with the human EBV− HIV− brain cell-line TK (JCRB1206, JCRB Cell-Bank, Japan), making 45. The study was approved by the relevant institutional regulatory boards in concordance with the Declaration of Helsinki.
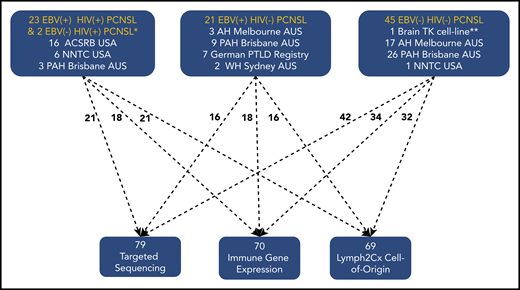
Consort diagram providing details of PCNSL subtype and sample testing performed. ACSRB, AIDS Cancer Specimen Resource Bank; AH, Austin Hospital; NNTC, NeuroAIDS National Tissue Collection; PAH, Princess Alexandra Hospital; WH, Westmead Hospital. *One of the 2 EBV− HIV+ PCNSL tissues underwent sequencing only, and both had COO. **Brain TK cell-line underwent targeted sequencing only.
Consort diagram providing details of PCNSL subtype and sample testing performed. ACSRB, AIDS Cancer Specimen Resource Bank; AH, Austin Hospital; NNTC, NeuroAIDS National Tissue Collection; PAH, Princess Alexandra Hospital; WH, Westmead Hospital. *One of the 2 EBV− HIV+ PCNSL tissues underwent sequencing only, and both had COO. **Brain TK cell-line underwent targeted sequencing only.
Sequencing
A customized, hybrid-capture SureSelect XT (Agilent, CA) panel of 54 genes was used to identify mutations in antigen presentation/processing (8 genes), immune function (7 genes), the BCR-dependent NF-κB pathway (8 genes), epigenetic regulation (10 genes), cell cycle/adhesion (12 genes), and B-cell differentiation (9 genes). These mutations were known to be of biological importance in EBV− HIV− PCNSL and/or tumor immunity, and were chosen on review of previous literature.3,4,6,7,19 The sequencing panel was also designed to enrich the targeted regions to obtain copy number variation (CNV) data for HLA class I/II alleles (known to be frequently absent in EBV− HIV− PCNSL),5,20,21 and the antigen presentation molecules CTSS, PSMB9, CIITA, CD80, B2M, CD58, and NLRC5. For mutations, the final probes covered 138 560 base pairs covering 97% breadth of coverage of the 532 targets. Polymerase chain reaction (PCR) assessment of PD-L1/PD-L2 copy number is outlined in the supplemental Data.
In SureSelectXT target enrichment, ultra-long 120-mer-biotinylated complementary RNA baits were hybridized with samples to capture region of interest and enrich them out of a next-generation sequencing genomic fragment. The KAPA Hyper Prep Kit were used in conjunction with Agilent SureSelect adapters and primers to improve the library quality and capture efficiency for FFPE samples with variable quality and limited quantity. Sequencing was performed using on an Illumina platform. The library pool was diluted and denatured according to the standard NextSeq protocol and sequenced to generate paired-end 76 base pair reads using a 150-cycle NextSeq500/550 Mid Output reagent Kit v2 (Illumina). After sequencing, fastq files were generated using bcl2fastq2 software (v2.18.0). The mean depth of coverage for the targeted DNA sequencing was >×200 for all subtypes: ×499 EBV− HIV− PCNSL, ×380 EBV− HIV+ PCNSL, ×236 EBV− HIV+ PCNSL, and ×1208 EBV+ HIV− PCNSL. Mean coverage per EBV-tissue status, is 492 (EBV tissue-negative) and 666 (EBV tissue-positive). All samples had mean coverage of target genes of at least ×50.
Mutations were “called” by 2 software programs: MuTect2 and Lofreq. Only nonsynonymous mutations that were predicted to have a high impact on the protein were included. The mutations were discarded if (1) they had a variant allele frequency <5%, or (2) they were present in at least 1 the 16 surgically excised nonmalignant lymph nodes samples (taken during diagnostic workup in patients without lymphoma that were being evaluated for potential breast cancer), or (3) were present in paired germline tissue (available in 8 EBV− HIV− PCNSL and 3 EBV+ HIV− PCNSL cases), or (4) they were present in the Genome Aggregation database (exomes release 2.0.2) at a frequency >1%. As expected, across subtypes, missense mutations (78.5%) predominated. Based on DNA availability, hotspot mutations detected by capture hybridization in MYD88 (1 hotspot, 8 cases) and CD79B (5 hotspots, 15 cases) underwent confirmatory Sanger sequencing of PCR-amplified gene products. In all 23 cases, the hotspots were validated. With regard to CNV, we used 3 tools to identify CNVs from the aligned read data including GATK, CNVPanelizer, and ONCOCN. Only CNVs identified by all 3 tools were included.
RNA quantification
Genes were digitally quantified for immune genes, Lymph2Cx and EBV genes using the nCounter platform (NanoString) as published.22-24 A targeted gene panel was chosen to permit analysis of selected clinically pertinent immune effectors (CD137, CD4, CD8, tumor necrosis factor-α [TNF-α]), macrophages (CD68, CD163), and immune-checkpoints (LAG-3, PD-1, PD-L1, PD-L2, TIM-3) similar to that previously outlined.22-24 EBV gene expression (EBER-1, LMP-1, EBNA2) to distinguish viral latency patterns (latency I: EBER+/LMP1−/EBNA2−; latency II: EBER+/LMP1+/EBNA2−; latency III: EBER+/LMP1+/EBNA2+)18 and cell-of-origin (COO) categorization (by NanoString Lymph2Cx assay) was performed.25 Normalized data are provided in the supplemental Data.
Statistical analysis
Values between groups of data were tested for statistical significance using the 2-tailed Mann-Whitney tests. Categorical data were compared using Fisher exact test or χ2 test. All tests were 2-sided at the threshold of P = .05. Benjamini and Hochberg false discovery rate was used to compare combined EBV+ vs combined EBV− tissues (threshold P = .05). All analyses were prepared using the GraphPad Prism platform (v7, GraphPad).
Results
Comparison of the mutational landscapes across PCNSL subtypes
Figure 2A shows results of individual samples grouped by PCNSL subtypes. First, EBV tissue-negative PCNSL: EBV− HIV− PCNSL and EBV− HIV+ PCNSL cases were analyzed. EBV− HIV− PCNSL was enriched in previously identified mutations.3-10 Mutations at high frequency (≥20%) include MYD88, CD79B, PIM1, KMT2D, TBL1XR1, TOX, and PRDM1. These involve BCR-NF-κB signaling, epigenetic regulation, cell cycle/adhesion, and B-cell differentiation. Mutations in antigen presentation/processing and immune function were less frequent. Only 1 EBV− HIV+ PCNSL had DNA suitable for sequencing. Although conclusions should not be overinterpreted, it is notable that this case had multiple (8) mutations.
![Mutational landscape of PCNSL, according to EBV tissue and HIV serological status. (A) Each column in this plot represents an individual case (with mutation[s] in the displayed genes) of the final sequencing cohort (n = 79), across the 4 tissue subtypes: EBV− HIV− PCNSL, EBV− HIV+ PCNSL, EBV+ HIV+ PCNSL, and EBV+ HIV− PCNSL. Mutated genes constitute individual rows and are sorted according to their mutational frequencies of mutated cases as provided on the far right. Mutation types are color coded as indicated in the key; red* brain lymphoma TK cell-line, red** patients without PTLD/iatrogenic immunosuppression. (B) Stacked histograms show the percentage (percentages rounded to whole numbers) of cases with mutations in MYD88, CD79B, PIM1 by EBV-tissue and HIV serological status across the 3 main subtypes. Because EBV− HIV+ PCNSL represented only 1 sequenced case, aggregate data are not shown. (C) Number of mutated genes observed using the targeted sequencing panel in EBV− HIV− PCNSL, EBV+ HIV+ PCNSL, and EBV+ HIV− PCNSL, with P values for paired subtypes: *P ≤ .05; **P ≤ .01; ***P ≤ .001; ****P ≤ .0001.](https://ash.silverchair-cdn.com/ash/content_public/journal/blood/137/11/10.1182_blood.2020008520/1/m_bloodbld2020008520f02.png?Expires=1768783479&Signature=JYJ2O0T6Z75abVkeCipB7QDHs5yxH7NVF91uPwUEMb9LHetcrKE6B0SXAF1KrsjP6Fph20NnoEWnG5ztPFTyR9UY9tCSIdA1GNkhFT7E00U3eqwr0SZ3IcQ7TRAqcsy83x~u9UGgMuzSHeUxkuTfq9ciLJziR-qNKTuz3Hmqf0TSp4EqbnDddc9MXonHmysduKlg4kk-yvyJxuxhKM62uyVxtfH0V4dazaBglibf~6MjfAKhdXfLcvkZPJpvWJXbaL92KxJM5DVfHnQn0EMZ5mBCCOzzKrCZFafFYTEhzhSDyykmMJPCerdIwo3U~ulQ6uF73S1tR1dm7QyP8FiGqQ__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
Mutational landscape of PCNSL, according to EBV tissue and HIV serological status. (A) Each column in this plot represents an individual case (with mutation[s] in the displayed genes) of the final sequencing cohort (n = 79), across the 4 tissue subtypes: EBV− HIV− PCNSL, EBV− HIV+ PCNSL, EBV+ HIV+ PCNSL, and EBV+ HIV− PCNSL. Mutated genes constitute individual rows and are sorted according to their mutational frequencies of mutated cases as provided on the far right. Mutation types are color coded as indicated in the key; red* brain lymphoma TK cell-line, red** patients without PTLD/iatrogenic immunosuppression. (B) Stacked histograms show the percentage (percentages rounded to whole numbers) of cases with mutations in MYD88, CD79B, PIM1 by EBV-tissue and HIV serological status across the 3 main subtypes. Because EBV− HIV+ PCNSL represented only 1 sequenced case, aggregate data are not shown. (C) Number of mutated genes observed using the targeted sequencing panel in EBV− HIV− PCNSL, EBV+ HIV+ PCNSL, and EBV+ HIV− PCNSL, with P values for paired subtypes: *P ≤ .05; **P ≤ .01; ***P ≤ .001; ****P ≤ .0001.
Mutational landscape of PCNSL, according to EBV tissue and HIV serological status. (A) Each column in this plot represents an individual case (with mutation[s] in the displayed genes) of the final sequencing cohort (n = 79), across the 4 tissue subtypes: EBV− HIV− PCNSL, EBV− HIV+ PCNSL, EBV+ HIV+ PCNSL, and EBV+ HIV− PCNSL. Mutated genes constitute individual rows and are sorted according to their mutational frequencies of mutated cases as provided on the far right. Mutation types are color coded as indicated in the key; red* brain lymphoma TK cell-line, red** patients without PTLD/iatrogenic immunosuppression. (B) Stacked histograms show the percentage (percentages rounded to whole numbers) of cases with mutations in MYD88, CD79B, PIM1 by EBV-tissue and HIV serological status across the 3 main subtypes. Because EBV− HIV+ PCNSL represented only 1 sequenced case, aggregate data are not shown. (C) Number of mutated genes observed using the targeted sequencing panel in EBV− HIV− PCNSL, EBV+ HIV+ PCNSL, and EBV+ HIV− PCNSL, with P values for paired subtypes: *P ≤ .05; **P ≤ .01; ***P ≤ .001; ****P ≤ .0001.
Findings in EBV tissue-negative cases markedly contrasted with EBV+ HIV− PCNSL and EBV+ HIV+ PCNSL (EBV tissue-positive) cases. In 44% of EBV+ HIV− PCNSL, no mutation was detected. In the remaining, only 1 mutation occurred in >1 patient (KMT2C, n = 2). For EBV+ HIV+ PCNSL cases, 20% had no mutation present in the targeted panel, with KMT2D the most common mutation (25%), and CREBBP, CD36, and MYC mutations each occurring in 15%. Only 5 other mutations occurred in >1 patient (TNFAIP3, KMT2C, CDH22, NFKBIE). Only 30% of EBV+ HIV+ PCNSL had ≥3 mutations detected.
Next, we examined the frequency with which MYD88, CD79B, and PIM1 mutations cooccurred (Figure 2B). These 3 mutations are contained in the recently proposed “C5” and “MCD” DLBCL molecular classifications.26-28 In the EBV− HIV− PCNSL samples, >80% possessed at least 1 mutation in either/or MYD88, CD79B, and PIM1, with cooccurrence of these mutations in 69%. Similarly, in EBV− HIV+ PCNSL, the single case sequenced had cooccurrence in MYD88 and PIM1. Strikingly, no MYD88, CD79B, and PIM1 mutations were seen in any of the EBV tissue-positive samples. Additionally, mutations in TBL1XR1, PRMD1, and TOX were also significantly overrepresented in EBV tissue-negative PCNSL by false discovery rate testing (supplemental Data).
The total number of mutated genes detected in the targeted panel were next compared (Figure 2C). There was a higher number of mutations observed in EBV− HIV− PCNSL compared with EBV+ HIV+ PCNSL and EBV+ HIV− PCNSL cases (P = 2.0 × 10−9, and P = 3.7 × 10−15, respectively). EBV+ HIV− PCNSL had a lower number of mutations compared with EBV+ HIV+ cases (P = .012).
COO differs by EBV-tissue status in PCNSL
To better understand the contrasting molecular underpinnings between the subtypes, we compared the COO classified by the Lymph2Cx assay. EBV− HIV− PCNSL (Figure 3A) mainly typed as activated B-cell (ABC) (69%). Fifty percent were ABC and 50% germinal center B cell (GCB) in the limited number (2 samples) of EBV− HIV+ PCNSL tested. However, COO was only 25% and 11% ABC in EBV+ HIV− PCNSL and EBV+ HIV+ PCNSL, respectively. ABC was higher in EBV− HIV− PCNSL compared with EBV-tissue positive PCNSL (P = .0001).
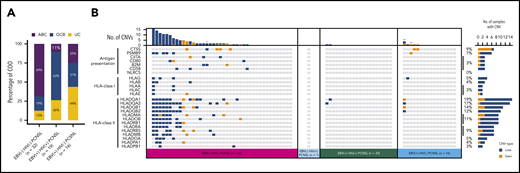
Molecular COO and copy number alterations in HLA class I/II alleles and antigen presentation/processing genes, separated into the PCNSL subtypes. (A) Stacked histograms show the relationship between the molecular COO classification (using the Lymph2CX assay) across 3 subtypes. Because EBV− HIV+ PCNSL represented only 2 COO cases, aggregate data are not shown. UC, unclassified. (B) Each column represents an individual case across the 4 PCNSL subtypes. Copy number altered genes constitute individual rows, with gains and losses shown in gold and blue, respectively. On the far right, percentages indicate the proportion of PCNSL tumors with copy number alterations of the specified gene; red* brain lymphoma TK cell-line, red** patients without PTLD/iatrogenic immunosuppression.
Molecular COO and copy number alterations in HLA class I/II alleles and antigen presentation/processing genes, separated into the PCNSL subtypes. (A) Stacked histograms show the relationship between the molecular COO classification (using the Lymph2CX assay) across 3 subtypes. Because EBV− HIV+ PCNSL represented only 2 COO cases, aggregate data are not shown. UC, unclassified. (B) Each column represents an individual case across the 4 PCNSL subtypes. Copy number altered genes constitute individual rows, with gains and losses shown in gold and blue, respectively. On the far right, percentages indicate the proportion of PCNSL tumors with copy number alterations of the specified gene; red* brain lymphoma TK cell-line, red** patients without PTLD/iatrogenic immunosuppression.
Comparison of copy number alterations in antigen presentation/processing and HLA class I/II alleles across PCNSL subtypes
Copy number (CN) alterations in HLA I/II alleles were characterized (Figure 3B). As anticipated, CN loss was frequent in HLA class I/II alleles of EBV− HIV− PCNSL (43%). However, CN loss in HLA class I/II was seen in only 10% EBV+ HIV+ PCNSL and 13% EBV+ HIV− PCNSL, respectively. The single EBV− HIV+ PCNSL had no HLA class I/II CN loss.
CN alterations in genes involved in antigen presentation/processing were also tested. A total of 31% EBV− HIV− PCNSL had CN loss for genes involved with antigen presentation/processing. Except in 8%, CN loss in these genes did not cooccur (ie, they were mutually exclusive). No CN loss in any of these genes was seen in the remaining PCNSL subtypes. CN gain of CTSS is a feature of follicular lymphoma,29,30 and was seen in 9% of cases overall. A subset of cases (27 EBV− HIV− PCNSL and 6 EBV+ HIV− PCNSL) had sufficient DNA available for PCR-based PD-L1, PD-L2 CN gain testing (supplemental Data). This showed there was no difference in CN gains between these PCNSL subtypes for PD-L1 or PD-L2 (supplemental Data; P = .87 and P = .08, respectively).
EBV tissue-positive PCNSL have distinct genetic pathway aberrations compared with EBV− HIV− PCNSL
To identify patterns within the 3 principal PCNSL subtypes (EBV− HIV+ PCNSL was excluded from the aggregate analysis because n = 1), we compared mutations and/or CN loss categorized by pathways (Figure 4). First, EBV tissue-positive PCNSL subtypes were evaluated. There were no differences in genetic pathway aberrations between EBV+ HIV− PCNSL and EBV+ HIV+ PCNSL for each of HLA class I/II CN loss, antigen presentation/processing (mutations combined with CN loss), or mutations in immune function, BCR-NF-κB, epigenetic regulation, cell cycle/adhesion, and B-cell differentiation. Next, EBV tissue-positive PCNSL was compared with EBV− HIV− PCNSL. There was a higher frequency of genetic pathway aberrations for all pathways (except immune function) in EBV− HIV− PCNSL relative to EBV tissue-positive PCNSL cases.
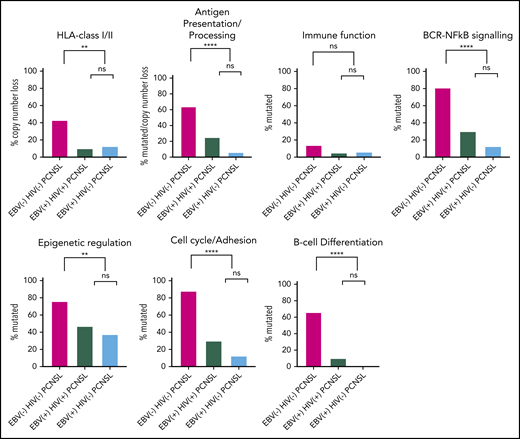
Combined mutations and copy number loss separated into the 2 EBV-tissue positive PCNSL subtypes and EBV−HIV−PCNSL. Mutations and/or CN loss were categorized into pathway categories. CN loss is shown for HLA class I/II. For antigen presentation/processing mutations were combined with CN loss. Mutations for immune function, BCR-NF-κB signaling, epigenetic regulation, cell cycle/adhesion and B-cell differentiation. Percentages are shown for each PCNSL subtype, with P values for paired subtypes: *P ≤ .05; **P ≤ .01; ***P ≤ .001; ****P ≤ .0001. There were 42, 20, and 16 cases of EBV− HIV−, EBV+ HIV+, and EBV+ HIV− PCNSL, respectively (data for the single EBV− HIV− PCNSL tissue is not shown).
Combined mutations and copy number loss separated into the 2 EBV-tissue positive PCNSL subtypes and EBV−HIV−PCNSL. Mutations and/or CN loss were categorized into pathway categories. CN loss is shown for HLA class I/II. For antigen presentation/processing mutations were combined with CN loss. Mutations for immune function, BCR-NF-κB signaling, epigenetic regulation, cell cycle/adhesion and B-cell differentiation. Percentages are shown for each PCNSL subtype, with P values for paired subtypes: *P ≤ .05; **P ≤ .01; ***P ≤ .001; ****P ≤ .0001. There were 42, 20, and 16 cases of EBV− HIV−, EBV+ HIV+, and EBV+ HIV− PCNSL, respectively (data for the single EBV− HIV− PCNSL tissue is not shown).
Differential gene expression of the TME across PCNSL subtypes
To delineate differences in TME between the 3 principal PCNSL subtypes (as before, EBV− HIV+ PCNSL was excluded from the aggregate analysis), we performed digital gene expression using a customized panel of immune genes (Figure 5). There was elevated TNF-α, CD68, CD163, PD-L1, PD-L2, LAG-3, and TIM-3 in EBV+ HIV− PCNSL compared with all other PCNSL subtypes, broadly indicative of a tolerogenic TME. EBV+ HIV+ PCNSL cases had low levels of CD4 and macrophage markers CD68 and CD163 compared with non-AIDS-related PCNSL.
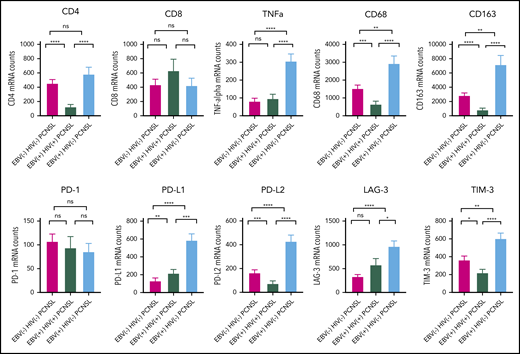
The tumor microenvironment in the 2 EBV tissue-positive PCNSL subtypes and EBV−HIV−PCNSL. A targeted gene expression panel was chosen of selected clinically pertinent immune effectors, macrophages, and immune-checkpoint markers. Gene counts are shown for each PCNSL subtype, with P values for paired subtypes: *P ≤ .05; **P ≤ .01; ***P ≤ .001; ****P ≤ .0001. There were 34, 18, and 18 cases of EBV− HIV−, EBV+ HIV+, and EBV+ HIV− PCNSL, respectively.
The tumor microenvironment in the 2 EBV tissue-positive PCNSL subtypes and EBV−HIV−PCNSL. A targeted gene expression panel was chosen of selected clinically pertinent immune effectors, macrophages, and immune-checkpoint markers. Gene counts are shown for each PCNSL subtype, with P values for paired subtypes: *P ≤ .05; **P ≤ .01; ***P ≤ .001; ****P ≤ .0001. There were 34, 18, and 18 cases of EBV− HIV−, EBV+ HIV+, and EBV+ HIV− PCNSL, respectively.
EBV-latency profiling/categorization was performed in 36 EBV tissue-positive PCNSL with sufficient RNA as previously outlined.18 The frequencies in 18 EBV+ HIV− PCNSL and 18 EBV+ HIV+ PCNSL of latency III/II/I were 56%/33%/11% and 78%/22%/0%, respectively.
Discussion
Using a combination of targeted sequencing and digital multiplex gene expression applied to an international series of an extremely rare and poorly categorized lymphoma subtype, we demonstrate that the genetic landscape typically observed in EBV− HIV− PCNSL is distinct from EBV-associated (tissue-positive) PCNSL. Notably, mutations in the BCR-dependent NF-κB pathway, the cell cycle/adhesion pathway, CN loss in HLA class I/II and mutations/CN loss in the antigen presentation/processing pathways all rarely occur in EBV-tissue positive cases. Immune evasion in patients with EBV+ HIV+ PCNSL (AIDS-related PCNSL) was mediated by reduced CD4, whereas in EBV+ HIV− PCNSL, the TME had increased expression of tolerogenic immune genes.
Among genetic aberrations known to be frequently observed in EBV− HIV− PCNSL are mutations in MYD88 and CD79B that activate NF-κB activation, which is a core pathway involved in the pathogenesis of PCNSL.4-8,26 Another central mechanism is mutation in the cell cycle/adhesion gene PIM1.9,10 Our observation that these genes frequently cooccurred and were enriched in the ABC COO,31 is in keeping with the recently proposed “C5” and “MCD” DLBCL molecular classifications.26-28 By marked contrast, in EBV tissue-positive cases, there were no mutations in PIM1, MYD88, and CD79B. Here, COO was typically either GCB or unclassified. This is similar to recent studies of systemic HIV+ and EBV+ DLBCL that used the Lymph2Cx classifier.32,33 Notably EBV induces an atypical germinal center reaction,34 and recent characterization of EBV-driven B-cell differentiation shows that segregation into GCB and ABC subtypes by a limited panel of immunohistochemical markers is overly simplistic.35 The combined mutational and COO data emphasize that the nature of the pathogenesis of PCNSL is tightly linked to presence or absence of EBV-tissue expression. This is consistent with Hodgkin lymphoma (HL), where EBV+ HL appears to have a lower mutation load than EBV− HL.36 EBV is known to play a key role in the pathogenesis of several types of B-cell lymphomas, and to transform and immortalize B cells.37-39 In our series, EBV tissue-positive cases typically (≥89%) exhibited latency II or III gene profiles associated with expression of EBV viral genes that have an established capacity for inducting B-cell proliferation and prevention of senescence.40,41 We found only ∼30% EBV tissue-positive cases had a detectable mutation in the targeted exome panel. However, given that in plasmablastic lymphomas some genes appear to be more frequently mutated in EBV+ than in EBV− cases,42 a comprehensive genomic analysis (eg, whole exome) is required to establish if other genetic aberrations are present, and to determine if these occur before and/or following EBV B-cell infection and malignant transformation.
EBV establishes a persistent infection and in the vast majority of individuals is asymptomatic. Lack of pathogenicity is in part maintained by a robust EBV-specific T-cell immune response.43 Both PTLD and AIDS are associated with a spectrum of B-cell lymphomas that are typically EBV associated.12,44-46 The emergence of EBV+ HIV+ PCNSL correlates with HIV-induced loss of EBV-specific CD4+ T cells rather than overall CD4+ T-cell loss.47 HIV was initially believed not to have oncogenic potential but rather increased cancer risk by allowing EBV to induce lymphomagenesis in coinfected subjects. More recently, this notion has been challenged.48 Interestingly, in our cohort, mutations were modestly but significantly more frequent in EBV+ HIV+ PCNSL than EBV+ HIV− PCNSL. This is in keeping with various lines of evidence that suggest that HIV itself contributes to the accumulation of DNA mutations.49-52
Immune evasion is a hallmark of cancer.53 Consistent with our observations in EBV− HIV− PCNSL, deletion involving the HLA locus has previously been observed.20 Losing the ability to present neoantigens through HLA loss facilitates immune evasion. By contrast, EBV tissue-positive cases typically had intact HLA and absent mutations/CN loss in antigen presentation/processing genes. To counter the presentation of an immunogenic virus, there were higher levels of genes for macrophage markers CD68/CD163 and immune checkpoints in EBV+ HIV− PCNSL compared with other subtypes, perhaps as an adaptive response to counter the elevated levels of the antiviral cytokine TNF-α. Given that EBV+ HIV− PCNSL mostly occurred in the setting of iatrogenic immunosuppression because of PTLD, the presence of a tolerogenic TME in these cases may appear counterintuitive. However, it is in line with previous observations comparing systemic DLBCL-PTLD with systemic DLBCL in the nonimmunocompromised, which have shown that immune gene expression clusters on the basis of EBV status rather than immune status. Specifically, EBV+ cases show both a tolerogenic TME and an increased T-cell signaling signature relative to EBV− DLBCL tissues irrespective of iatrogenic immunosuppression.54 Similarly, studies restricted to systemic DLBCL in the nonimmunocompromised confirm that EBV+ DLBCL has a tolerogenic TME, and are associated with higher levels of antigen-presenting molecules.32,55 Put together, it seems likely that the alterations in the TME are modulated by the virus to promote tumor escape. Interestingly, the proportion of PD-L1, PD-L2 gene amplification was similar between EBV− HIV− PCNSL and EBV+ HIV− PCNSL, suggesting that the increase in expression of these ligands in EBV+ HIV− PCNSL may be due to macrophage expression, as previously observed.56 CD4+ T cells and CNS-associated macrophages are key targets of HIV-infection,57 and as expected EBV+ HIV+ PCNSL (AIDS-related PCNSL) had low levels of CD4 and macrophage markers compared with non-AIDS-related PCNSL.
The distinct immunobiology of EBV tissue-positive PCNSL has therapeutic implications (Figure 6). The BTK-inhibitor ibrutinib is known to cross the blood–brain barrier (BBB) and has impressive phase 1/2 efficacy in EBV− HIV− PCNSL,58-61 and may have beneficial effects on components of the TME in the non-PCNSL setting.62,63 Mutations such as CARD11 and PIM1 that are associated with complete or partial ibrutinib resistance in PCNSL and/or systemic DLBCL59,64 were rarely observed in EBV tissue-positive cases. However, the relevance of these mutations was established in the context of an intact B-cell receptor signaling complex, and it is unclear what the impact of absent CARD11 and PIM1 mutations makes on EBV-transformed B-cell sensitivity. Also, other BTK inhibitors are possibly better tolerated.65 Although upregulation of immune checkpoints is observed, checkpoint blockade is potentially contraindicated in PTLD because of the risk of graft rejection and graft-versus-host-disease. By contrast, checkpoint-blockade would not be contraindicated in EBV− HIV− PCNSL and AIDS-related PCNSL.10,56,66 The retained ability of malignant B cells to present antigens in EBV-associated PCNSL is notable. Restoration of EBV-specific T-cell immunity, has been shown to induce clinical response in EBV+ lymphomas.67-69 Adoptive transfer of EBV-specific third-party virus specific T cells (VST) chosen on the basis of the best HLA match and in vitro effector function has previously been shown to induce high response rates in EBV+ PTLD and can cross the BBB.70,71
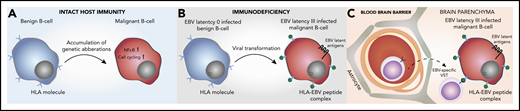
Pathogenesis of EBV tissue-positive PCNSL in the immunosuppressed, and potential implications for immunotherapy. (A) Mutations in the NF-κB and cell-cycling pathways and deletion of HLA class I/II molecules in EBV− HIV− PCNSL. (B) Viral transformation in EBV tissue-positive PCNSL, with the loss of host immunity providing no selection pressure to prevent presentation of EBV-immunogenic peptides by HLA I/II molecules. (C) EBV-specific viral specific T cells (VST) cross the blood–brain barrier to target the EBV+ malignant B cell.
Pathogenesis of EBV tissue-positive PCNSL in the immunosuppressed, and potential implications for immunotherapy. (A) Mutations in the NF-κB and cell-cycling pathways and deletion of HLA class I/II molecules in EBV− HIV− PCNSL. (B) Viral transformation in EBV tissue-positive PCNSL, with the loss of host immunity providing no selection pressure to prevent presentation of EBV-immunogenic peptides by HLA I/II molecules. (C) EBV-specific viral specific T cells (VST) cross the blood–brain barrier to target the EBV+ malignant B cell.
In summary, EBV-associated PCNSL in the immunosuppressed is immunobiologically distinct from EBV− HIV− PCNSL, and, despite expressing an immunogenic virus, retains the ability to present EBV. Results support combination strategies that cross the BBB, block EBV-driven oncogenesis and target EBV antigens. Based on the cumulative biological data, a phase 1 Australasian Leukaemia/Lymphoma Group clinical trial incorporating EBV-specific third-party VST (ACTRN12618001541291) has commenced.
Presented in part in oral form at the 15th International Conference on Malignant Lymphoma, Lugano, Switzerland, June 12, 2019.
Study data is included in the supplemental file.
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Acknowledgments
This work was supported by the Leukaemia Foundation and the Mater Foundation (M.K.G.), the University of Queensland Mai Lan Kunzy Scholarship (T.H.), a National Health and Medical Research Council Early Career Fellowship, and a Princess Alexandra Hospital Award and a Cancer Australia and Cancer Cure Grant (ID1161139) (C.K.). The Translational Research Institute is supported by the Australian government. Specimens were provided by AIDS and Cancer Specimen Resource, funded by the National Institutes of Health, National Cancer Institute (UM1CA181255). This publication was also made possible from National Institutes of Health funding through the National Institute of Mental Health and National Institute of Neurological Disorders and Stroke by the following grants: Manhattan HIV Brain Bank (U24MH100931), Texas NeuroAIDS Research Center (U24MH100930), National Neurological AIDS Bank (U24MH100929), California NeuroAIDS Tissue Network (U24MH100928), and Data Coordinating Center (U24MH100925). The contents are solely the responsibility of the authors and do not necessarily represent the official view of the NeuroAIDS National Tissue Collection or National Institutes of Health.
Authorship
Contribution: M.K.G., T. H., and C. K. conceived and designed the study; M.K.G. provided financial support; M.K.G. provided administrative support; M.K.G., S.D., R.U.T., G.B., G.H., E.B., L.C., J.W., E.H., and C.K. provided study materials or patients; M.K.G., T.H., S.B., K.O., J.W.D.T., L.F., H.O., K.B., L.M.d.L., M.B.S., E.B., L.C., and C.K. collected and assembled data; M.K.G., T.H., S.C.L., S.B., K.O., J.W.T., F.V., V.M., J.G., C.G., E.B., L.C., L.M.R., A.M., K.B., B.C., and C.K. provided data analysis and interpretation; and all authors undertook manuscript writing, final approval of manuscript, and are accountable for all aspects of the work.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: M. K. Gandhi, Level 7, Translational Research Institute, Brisbane, QLD 4102, Australia; e-mail: maher.gandhi@mater.uq.edu.au; and C. Keane, Level 7, Translational Research Institute, Brisbane, QLD 4102, Australia; e-mail: colm.keane@mater.uq.edu.au.
