

Key Points
Bone marrow FGF-23 from erythroblasts promotes G-CSF–induced progenitor mobilization.
FGF-23 interferes with the chemoattraction of progenitors toward CXCL-12.

Abstract
Fibroblast growth factor 23 (FGF-23) hormone is produced by bone-embedded osteocytes and regulates phosphate homeostasis in kidneys. We found that administration of granulocyte colony-stimulating factor (G-CSF) to mice induced a rapid, substantial increase in FGF-23 messenger RNA in bone marrow (BM) cells. This increase originated mainly from CD45−Ter119+CD71+ erythroblasts. FGF-23 protein in BM extracellular fluid was markedly increased during G-CSF–induced hematopoietic progenitor cell (HPC) mobilization, but remained stable in the blood, with no change in the phosphate level. Consistent with the BM hypoxia induced by G-CSF, low oxygen concentration induced FGF-23 release from human erythroblast HUDEP-2 cells in vitro. The efficient mobilization induced by G-CSF decreased drastically in both FGF-23−/− and chimeric mice with FGF-23 deficiency, only in hematopoietic cells, but increased in osteocyte-specific FGF-23−/− mice. This finding suggests that erythroblast-derived, but not bone-derived, FGF-23 is needed to release HPCs from BM into the circulation. Mechanistically, FGF-23 did not influence CXCL-12 binding to CXCR-4 on progenitors but interfered with their transwell migration toward CXCL-12, which was canceled by FGF receptor inhibitors. These results suggest that BM erythroblasts facilitate G-CSF–induced HPC mobilization via FGF-23 production as an intrinsic suppressor of chemoattraction.
Introduction
Granulocyte colony-stimulating factor (G-CSF) is clinically used for hematopoietic stem/progenitor cell (HSC/HPC) mobilization from the bone marrow (BM) into the circulation. G-CSF stimulation and subsequent marrow hypersympathetic tone suppress the hematopoietic microenvironment, such as osteolineage cells,1-5 mesenchymal stem cells,6 and macrophages,7-9 resulting in the release of HSCs/HPCs from the BM. However, the mechanism for mobilization is still unclear. The contribution of the CXCR-4/CXCL-12 axis in this process is not fully understood. Although the CXCL-12 protein level in BM extracellular fluid is drastically decreased, probably because of osteoblast suppression and increased protease activity,2,10-12 total CXCL-12 expression in CXCL-12-abundant reticular (CAR) cells is only minimally affected by G-CSF.13 A CXCR-4 antagonist, plerixafor, is also clinically applied to facilitate G-CSF mobilization, particularly in patients with poor mobilization efficiency (eg, multiple myeloma).14 There may be an unknown mechanism to counteract the function of CXCR-4, rather than CXCL-12 downregulation in BM, during G-CSF mobilization.
During our study on the role of a calcium-regulating hormone (vitamin D) in mobilization,3 we noted a strong induction, not only in bone tissue, but also in BM cells, of another major mineral (phosphate)-regulating hormone, fibroblast growth factor 23 (FGF-23). FGF-23 is known to be mainly produced by bone-embedded osteocytes and, together with an FGF receptor (FGFR) coreceptor α-klotho, regulates phosphate homeostasis in the kidneys.15 However, the strong induction of FGF-23 in the local tissue and its unique function is unknown.
In this study, we showed that FGF-23 is dramatically increased in BM, mainly by erythroblasts during G-CSF treatment, and promotes HPC mobilization by inhibiting CXCR-4–mediated chemoattraction.
Methods
Mice for the experiments
Mice were under the husbandry care of the Institute for Experimental Animals, Kobe University Graduate School of Medicine. FGF-23−/− and FGF-23flox/flox mice were generated on a C57BL/6 background, as described in the supplemental Methods (available on the Blood Web site). DMP-1-cre mice were generated, as described previously,16 backcrossed onto a C57BL/6 background for >10 generations, and crossed with FGF-23flox/flox mice to generate mice with conditional FGF-23 deletion in osteocytes. C57BL/6 mice were purchased from CLEA Japan (Chiba, Japan) 1 to 2 weeks before the experiments. C57BL/6-CD45.1 congenic mice were purchased from The Jackson Laboratory (Bar Harbor, ME). Animals were maintained under specific pathogen–free conditions in a 12-hour light/12-hour dark cycle. All mice were used at 7 to 8 weeks of age, except for experiments with FGF-23+/+ and FGF-23−/− mice, for which 5-week-old littermates were used. Male mice were used in all experiments, except for the characterization of FGF-23−/− mice (supplemental Figures 5-11) and transplant donors for BM chimera (see Figure 5; supplemental Figures 12-13 and 15), for which female FGF-23+/+ and FGF-23−/− littermates were also used in some experiments. All animal studies were approved by the Animal Care and Use Committee of Kobe University.
G-CSF mobilization and isoproterenol treatment
HPC mobilization by G-CSF was induced, as described previously.3 In brief, mice were injected with recombinant human G-CSF (filgrastim; kindly provided by Kyowa Kirin, Tokyo, Japan; 125 μg/kg per dose every 12 hours in 8 divided doses, administered subcutaneously) in phosphate-buffered saline (PBS) supplemented with 0.1% bovine serum albumin (PBS/BSA). Blood and BM were harvested 3 hours after the last dose of G-CSF, unless otherwise indicated.
Treatment with single-dose isoproterenol (dl-isoproterenol hydrochloride, 100 mg/kg, administered intraperitoneally; Sigma-Aldrich, St Louis, MO) was performed as described previously.3 BM was harvested at time points established in the protocol.
Competitive repopulation assay
The stem cell activities of blood mobilized by G-CSF were assessed by long-term competitive reconstitution, as described previously,3 with minor modifications. In brief, 150 μL mobilized blood (CD45.2), together with 2 × 105 BM nucleated cells from a CD45.1 competitor donor, was injected into lethally irradiated (14 Gy, split dose) CD45.1-recipient mice. The proportion of peripheral blood leukocytes bearing CD45.1 or CD45.2 antigen was determined monthly after transplantation by flow cytometry. Repopulating units (RUs) were calculated by using the following standard formula: RUs = %(C)/(100 − %), where % is the measured percentage of donor cells (CD45.2+ cells derived from mobilized blood), and C is the number of competitor marrow cells per 105, which was 2 in this study.
ELISA for FGF-23 and CXCL-12
For the enzyme-linked immunosorbent assays (ELISAs), BM extracellular fluid was obtained by flushing femoral BM with 500 μL PBS, followed by centrifugation after several pipetting steps. FGF-23 quantification in serum, BM extracellular fluid, culture supernatants, and cell lysates was performed with an FGF-23 ELISA kit for mouse/human intact FGF-23 (iFGF-23; Kainos Laboratories, Tokyo, Japan) and an FGF-23 ELISA kit for mouse/rat C-terminal FGF-23 (cFGF-23; Immutopics, San Clemente, CA) according to the manufacturers’ recommendations. CXCL-12 in BM extracellular fluid was evaluated with a CXCL-12 ELISA kit (R&D Systems, Minneapolis, MN).
Flow cytometry and cell sorting
The reagents for flow cytometry, including the antibodies PE anti-mouse CD45, biotin anti-mouse lineage panel (CD11b, Gr-1, Ter119, CD3e, and B220), FITC anti-mouse Sca-1, PE anti-mouse Sca-1, FITC anti-mouse c-kit, PE anti-mouse c-kit, APC anti-mouse c-kit, APC anti-mouse CXCR-4, PerCP/Cy5.5 anti-mouse CD8a, and FITC anti-mouse CD48 were from BD PharMingen (San Diego, CA). FITC anti-mouse CD71, PerCP streptavidin, PerCP/Cy5.5 anti-mouse Ter119, PerCP/Cy5.5 anti-mouse CD45R/B220, PerCP/Cy5.5 anti-mouse Ly-6G/Ly-6C (Gr-1), PerCP/Cy5.5 anti-mouse CD11b, PerCP/Cy5.5 anti-mouse CD4, BV421 anti-mouse Sca-1, PE anti-mouse CD127, APC/Cy7 anti-mouse CD16/32, PE/Cy7 anti-mouse Sca-1, PE anti-mouse CD135, BV421 anti-mouse CD150, PE anti-mouse CD45R/B220, and PE anti-mouse CD3e were from BioLegend (San Diego, CA). FITC anti-mouse CD34, PE/Cy5 streptavidin, and APC/eFluor780 streptavidin were from eBioscience (San Diego, CA). FITC anti-mouse IgM was from SouthernBiotech (Birmingham, AL). Cells were suspended in 0.5% BSA/2 mM EDTA/PBS. Cell analyses were performed on a FACScan, Accuri C6 (BD Biosciences, San Jose, CA) or a CytoFLEX S flow cytometer (Beckman Coulter, Brea, CA). Data analysis was performed with FlowJo (Ashland, OR).
BM mononuclear cells (MNCs) were obtained by density gradient centrifugation of Lympholyte-M (Cedarlane, Burlington, ON, Canada) and CD45+ cells, CD45−Ter119+CD71+ erythroblasts, and CD45−Ter119− nonhematopoietic (stromal) cells were sorted using the MoFlo XDP flow cytometer with Summit software (Beckman Coulter, Brea, CA).
HUDEP-2 culture
The human umbilical cord blood–derived erythroid progenitor cell line HUDEP-2 was generated as described previously.17 HUDEP-2 cells were cultured in serum-free medium for expansion of hematopoietic cells (STEMCELL Technologies, Vancouver, BC, Canada) supplemented with 50 ng/mL human stem cell factor (R&D Systems), 3 IU/mL human erythropoietin (epoetin-α [epo-α]; kindly provided by Kyowa Kirin), 1 μg/mL doxycycline (Sigma-Aldrich), and 10−6 M dexamethasone (Sigma-Aldrich) in a 24-well nonculture treatment plate (Falcon; Corning, Corning, NY) at 1 × 107 cells per 500 μL per well. After incubation in 21% and 5% O2 conditions with 5% CO2 at 37°C for 4 hours, the supernatants were used for FGF-23 ELISA.
Cell lysates
Sorted erythroblasts were lysed in a buffer containing 50 mM Tris-HCl (pH 7.5), 150 mM NaCl, 1% Triton X-100 (Wako, Osaka, Japan), 0.2 M sodium orthovanadate (Sigma-Aldrich), 0.2 M phenylmethylsulfonyl fluoride (Sigma-Aldrich), 1 M sodium fluoride (Nacalai Tesque, Kyoto, Japan), and protease inhibitor cocktail (Sigma-Aldrich). After centrifugation, the supernatants were directly used for FGF-23 ELISA.
Transwell migration
BM MNCs in Iscove’s modified Dulbecco’s medium (IMDM) supplemented with 2% fetal bovine serum (FBS; 1 × 106 cells per 100 μL) were added to the upper chamber of a transwell insert (pore size 5 μm; Corning). They migrated into the lower chamber, which contained 600 μL IMDM/2% FBS with 300 ng/mL mouse CXCL-12 (R&D Systems) in a 24-well nonculture treatment plate (Falcon), at 37°C, 5% CO2. Various combinations of 3 μg/mL mouse FGF-23 (R&D Systems), 3 μg/mL mouse α-klotho (R&D Systems), and 10 μg/mL heparin (STEMCELL Technologies) were added to the upper or lower chamber. After 4 hours, the migration efficiency of the HPCs was evaluated by an assay of colony-forming unit cells (CFU-Cs) in a culture of input cells without loading into the transwell chamber and cells in the lower chamber.
To assess the effect of FGFR antagonists, the cells were incubated with 10 nM BGJ-398 (infigratinib; ChemScene, Monmouth Junction, NJ) or 10 nM JNJ-42756493 (erdafitinib; Cayman Chemical, Ann Arbor, MI) starting at 30 minutes before the transwell migration assay.
Binding assay of fluorochrome-conjugated CXCL-12
BM MNCs were incubated in IMDM/2% FBS for 2 hours, with or without FGF-23comb (3 μg/mL mouse FGF-23 [R&D Systems]+3 μg/mL mouse α-klotho (R&D Systems)+10 μg/mL heparin [STEMCELL Technologies]), and 20 μM Alexa647-conjugated mouse CXCL-12 (Almac, Nr Penicuik, United Kingdom) was added for the last 30 minutes of the period. Cells were stained with PE anti-Sca-1, FITC anti-c-kit, and biotin lineage panel, followed by PerCP streptavidin. CXCL-12 binding to lineage–Sca-1+c-kit+ cells was analyzed by flow cytometry. As a control for this experiment, cells without FGF-23comb were incubated with mouse CXCL-12 (nonfluorescent, 300 ng/mL; R&D Systems), starting 30 minutes before the addition of Alexa647-conjugated mouse CXCL-12.
Actin polymerization
Actin polymerization in HPCs was assessed as described previously,18 with minor modifications. In brief, BM MNCs (2 × 106 cells per 100 μL PBS) were stimulated with 500 ng/mL mouse CXCL-12 (R&D Systems) for the indicated seconds, fixed, and permeabilized with BD Cytofix/Cytoperm Fixation/Permeabilization Kit (BD Biosciences), according to the manufacturer’s recommendations. The cells were stained with 3 U/mL Alexa488-conjugated phalloidin (Invitrogen, Carlsbad, CA) for 1 hour and further stained with APC anti-c-kit and a biotin lineage panel, followed by PerCP streptavidin. Actin polymerization in lineage c-kit+ cells was analyzed by flow cytometry.
Statistical analysis
All data were pooled from at least 3 independent experiments, except those shown in supplemental Figure 16E, where data were from 2 experiments. All center values shown in the graphs refer to the mean. All values were reported as the mean ± standard error of the mean (SEM). Statistical analysis was conducted with the 2-tailed, unpaired Student t test, Mann-Whitney U test, and 1-way analysis of variance (ANOVA) with Tukey’s post hoc test. No samples or animals were excluded from the analysis. The animals were randomly assigned to groups. Statistical significance was assessed with Prism (GraphPad Software, San Diego, CA) and defined as P < .05.
Complete descriptions of other procedures (generation of FGF-23−/− and FGF-23flox/flox mice, generation of chimeric mice, blood biochemistries, RNA extraction and quantitative reverse transcription-polymerase chain reaction, bone histomorphometry, and pimonidazole staining) are provided in the supplemental Methods.
Results
FGF-23 is induced in BM cells during G-CSF–induced mobilization
FGF-23 messenger RNA (mRNA) in bone tissue was increased during G-CSF mobilization (every 12 hours for 8 doses; Figure 1A) and after a short time (1 hour) by single-dose G-CSF (Figure 1B). Although FGF-23 is known as a bone-derived hormone, BM cells similarly showed a sharp increase in FGF-23 mRNA (Figure 1C). Because G-CSF is a strong inducer of the sympathetic nervous system (SNS) signals,1-5 FGF-23 mRNA induction in BM cells was examined by using a single-dose pan-β-adrenergic receptor agonist isoproterenol, displaying similar or even stronger induction of FGF-23 mRNA (Figure 1D). A rapid (2 hours) increase in intact FGF-23 (iFGF-23) protein was observed in BM extracellular fluid (BM flushed from 1 femur with 500 μL PBS) and was maintained for 12 hours by single-dose G-CSF (Figure 2A). Alteration of the iFGF-23 protein level in BM extracellular fluid was assessed during G-CSF mobilization. Without G-CSF, most of the mice showed lower than detectable levels by ELISA. The level increased with the number of G-CSF doses, which peaked at 6 (Figure 2B), but remained stable in the blood with unchanged phosphate levels during G-CSF mobilization (Figure 2C-D). A rough estimation of the actual iFGF-23 concentration in the BM extracellular cavity during G-CSF mobilization (supplemental Figure 1) was extremely high (∼1-3 μg/mL; ∼20 000 times higher than levels in the blood). Thus, FGF-23 is a G-CSF–inducible local factor in BM.
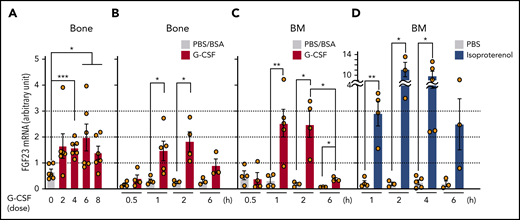
Expression of FGF-23 mRNA in bone and BM. (A-B) FGF-23 mRNA in bone tissue during G-CSF mobilization (A) (every 12 hours for 8 doses, harvested at 3 hours after each dose, n = 6) and after single-dose G-CSF (B) at the indicated hours (n = 3-5). (C-D) FGF-23 mRNA in BM cells after single-dose G-CSF (C) at the indicated hours (n = 3-5) and after single-dose isoproterenol (D) at the indicated hours (n = 3-5). Combined data of at least 3 independent experiments are shown. Data are means ± SEM. *P < .05; **P < .01; ***P < .001 (ANOVA).
Expression of FGF-23 mRNA in bone and BM. (A-B) FGF-23 mRNA in bone tissue during G-CSF mobilization (A) (every 12 hours for 8 doses, harvested at 3 hours after each dose, n = 6) and after single-dose G-CSF (B) at the indicated hours (n = 3-5). (C-D) FGF-23 mRNA in BM cells after single-dose G-CSF (C) at the indicated hours (n = 3-5) and after single-dose isoproterenol (D) at the indicated hours (n = 3-5). Combined data of at least 3 independent experiments are shown. Data are means ± SEM. *P < .05; **P < .01; ***P < .001 (ANOVA).
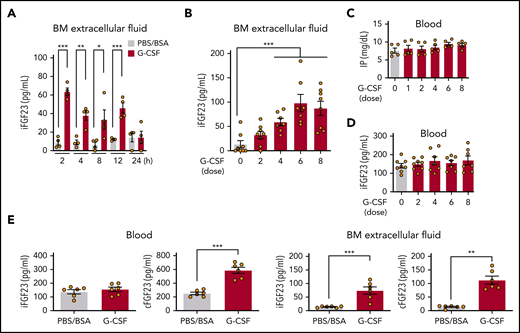
Expression of FGF-23 protein in BM. iFGF-23 protein in BM extracellular fluid after single-dose G-CSF (A) at the indicated hours (n = 4) and during G-CSF mobilization (B) (n = 7-8). Inorganic phosphate (C) (IP; n = 5) and iFGF-23 protein (D) (n = 7-8) in the blood in G-CSF mobilization. (E) iFGF-23 and cFGF-23 protein levels in the blood and BM extracellular fluid in mice treated with 6 doses of G-CSF (n = 6). Combined data of at least 3 independent experiments are shown. Data are means ± SEM. *P < .05; **P < .01; ***P < .001 (ANOVA and Student t test).
Expression of FGF-23 protein in BM. iFGF-23 protein in BM extracellular fluid after single-dose G-CSF (A) at the indicated hours (n = 4) and during G-CSF mobilization (B) (n = 7-8). Inorganic phosphate (C) (IP; n = 5) and iFGF-23 protein (D) (n = 7-8) in the blood in G-CSF mobilization. (E) iFGF-23 and cFGF-23 protein levels in the blood and BM extracellular fluid in mice treated with 6 doses of G-CSF (n = 6). Combined data of at least 3 independent experiments are shown. Data are means ± SEM. *P < .05; **P < .01; ***P < .001 (ANOVA and Student t test).
To assess the processing of the FGF-23 protein, iFGF-23 and C-terminal FGF-23 (cFGF-23 [iFGF-23+processed C-terminal FGF-23 fragment]) were measured in serum and BM extracellular fluid at 6 doses of PBS/BSA or G-CSF. As shown in Figure 2E, G-CSF treatment did not alter the iFGF-23 level, whereas cFGF-23 was clearly increased in the circulation. In BM extracellular fluid, most of the increased FGF-23 protein was iFGF-23, but the C-terminal FGF-23 fragment was most likely also increased (Figure 2E). These results suggest that a portion of the increased FGF-23 in BM was cleaved and that only the processed C-terminal FGF-23 fragment goes through the marrow-blood barrier during G-CSF treatment.
Erythroblasts produce FGF-23
FGF-23 mRNA was induced in the sorted CD45−, but not CD45+, cell population using single-dose G-CSF (Figure 3A) and isoproterenol (Figure 3B). Most of the CD45− BM cells were Ter119+ erythroid lineage cells (the ratio of CD45−Ter119+CD71+ erythroblasts to CD45−Ter119− nonhematopoietic [stromal] cells was ∼7:1 [supplemental Figure 2]). These cells were sorted by using single-dose G-CSF or isoproterenol. FGF-23 mRNA was strongly induced in both erythroblasts and stromal cells (Figure 3C). CD45−Ter119+CD71+ erythroblasts were also sorted using single-dose G-CSF; cell lysates contained an increased iFGF-23 protein level similar to that obtained with single-dose isoproterenol (Figure 3D). Because these data were almost equivalent to cFGF-23 levels (Figure 3E), erythroblasts most likely produced iFGF-23 without a major contribution to its cleavage during G-CSF treatment.
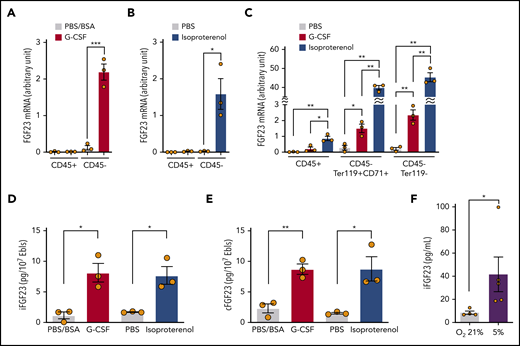
Expression of FGF-23 mRNA and protein in erythroblasts. (A-B) FGF-23 mRNA in sorted CD45+ or CD45− BM cells at 3 hours after single-dose G-CSF (A) or isoproterenol (B) (n = 3). (C) FGF-23 mRNA in sorted CD45+ BM cells or CD45−Ter119+CD71+ erythroblasts at 3 hours after single-dose G-CSF or isoproterenol (n = 3). (D-E) iFGF-23 (D) and cFGF-23 (E) protein levels in cell lysates of sorted CD45−Ter119+CD71+ erythroblasts at 2 hours after single-dose G-CSF or 1 hour after single-dose isoproterenol (n = 3). (F) iFGF-23 protein from the human erythroblast cell line HUDEP-2 after 4 hours of culture under the indicated oxygen conditions in the culture supernatants (n = 4-5). Combined data of at least 3 independent experiments are shown. Data are means ± SEM. *P < .05; **P < .01; ***P < .001 (ANOVA, Student t test, and Mann-Whitney U test). Ebls, erythroblasts.
Expression of FGF-23 mRNA and protein in erythroblasts. (A-B) FGF-23 mRNA in sorted CD45+ or CD45− BM cells at 3 hours after single-dose G-CSF (A) or isoproterenol (B) (n = 3). (C) FGF-23 mRNA in sorted CD45+ BM cells or CD45−Ter119+CD71+ erythroblasts at 3 hours after single-dose G-CSF or isoproterenol (n = 3). (D-E) iFGF-23 (D) and cFGF-23 (E) protein levels in cell lysates of sorted CD45−Ter119+CD71+ erythroblasts at 2 hours after single-dose G-CSF or 1 hour after single-dose isoproterenol (n = 3). (F) iFGF-23 protein from the human erythroblast cell line HUDEP-2 after 4 hours of culture under the indicated oxygen conditions in the culture supernatants (n = 4-5). Combined data of at least 3 independent experiments are shown. Data are means ± SEM. *P < .05; **P < .01; ***P < .001 (ANOVA, Student t test, and Mann-Whitney U test). Ebls, erythroblasts.
To identify the possible triggering signal for secretion of FGF-23 from erythroblasts, we used the human erythroblast cell line HUDEP-2.17 HUDEP-2 incubation with various G-CSF or isoproterenol concentrations resulted in the no iFGF-23 protein increase in culture supernatants (not shown). Because the BM microenvironment was reported to become hypoxic during G-CSF mobilization,19,20 HUDEP-2 cells were cultured in normoxic (21%) and hypoxic (5%) conditions. iFGF-23 protein was significantly increased in supernatants under hypoxia (Figure 3F).
Indeed, hypoxia induction in BM, as assessed by pimonidazole incorporation, was rapid, occurring 2 hours after single-dose G-CSF and 1 hour after single-dose isoproterenol (supplemental Figure 3).
Thus, during G-CSF mobilization, BM erythroblasts produce iFGF-23 protein in the BM cavity, at least partially triggered by hypoxia.
G-CSF–induced mobilization is impaired in the absence of FGF-23 production by hematopoietic cells
To analyze the function of BM FGF-23 in mobilization, mouse models deficient in FGF-23 were generated. Similar to a previous model,21 FGF-23−/− mice, which lacked both iFGF-23 and cFGF-23 in serum, displayed a senescencelike phenotype, such as growth retardation, short life-span, and fragility of the bone, with altered mineral metabolism (supplemental Figures 4-7). Five-week-old FGF-23−/− mice used for G-CSF mobilization showed a slight decrease in platelet count in the blood, with normal cellularity and number of CFU-Cs in the BM (supplemental Figure 8). A detailed flow cytometric analysis of BM cells in the mice revealed normal long- and short-term HSCs, with some alterations in the number of progenitors (supplemental Figure 9-11). To assess the effect of FGF-23 deficiency only in the hematopoietic system, chimeric mice were generated by transplanting FGF-23+/+ or FGF-23−/− BM cells into lethally irradiated wild-type mice (supplemental Figure 12A-B). Low body weight and hematopoietic phenotypes observed in FGF-23−/− mice were all corrected in FGF-23−/− BM chimera (supplemental Figures 12C-F and 13), suggesting that FGF-23 deficiency only in hematopoietic cells, including erythroblasts, does not alter steady-state hematopoiesis.
The mobilization efficiency of HPCs (LSKs and CFU-Cs) and HSCs (RUs at 6 months after competitive transplantation) by G-CSF was drastically suppressed in FGF-23−/− mice (Figure 4; supplemental Figure 14). Mobilization was also significantly suppressed in FGF-23−/− BM chimeric mice (Figure 5A; supplemental Figure 15A). The level of iFGF-23 protein in BM extracellular fluid after G-CSF was significantly lower in chimeric mice with FGF-23−/− BM than in FGF-23+/+ BM chimera, although the decrease was rather mild, probably because of FGF-23 production by nonhematopoietic cells (Figure 5B). In contrast, the downregulation of CXCL-12 protein in BM extracellular fluid by G-CSF was similar between chimeric mice with FGF-23+/+ and FGF-23−/− BM, with a trend in lower levels of CXCL-12 in FGF-23−/− BM chimera, with or without G-CSF (supplemental Figure 15B), suggesting that the impaired mobilization in FGF-23−/− BM chimera was not likely to be related to the altered suppression of CXCL-12.
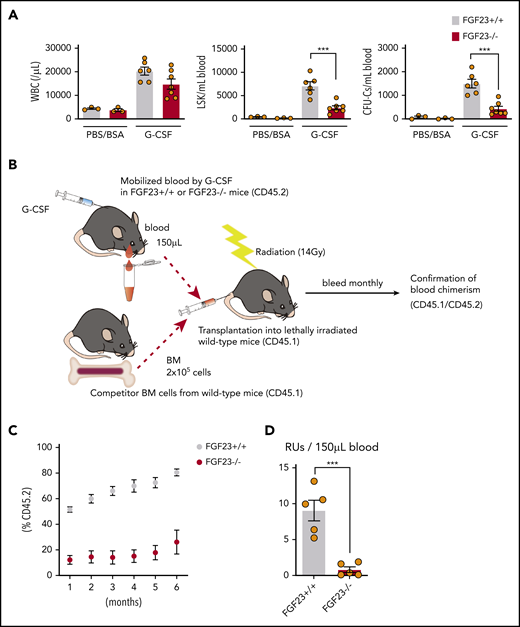
Impaired mobilization in FGF-23−/− mice. (A) Efficiency of G-CSF mobilization as assessed by white blood cells (WBCs), LSKs, and CFU-Cs in the blood in FGF-23+/+ and FGF-23−/− mice (n = 3 for the PBS/BSA group; n = 6-7 for the G-CSF group). (B-D) Assessment of competitive repopulating activity in G-CSF mobilized blood from FGF-23+/+ or FGF-23−/− mice. Competitive repopulation assay (B), %CD45.2 level in leukocyte chimerism (C), and RUs at 6 months after transplantation (D) (n = 5). Combined data of at least 3 independent experiments are shown. Data are means ± SEM. ***P < .001 (Student t test).
Impaired mobilization in FGF-23−/− mice. (A) Efficiency of G-CSF mobilization as assessed by white blood cells (WBCs), LSKs, and CFU-Cs in the blood in FGF-23+/+ and FGF-23−/− mice (n = 3 for the PBS/BSA group; n = 6-7 for the G-CSF group). (B-D) Assessment of competitive repopulating activity in G-CSF mobilized blood from FGF-23+/+ or FGF-23−/− mice. Competitive repopulation assay (B), %CD45.2 level in leukocyte chimerism (C), and RUs at 6 months after transplantation (D) (n = 5). Combined data of at least 3 independent experiments are shown. Data are means ± SEM. ***P < .001 (Student t test).
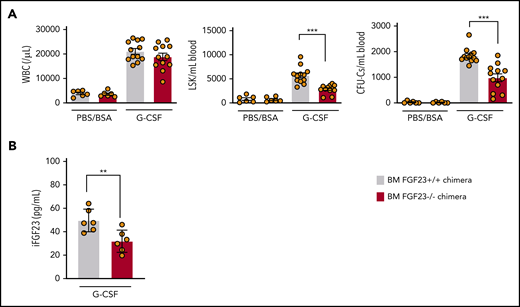
Impaired mobilization in chimeric mice with FGF-23−/− BM. (A) Efficiency of G-CSF mobilization as assessed by WBCs, LSKs, and CFU-Cs in the blood in wild-type mice that harbor FGF-23+/+ and FGF-23−/− BM (chimeric mice: n = 6 for the PBS/BSA group and n = 12 for the G-CSF group). (B) iFGF-23 protein in BM extracellular fluid of chimeric mice after G-CSF mobilization (n = 6). Combined data of at least 3 independent experiments are shown. Data are means ± SEM. **P < .01; ***P < .001 (Student t test).
Impaired mobilization in chimeric mice with FGF-23−/− BM. (A) Efficiency of G-CSF mobilization as assessed by WBCs, LSKs, and CFU-Cs in the blood in wild-type mice that harbor FGF-23+/+ and FGF-23−/− BM (chimeric mice: n = 6 for the PBS/BSA group and n = 12 for the G-CSF group). (B) iFGF-23 protein in BM extracellular fluid of chimeric mice after G-CSF mobilization (n = 6). Combined data of at least 3 independent experiments are shown. Data are means ± SEM. **P < .01; ***P < .001 (Student t test).
A previous report on conditional FGF-23 deletion in DMP-1+ osteocytes showed no major decrease in serum FGF-23 with normal mineral metabolism,22 probably because of the complementary FGF-23 production from cells other than osteocytes. Consistent with this model, DMP-1-cre+ FGF-23flox/flox mice had a normal appearance, with body weight and blood cell counts comparable to those of DMP-1-cre−FGF-23flox/flox control mice (supplemental Figure 16A-B), resulting in the nonsuppression of both mobilization (rather an increase; supplemental Figure 16C-D) and iFGF-23 protein in BM extracellular fluid (supplemental Figure 16E). Thus, hematopoietic cell (practically erythroblast)–derived BM FGF-23 is indispensable for normal mobilization.
FGF-23 counteracts the CXCR-4–mediated chemoattraction of HPCs through FGFRs
That strong BM FGF-23 promotes mobilization was among several possibilities tested by using 2 hypotheses: (1) as a direct chemorepellent that pushes out HPCs from BM into the blood, according to the FGF-23 gradient, and (2) as a suppressor of chemoattraction that anchors HPCs in BM. The transwell migration of CFU-Cs was assessed in the presence of a high level of FGF-23 (recombinant mouse FGF-23 that contained both an N-terminal FGFR binding region and a C-terminal α-klotho binding region), almost equivalent to the concentration in the BM extracellular cavity in vivo, after 6 to 8 doses of G-CSF. To stabilize the binding of FGF-23 to FGFRs, heparin was added, with or without α-klotho.23 Incubation with an FGF-23/α-klotho/heparin combination (FGF-23comb) did not alter the proliferation potential of HPCs, because the size (not shown) and number (Figure 6A) of colonies were unchanged. A simple gradient of FGF-23comb in the upper or lower chamber, did not induce CFU-C migration into the lower chamber, suggesting that FGF-23 does not function as a chemorepellent or chemoattractant [Figure 6B (no CXCL-12)]. With CXCL-12 in the lower chamber as a chemoattractant, FGF-23 in the upper chamber significantly suppressed CFU-C migration toward CXCL-12. This effect was stronger in combination with α-klotho (Figure 6B). A similar, but lesser, effect was observed when FGF-23comb was present in the lower chamber (Figure 6B). Thus, a high FGF-23 concentration suppresses the chemoattraction of HPCs toward CXCL-12.
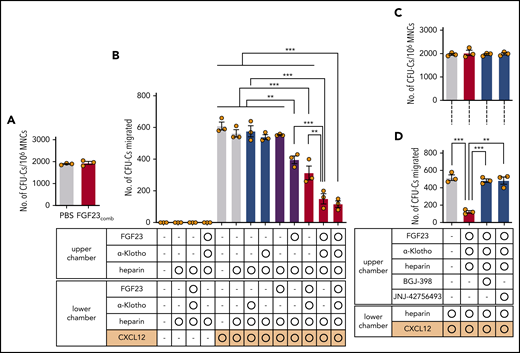
Function of FGF-23 in CFU-C transwell migration toward CXCL-12. (A) Colony formation of BM MNCs incubated with FGF-23comb (n = 3). (B) Transwell migration of CFU-Cs toward CXCL-12 in the presence of FGF-23comb (n = 3). (C) Colony formation of BM MNCs incubated with FGF-23comb and FGFR inhibitors (n = 3). (D) Transwell migration of CFU-Cs toward CXCL-12 in the presence of FGF-23comb and FGFR inhibitors (n = 3). Combined data of 3 independent experiments are shown. Data are means ± SEM. **P < .01; ***P < .001 (ANOVA and Student t test).
Function of FGF-23 in CFU-C transwell migration toward CXCL-12. (A) Colony formation of BM MNCs incubated with FGF-23comb (n = 3). (B) Transwell migration of CFU-Cs toward CXCL-12 in the presence of FGF-23comb (n = 3). (C) Colony formation of BM MNCs incubated with FGF-23comb and FGFR inhibitors (n = 3). (D) Transwell migration of CFU-Cs toward CXCL-12 in the presence of FGF-23comb and FGFR inhibitors (n = 3). Combined data of 3 independent experiments are shown. Data are means ± SEM. **P < .01; ***P < .001 (ANOVA and Student t test).
Among several possibilities, how FGF-23 inhibits HPC migration toward CXCL-12 was tested using 2 hypotheses: (1) as a modulator of CXCL-12 binding to CXCR-4 on the HPC surface and (2) as a signaling molecule via FGFR that counteracts CXCR-4 function. FGF-23comb did not suppress the surface expression of CXCR-4 on lineage–c-kit+ (LK; not shown) and LSK cells (supplemental Figure 17A) and did not inhibit the binding of fluorochrome-conjugated CXCL-12 in LK (not shown) and LSK cells (supplemental Figure 17B). Furthermore, as a rapid (in seconds) response to CXCL-12, actin polymerization in LK cells was measured using fluorochrome-conjugated phalloidin, resulting in a noninhibitory response in the presence of FGF-23comb (supplemental Figure 17C). Thus, FGF-23 is not a modulator of CXCL-12/CXCR-4 binding on the cell surface, whereas the FGF-23comb–mediated inhibitory effect on the transwell migration of CFU-Cs toward CXCL-12 was canceled by 2 different FGFR antagonists: BGJ-398 (infigratinib) and JNJ-42756493 (erdafitinib) (Figure 6C-D). Thus, FGF-23 counteracts the chemoattraction of CXCR-4 through FGFRs.
Discussion
FGF-23 actions, other than as a phosphate-regulating hormone, such as in cardiac left ventricular hypertrophy and the increased production of inflammatory proteins from hepatocytes, have been studied as complications of chronic kidney disease (CKD).23-25 FGF-23 plasma levels can reach 1000-fold above normal in advanced CKD, and high FGF-23 impairs neutrophil activation, such as integrin clustering and low rolling velocity on selectins, after stimulation with CXCL-1 and -8 via FGFR-2 by activating protein kinase A and inhibiting the small GTPase Rap-1, resulting in decreased recruitment to inflammatory sites.26 FGF-23 in BM may similarly modulate these molecules and impair the activation and deregulate the determination of the trafficking direction of HSCs/HPCs upon stimulation by CXCL-12. This notion may explain the inactivated status of very late antigen-4 (VLA-4) integrin in human mobilized CD34+ cells.27 It is possible that the high level of FGF-23 is a common inhibitory factor for signaling from a wide range of chemokine receptors in various cell types in BM, such as mesenchymal and endothelial cells, in addition to hematopoietic cells. The FGF-23 action on neutrophils does not require α-klotho.23,26 However, our current study showed that the inhibitory effect of FGF-23 on CXCR-4–mediated HPC transwell migration was active without it but was significantly enhanced with it, suggesting that FGFR-1, which can act as an α-klotho–dependent FGF-23 receptor,28 in addition to α-klotho–independent FGFR-2–like neutrophils, may mainly contribute to this effect.
According to accumulated studies on CXCL-12 in mobilization, the drastic decrease in CXCL-12 mRNA in BM cells and protein in BM extracellular fluid at the time of massive mobilization (after 4-5 consecutive days of G-CSF treatment) seems to be a simple and critical event for mobilization.2,10-12 However, decreased CXCL-12 mRNA in BM is achieved, even after 2 days of G-CSF treatment during which the mobilization efficiency is still minute.29 Day et al reported that 7 days of treatment with G-CSF decreased CXCL-12 mRNA in sorted CAR cells, but the number of these cells per femur increased.13 This result suggests that total CXCL-12 expression in BM is relatively preserved, even after G-CSF treatment of >4 days, which is consistent with the efficient enhancement (>3 times) of mobilization by the addition of CXCR-4 antagonist at the end of 4 days of G-CSF treatment in both human and mouse.30 Thus, together with the new finding that mobilization efficiency is greatly impaired in the absence of FGF-23 production from hematopoietic cells as an endogenous counteraction for CXCR-4, the major contribution of the CXCR-4/CXCL-12 axis in G-CSF–induced mobilization may be the inhibition of the function of CXCR-4, rather than the decrease of CXCL-12 in the BM microenvironment.
In our study of HUDEP-2 cells, hypoxia seemed to trigger partial FGF-23 release. BM in G-CSF–treated mice became hypoxic, and hypoxia-inducible factor-1α was necessary for mobilization.19,20 Another possible pathway for FGF-23 release is hemolysis or apoptosis of erythroblasts, as the number of erythroblasts in BM has been reported to be significantly decreased during G-CSF treatment by the depletion of erythroid island macrophages.31 Because erythroblasts are reported to be positioned near HSCs,32 FGF-23 produced from erythroblasts may effectively promote HSC mobilization at local sites. Patients with multiple myeloma or other hematological malignancies after repeated chemotherapy often show anemia and poor mobilization. Thus, the decreased number of BM erythroblasts may be a reason for poor mobilization in these patients because of the insufficient supply of BM FGF-23.
This study showed that G-CSF triggers FGF-23 production from erythroblasts, mainly in intact form, and only the C-terminal FGF-23 fragment, which is probably cleaved in the marrow cavity, but not iFGF-23, likely goes through the marrow-blood barrier into the circulation. It is reported that administration of Epo can induce FGF-23 production from erythroid progenitors33,34 and increase the blood C-terminal FGF-23 fragment level as a dominant form,35 suggesting that Epo may trigger a strong intracellular cleavage of FGF-23. FGF-23 cleavage in erythroid progenitors may be tightly regulated, depending on stimulation, and the signal to erythroblasts during G-CSF treatment, possibly hypoxia, may induce a different FGF-23 regulation from Epo.
B lymphopoiesis is strongly suppressed by mobilizing doses of G-CSF via the reprogramming of BM stromal cells.13,29 CXCL-12, also known as the pre-B-cell growth-stimulating factor,36 is necessary for the survival and activation of B-cell precursors.37 In our current study, it is also possible that increased FGF-23 by G-CSF inhibits CXCR-4 function in B-cell precursors in BM. The CXCR-4/CXCL-12 axis is also important for the survival, chemoattraction, and integrin activation of myeloma cells.37,38 Also, myeloma cells have been reported to directly induce apoptosis of immature erythroblasts via the expression of Fas ligand and tumor necrosis factor–related apoptosis-inducing ligand.39,40 The reduction of erythroblasts and the inhibition of the productive potential for BM FGF-23 may be advantageous for myeloma survival.
In addition to erythroblasts, we found a partial contribution of BM stromal cells to the increase in marrow FGF-23 during G-CSF treatment. That no major reduction was found in the FGF-23 protein level in the blood and BM in osteocyte-specific, FGF-23-deficient mice in this and another study22 indicates that many cells other than osteocytes can produce FGF-23 in the absence of osteocytic FGF-23 or upon certain stresses such as G-CSF/SNS stimulation. Increased G-CSF–induced mobilization in the absence of osteocytic FGF-23 may be related to the strong complementary production of FGF-23 from osteoblast lineage cells, and possibly erythroblasts as well, at the local endosteal region.
This study proposes that G-CSF–induced mobilization could be achieved in 2 major steps (Figure 7): (1) the SNS-mediated suppression of the microenvironment, such as osteolineage cells and macrophages, generates a “ready-to-be-mobilized” state for HSCs/HPCs, and (2) the remaining anchoring pathway by CXCL-12+ cells, such as CAR cells, is suppressed by FGF-23, mainly from erythroblasts, which counteracts CXCR-4 function via FGFRs.
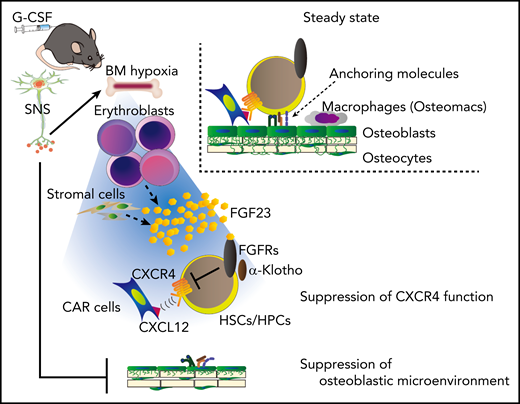
A model for G-CSF–induced mobilization by the cooperative regulation of the SNS and erythroblasts. The SNS-mediated suppression of the microenvironment, such as osteolineage cells and macrophages, generates a “ready-to-be-mobilized” state for HSCs/HPCs. G-CSF, probably via SNS, induces BM hypoxia, which triggers the release of FGF-23 mainly from erythroblasts (and also partially from stromal cells). The residual anchoring pathway, chemoattraction toward CXCL-12+ cells, such as CAR cells, is suppressed by the high concentration of BM FGF-23, which counteracts the CXCR-4 function via FGFRs.
A model for G-CSF–induced mobilization by the cooperative regulation of the SNS and erythroblasts. The SNS-mediated suppression of the microenvironment, such as osteolineage cells and macrophages, generates a “ready-to-be-mobilized” state for HSCs/HPCs. G-CSF, probably via SNS, induces BM hypoxia, which triggers the release of FGF-23 mainly from erythroblasts (and also partially from stromal cells). The residual anchoring pathway, chemoattraction toward CXCL-12+ cells, such as CAR cells, is suppressed by the high concentration of BM FGF-23, which counteracts the CXCR-4 function via FGFRs.
Original data are available by e-mail request to the corresponding author (Yoshio Katayama; katayama@med.kobe-u.ac.jp).
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Acknowledgments
The authors thank Lynda F. Bonewald (Indiana Center for Musculoskeletal Health, Indiana University School of Medicine, Indianapolis, IN) for DMP-1-Cre mice.
This work was supported by grants from the Japan Science and Technology Agency (PRESTO; JPMJPR12M7), the Japan Agency for Medical Research (AMED) (CREST; JP18gm0910012h2), and the Japan Society for the Promotion of Science (Grants-in-Aid for Scientific Research 15H04856 and 18H02837 and Challenging Research grant 17K19660) (all Y. Katayama).
Authorship
Contribution: S.I. performed all experiments and wrote the manuscript; T.S., K.W., N.A., Y. Kawano, H.K., A.S., and K.M. helped with animal maintenance and tissue sample preparation; Y.N. supervised the study of HUDEP-2 cells; S.M., S.T., and T.M. supervised the studies involving FGF-23−/− and FGF-23 floxed mice; and Y. Katayama supervised all experiments and wrote the manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
The current affiliation for K.W. is the Area of Cell and Developmental Biology, Centro Nacional de Investigaciones Cardiovasculares Carlos III (CNIC), Madrid, Spain.
The current affiliation for N.A. is the Department of Hematology and Oncology, Okayama University Hospital, Okayama, Japan.
The current affiliation for Y. Kawano is the Endocrine/Metabolism Division, Wilmot Cancer Institute, University of Rochester Medical Center, Rochester, NY.
The current affiliation for H.K. is the Endocrine/Metabolism Division, Wilmot Cancer Institute, University of Rochester Medical Center, Rochester, NY.
The current affiliation for K.M. is the Hematology and Oncology Division, Penn State College of Medicine, Hershey, PA.
Correspondence: Yoshio Katayama, Department of Medicine, Kobe University Graduate School of Medicine, 7-5-1 Kusunoki-cho, Chuo-ku, Kobe 650-0017, Japan; e-mail: katayama@med.kobe-u.ac.jp.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal