

Key Points
PF4 bound to VWF strings form antigenic complexes that are recognized by HIT antibodies.
PF4-VWF-HIT antibody complexes enhance platelet adhesion via platelet FcγRIIA and glycoprotein Ib-IX receptors.

Abstract
Heparin-induced thrombocytopenia (HIT) is a prothrombotic disorder mediated by complexes between platelet factor 4 (PF4) and heparin or other polyanions, but the risk of thrombosis extends beyond exposure to heparin implicating other PF4 partners. We recently reported that peri-thrombus endothelium is targeted by HIT antibodies, but the binding site(s) has not been identified. We now show that PF4 binds at multiple discrete sites along the surface of extended strings of von Willebrand factor (VWF) released from the endothelium following photochemical injury in an endothelialized microfluidic system under flow. The HIT-like monoclonal antibody KKO and HIT patient antibodies recognize PF4-VWF complexes, promoting platelet adhesion and enlargement of thrombi within the microfluidic channels. Platelet adhesion to the PF4-VWF-HIT antibody complexes is inhibited by antibodies that block FcγRIIA or the glycoprotein Ib-IX complex on platelets. Disruption of PF4-VWF-HIT antibody complexes by drugs that prevent or block VWF oligomerization attenuate thrombus formation in a murine model of HIT. Together, these studies demonstrate assembly of HIT immune complexes along VWF strings released by injured endothelium that might propagate the risk of thrombosis in HIT. Disruption of PF4-VWF complex formation may provide a new therapeutic approach to HIT.
Introduction
Heparin-induced thrombocytopenia (HIT) is initiated by antibodies that target antigenic complexes between human (h) platelet factor 4 (PF4, CXCL4) and heparin.1 The resultant immune complexes activate platelets,2 monocytes,3 neutrophils,4 and endothelial cells,5 generating an intensely prothrombotic environment. However, the risk of thromboembolism in HIT extends beyond the duration of heparin exposure, which may implicate other polymeric molecules capable of binding hPF4 in antigen formation, including cell surface glycosaminoglycans (GAGs),2 DNA,6 polyphosphates,7 and bacterial components.8 Prothrombotic pathways initiated by these alternative mechanisms of antigen formation may be relatively resistant to current antithrombotic therapies, but may offer new therapeutic targets for patients with HIT.9
The endothelium has been identified as a potential target for HIT antibodies.5 We observed extensive binding of HIT antibodies to endothelium beneath and immediately downstream of thrombi induced within endothelialized microfluidic channels and in cremaster vessels in a murine model of HIT,10 but the site of antigen assembly has not been identified. Retraction of injured endothelium exposes potential ligands recognized by platelets,11 but cell activation also changes the composition and thickness of the glycocalyx12,13 and induces expression of receptors and ligands, including tissue factor and P-selectin,14,15 on the endothelium.
Activated endothelium also releases ultra-large multimers of von Willebrand factor (VWF) from Weibel-Palade bodies.16,17 These multimers have a low threshold for shear-induced unfolding and can self-associate into VWF strings along the endothelium18 that readily bind platelets.19 However, neither an interaction between hPF4 and VWF nor involvement of VWF in the propagation of HIT has been reported. In this study, we addressed these questions using an endothelialized microfluidic model wherein localized photochemical injury using hematoporphyrin (Figure 1A 20 ) causes the release of ultralarge multimers of VWF that form strings under flow.10 We found that hPF4 binds to these VWF strings, forming complexes recognized by HIT antibodies. Platelets bind extensively to these complexes, and the binding is inhibited by monoclonal antibodies blocking either FcγRIIA21 or glycoprotein (GP) Ib-IX.22 We also demonstrate the contribution of hPF4-VWF-HIT antibody complexes to thrombosis following injury to the vasculature in a passive immunization murine model of HIT.10 These studies extend the diversity of polymeric molecules that might contribute to the propagation of thrombosis in HIT in the absence of heparin through pathways not targeted by inhibitors of thrombin or factor Xa (FXa). The clinical implications of these findings are discussed.
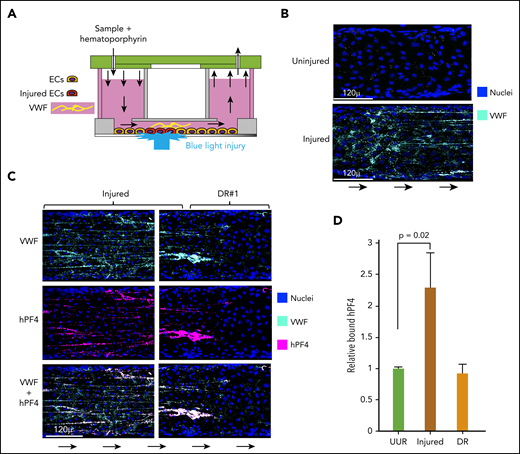
hPF4 binds to elongated strings of VWF released from injured HUVECs in a microfluidic system. (A) Schematic of the endothelialized microfluidic system used to study the effect of photochemical injury to targeted regions of the endothelium. (B) Comparison of a representative area of uninjured endothelium and the immediate upstream hematoporphyrin-injured area showing pyknotic nuclei and VWF strings only released after injury. (C) Appearance at representative injured site within the microfluidic channel and at site DR, the first 250 µ downstream of the edge of the injured site. Direction of flow is indicated by arrows. Channels were stained for PF4 using a polyclonal anti-hPF4 antibody as well as staining for nuclei and VWF. (D) Relative binding of PF4 to either 250 µ of the upstream uninjured region (UUR), the injured region, or the downstream of injury region (DR). Results are expressed as the mean ± 1 standard error of the mean (SEM) from N ≥ 4 separate microfluidic channels. P values were determined using a 1-way ANOVA analysis to compare binding to the UUR of the channel. EC, endothelial cells.
hPF4 binds to elongated strings of VWF released from injured HUVECs in a microfluidic system. (A) Schematic of the endothelialized microfluidic system used to study the effect of photochemical injury to targeted regions of the endothelium. (B) Comparison of a representative area of uninjured endothelium and the immediate upstream hematoporphyrin-injured area showing pyknotic nuclei and VWF strings only released after injury. (C) Appearance at representative injured site within the microfluidic channel and at site DR, the first 250 µ downstream of the edge of the injured site. Direction of flow is indicated by arrows. Channels were stained for PF4 using a polyclonal anti-hPF4 antibody as well as staining for nuclei and VWF. (D) Relative binding of PF4 to either 250 µ of the upstream uninjured region (UUR), the injured region, or the downstream of injury region (DR). Results are expressed as the mean ± 1 standard error of the mean (SEM) from N ≥ 4 separate microfluidic channels. P values were determined using a 1-way ANOVA analysis to compare binding to the UUR of the channel. EC, endothelial cells.
Methods
Human blood samples
Deidentified whole blood samples from volunteers with no history of a bleeding diathesis or known use of medications with antiplatelet or anticoagulant effects were drawn through a 19-gauge needle into sodium citrate (0.38% final concentration, Fisher). Blood samples were maintained at room temperature and studied within 1 hour. Deidentified plasma samples were also obtained from patients determined to have HIT based on clinical assessment, a positive HIT enzyme-linked immunosorbent assay, and positive serotonin-release assay.2 Samples collected in the same manner from patients who had been evaluated for HIT, but with a low pretest probability of HIT based on history, enzyme-linked immunosorbent assay and SRA results were used as negative controls. Human umbilical endothelial cells (HUVECs) were purchased (Lonza). The Children’s Hospital of Philadelphia institutional human review board approved these studies in accord with the Helsinki Principles.
Mouse genotype and blood sampling
Wild-type C57Bl6 mice and hPF4/FcγRIIA-double transgenic mice on a cxcl4−/− background23 or “HIT mice” were studied. These mice develop thrombocytopenia and thromboses when injected with the HIT-like murine monoclonal antibody KKO24 or with immunoglobulin G (IgG) isolated from patients with HIT.2,25,26 Approval for these animal studies was obtained from the Children’s Hospital of Philadelphia Institutional Animal Care and Use Committee.
Endothelialized microfluidic studies
Endothelialized microfluidic studies were performed as described previously10 using the Bioflux 1000 Controller system. The microscope was equipped with a 37°C heating motorized stage and an HXP120C metal-halide illumination source. Forty-eight-well Bioflux plates were coated with fibronectin (50 µg/mL, Sigma-Aldrich), seeded with HUVECs (passage 3-4, 6-8 × 106 cells/channel) that were allowed to adhere and cultured at 37°C under 5% CO2 in endothelial growth media (Lonza; basal culture medium [BCM] containing optimized concentrations of bovine brain extract with heparin, human endothelial growth factor, hydrocortisone, ascorbic acid, gentamicin, amphotericin B, and fetal bovine serum) until confluent. The microfluidic chambers were then washed with BCM alone, containing no heparin for 4 hours before usage. Photochemical injury was induced by adding hematoporphyrin (50 µg/mL final concentration, Sigma-Aldrich). The cell linings were injured using a HXP120C light source with a 475-nm excitation and 530-nm emission filter for 20 to 30 seconds.10,20 hPF4 and VWF were detected using Alexa Fluor-labeled antibodies. HUVEC nuclei were labeled with Hoeschst 33342 (Fisher) for 15 minutes at room temperature. The analysis of the released VWF is described in the supplemental Methods, available on the Blood Web site.
Analysis of hPF4-VWF-HIT antibody complexes
All experimental solutions were made in BCM without heparin. Localized photochemical injury was induced using hematoporphyrin. Channels were washed with BCM and then infused with hPF4 (0-25 µg/mL) in BCM for 10 minutes. In some experiments, KKO or an isotype control TRA (each at 10 µg/mL, see preparation in supplemental Methods), or plasma from HIT patients (1:100 dilution in BCM), or a polyclonal rabbit anti-hPF4 antibody (2 µg/mL, PeproTech) was added to the hPF4 solution at the 10-minute mark and allowed to flow continuously for an additional 10 minutes. After a total flow time of 20 minutes, channels were washed briefly with BCM and then fixed for 15 minutes at room temperature with Cytofix Fixation Buffer (BD Biosciences).
Experiments were performed to determine if hPF4-VWF-HIT antibody complexes would bind selectively to VWF or could be eluted by bovine serum albumin (BSA, Sigma-Aldrich) or plasma-derived hVWF. Once VWF-PF4-KKO complexes were allowed to form, a solution of BCM containing BSA, equimolar to the monomer concentration in 10 µg/mL of VWF, or purified plasma-derived hVWF (10 or 20 µg/mL final concentration, Haematologic Technologies) was perfused through the channels. Channels were fixed with 2% paraformaldehyde (BD Biosciences).10 hPF4, VWF, and KKO binding was quantified via confocal imaging.
Experiments were also performed to examine the effects of drugs known to prevent formation or to disrupt strings of VWF or with heparin to dissociate hPF4 from vascular surfaces. To accomplish this, hPF4-VWF-HIT antibody complexes were allowed to form and then exposed to the following interventions flowed through the channels at 10 dynes/cm2 for 10 minutes: unfractionated porcine heparin (heparin, 0-50 units/mL final concentration, Novaplus), a disintegrin and metalloproteinase with a thrombospondin type 1 motif, member 13 (ADAMTS13, 0-7 nM, R&D Systems), and N-acetylcysteine (NAC, 0-10 mg/mL final concentration, AuroMedics). In some experiments, a solution containing heparin (0-40 units/mL), hPF4 (0-100 µg/mL), and KKO (10 µg/mL) was infused into channels for 10 minutes after hPF4-VWF-HIT antibody complexes had been allowed to form. The channels were washed with BCM, fixed with Cytofix Fixation Buffer (BD Biosciences), and imaged to quantify the presence of hPF4, KKO, and/or extended VWF strings.
Platelet accumulation on hPF4-VWF-HIT antibody complexes
To track platelet adhesion to hPF4-VWF-HIT antibody complexes, whole blood from healthy volunteers was labeled with calcein AM (2 µg/mL final concentration, Fisher) at room temperature for 15 minutes, recalcified immediately before infusion using 2M CaCl2 (final concentration of 12 mM, Sigma-Aldrich), and perfused through the channels at 10 dynes/cm2. Platelet accumulation was quantified based on total fluorescent intensity detected within the localized area of injury. In other experiments, IV.321 (20 µg/mL) and/or AK222 (60 µg/mL) were added to whole blood for 30 minutes, and platelets adherent to hPF4-VWF-HIT antibody complexes were quantified.
Role of VWF in thrombosis in HIT in vivo
Thrombosis was induced using a previously described murine model of HIT25 by infusing KKO following rose bengal photochemical injury to the carotid artery.3 HIT mice were anesthetized with Nembutal (80 mg/kg). Either TRA (2 mg/kg) or KKO (2 mg/kg) or KKO (2 mg/kg) + NAC (0.8 mg/g, AuroMedics) or KKO (2 mg/kg) + ADAMTS13 (0.1 mg/kg), each in phosphate-buffered saline brought to a total volume of 250 to 280 µL was infused via the left jugular vein. These concentrations of NAC and ADAMTS13 were shown by others to exert antithrombotic activity in mice without causing bleeding or impairing viability.27,28 The injected materials were allowed to circulate for 10 to 15 minutes during which time the right common carotid artery was surgically exposed. Rose bengal (50 mg/kg) was then infused and a miniature Doppler flow probe (Model 0.5VB; Transonic Systems) positioned around the artery. After 5 minutes of measuring baseline flow, a 540-nm laser was targeted to the carotid artery. Using the data points from the Doppler flow probe, the time to occlusion (blood flow <15% of original for ≥10 minutes) was determined and the relative area under the curve (AUC) of total blood flow calculated. If the vessel did not occlude, experiments were ceased at 2 hours.
Mice were injected by the left jugular vein with Alexa Fluor647-labeled anti-mouse CD41 and Alexa Fluor541-labeled anti-mouse fibrin antibodies via the jugular vein immediately before cremaster arteriole laser injuries done as described previously.10 The size of the resultant clots was noted by fibrin and platelet accumulation at 5 minutes postinjury as described.10 After 5 injuries were done and recorded, mice received 1 mg/kg of either KKO or TRA intravenously with or without 50 µg/kg of ADAMTS13. Fibrin and platelet accumulation at each of the injury sites was then recorded 15 minutes later and analyzed. Final data were collected and calculated using Slidebook 6.0 (Intelligent Imaging Innovations).
The recombinant proteins and antibodies used in these studies, analysis of the VWF released in the microfluidic channels, and the use of dynamic light scattering to analyze PF4-VWF complexes are detailed in the supplemental Methods.
Statistics
Differences between 2 groups were compared using a 2-sided Student t test. Differences between more than 2 groups were determined by Kruskal-Wallis 1-way analysis of variance (ANOVA). All analyses were performed on GraphPad Prism 6.0 (GraphPad Software). Differences were considered significant when the P values were ≤.05.
Results
hPF4 binds to extended strings of VWF that form after its release from injured endothelium
We previously reported that PF4 released from activated platelets localized predominantly along endothelium beneath or immediately downstream of a site of injury in both an endothelialized microfluidic model system and in vivo in a murine model of HIT10 ; however, the target(s) to which the hPF4 bound, and subsequently the HIT-like antibodies bound, was not identified. To begin to explore the potential PF4-binding sites on injured endothelium, we used the hematoporphyrin injury model in an endothelialized microfluidic model that does not cause endothelial cells to detach or to lose cell–cell contact.10,20 This model enables us to explore a human endothelial surface, to control the composition of the thrombus, and to contrast binding to injured and adjacent uninjured endothelium (Figure 1A). Within the injured zone, there was extensive expression of VWF along the luminal surfaces of the injured endothelial cells (Figure 1B). Postinjury, we infused recombinant hPF4. The most extensive binding of hPF4 was restricted to the injured area with less seen immediately downstream (Figure 1C-D). The prominent binding of hPF4 within these regions followed a linear pattern similar to the distribution of the strings of VWF emanating from the endothelium. Therefore, we asked whether hPF4 was binding to the released VWF. VWF and hPF4 colocalized29 (P = .02 VWF strand-containing regions vs regions without VWF) as seen in enlarged images (supplemental Figures 1A-B) and in video images (supplemental Video 1). hPF4 also showed significantly greater binding to extended strings of VWF compared with unelongated vWF (P < .01, supplemental Figure 1C).
KKO and HIT patient IgGs bind PF4-VWF complexes in vitro
PF4 binds to polyanions in addition to heparin, including GAGs,30 DNA aptamers,6 and polyphosphates,7 each of which forms complexes that are recognized by HIT or HIT-like antibodies. Binding of hPF4 to heparin leads to the formation of high-molecular-weight oligomers postulated to increase antibody avidity,31,32 and it is possible that the repeating VWF structures that align along the strings provide a similar template for antigen assembly. To determine if hPF4-VWF complexes are also recognized by HIT-like antibodies, we induced injury in the microfluidic chambers and introduced PF4 and KKO, a monoclonal antibody known to replicate key features of patient-derived HIT antibodies.24 Binding of KKO followed the same distribution seen with hPF4, with both colocalizing together along the strings of VWF released by the injured endothelium (Figure 2A). This binding localized primarily to the area of endothelial injury and to the immediately downstream region (Figure 2B) and was significantly associated with the VWF strings (supplemental Figure 2). Moreover, bound KKO appeared to be aligned at regular intervals along the VWF strings, with little KKO seen in association with smaller, unelongated deposits of VWF (P < .0005; supplemental Figure 3; supplemental Video 2).

KKO and HIT patient IgGs bind to PF4-VWF complexes ex vivo. (A) Representative injured area and downstream of injury region (DR) in the microfluidic channels as in Figure 1C stained for VWF and KKO. (B) Comparison as in Figure 1D for total KKO bound relative to the UUR and DR. Means ± 1 SEM are shown for N ≥ 5 separate studies. P values were determined by a 1-way ANOVA. Panels C-D are similar to panels A-B, respectively, but show only the injured areas stained for either VWF or for anti-human Fab fragments (αHm-Fab). (C) Diluted plasma from patients at high risk of having HIT or low-risk controls (Ctl)33 were used. (D) Relative binding of IgG from the HIT or control samples (N = 6 normal controls) are shown. Means ± 1 SEM are shown from N ≥ 5 separate studies. P values were determined using a 1-way ANOVA to compare relative binding of each IgG in the absence of hPF4 vs in the presence of infused PF4 (25 µg/mL) with control samples in the absence of PF4 set to 1.
KKO and HIT patient IgGs bind to PF4-VWF complexes ex vivo. (A) Representative injured area and downstream of injury region (DR) in the microfluidic channels as in Figure 1C stained for VWF and KKO. (B) Comparison as in Figure 1D for total KKO bound relative to the UUR and DR. Means ± 1 SEM are shown for N ≥ 5 separate studies. P values were determined by a 1-way ANOVA. Panels C-D are similar to panels A-B, respectively, but show only the injured areas stained for either VWF or for anti-human Fab fragments (αHm-Fab). (C) Diluted plasma from patients at high risk of having HIT or low-risk controls (Ctl)33 were used. (D) Relative binding of IgG from the HIT or control samples (N = 6 normal controls) are shown. Means ± 1 SEM are shown from N ≥ 5 separate studies. P values were determined using a 1-way ANOVA to compare relative binding of each IgG in the absence of hPF4 vs in the presence of infused PF4 (25 µg/mL) with control samples in the absence of PF4 set to 1.
We then asked whether diluted plasma from patients with HIT express IgGs that bind to hPF4-VWF complexes. A representative field of IgG binding from 1 such HIT plasma compared with plasma from a patient with a low probability of having HIT is shown (Figure 2C). IgG from 5 of 6 plasma samples from patients with HIT selectively bound to the extended VWF strings released from injured endothelium in the presence of added hPF4 compared with none of the 6 control samples (Figure 2D).
Physical characterization of the hPF4-VWF complexes in solution based on dynamic light scattering
We used dynamic light scattering7 as an independent method to study the binding of hPF4 to VWF. We first examined the particle size of hPF4 and VWF individually. The size distribution of hPF4 molecules followed a Gaussian-size distribution, with no particles >10 nm in size. In contrast, VWF displayed a more complex size distribution consistent with polymers composed of a variable number of monomer subunits (supplemental Figure 4A). When hPF4 was added to VWF at ratio of 10 µg/mL:1 µg/mL and at 10 µg/mL:5 µg/mL, single narrow Gaussian distributions of particle sizes were seen (supplemental Figure 4B). When the ratio of hPF4 relative to VWF was further decreased (hPF4:VWF = 10 µg/mL:10 µg/mL), the apparent mean mass of the hPF4-VWF complexes widened with an appearance similar to that of VWF alone. These data provide additional support for our interpretation of the colocalization studies, showing that PF4 binds directly to VWF.
Effect of plasma VWF on hPF4-VWF-HIT antibody complex formation
We next asked whether plasma-derived VWF contributes to antigen formation. It has been reported previously that PF4 did not enhance binding of VWF to platelets directly,34 but the effect of plasma VWF on hPF4-VWF-HIT antibody immune complex formation is unknown. Plasma VWF can self-associate with immobilized VWF under flow.35,36 We therefore injured the endothelial lining in the microfluidic channels and introduced hPF4 and KKO to permit hPF4-VWF-KKO complexes to assemble. The channels were then infused with plasma hVWF at physiologic plasma levels (low, 10 µg/mL), at a twofold higher level (20 µg/mL),37 or with BSA at an equimolar level relative to the number of VWF monomeric equivalency in plasma or at a twofold higher concentration to assess specificity. Heparin, which removes hPF4-HIT antibody complexes from cell surfaces2 was used as a positive control, expecting it to elute hPF4 from the VWF strings. Perfusing soluble VWF across the injured endothelium increased the binding of hPF4, albeit the increase did not reach statistical significance, but did not affect binding of KKO (Figure 3A-B, respectively). We speculate that perhaps at low VWF concentrations, circulating VWF may be binding to PF4 complexed to the VWF strings and that at higher concentration, this increased VWF binding may be balanced by PF4 washout.

Effect of soluble VWF on hPF4 and KKO binding to endothelium. Expression of (A) hPF4 and (B) KKO on VWF strings in the microfluidic chamber after elutriation with low (10 µg/mL) or high (20 µg/mL) concentrations of plasma VWF or BSA. Heparin (4 U/mL) was also studied as a positive control. Means ± 1 SEM are shown for N ≥ 4 separate channels in the case of control and N ≥ 5 for all experimental additions. P values were determined using a 1-way ANOVA to compare residual relative binding in the absence vs the presence of added protein or heparin.
Effect of soluble VWF on hPF4 and KKO binding to endothelium. Expression of (A) hPF4 and (B) KKO on VWF strings in the microfluidic chamber after elutriation with low (10 µg/mL) or high (20 µg/mL) concentrations of plasma VWF or BSA. Heparin (4 U/mL) was also studied as a positive control. Means ± 1 SEM are shown for N ≥ 4 separate channels in the case of control and N ≥ 5 for all experimental additions. P values were determined using a 1-way ANOVA to compare residual relative binding in the absence vs the presence of added protein or heparin.
Perfusing BSA as a negative control had little effect. Heparin, with its known high affinity for PF4,30 markedly reduced the binding of both hPF4 and of KKO, as expected. These studies support the concept that the shear-induced changes in VWF approximate epitopes for PF4, which generates the spatial periodicity of the observed PF4-VWF complexes. Soluble VWF could not compete away PF4 bound to VWF strings, but may incorporate within the strings, enhancing PF4 binding.
hPF4-VWF-HIT antibody complexes are prothrombotic in vitro
Do the hPF4-VWF-HIT antibody complexes contribute to the prothrombotic state seen in HIT? To address this question, we first asked whether binding of platelets to the hPF4-VWF strings on the endothelium was enhanced by KKO in the microfluidic system. Endothelial injury was required to initiate the adhesion of human platelets in whole blood (Figure 4A). Platelet adhesion was greatest when KKO as well as hPF4 were present (Figure 4B-C), whereas neither KKO alone nor hPF4 plus the isotype control monoclonal antibody TRA24 affected platelet adhesion.

hPF4-VWF-HIT antibody complexes are prothrombotic in vitro. (A) Representative fields showing (top) uninjured and (bottom) injured endothelium as in Figure 1B, but after exposure to the flow of recalcified whole human blood for 10 minutes. Binding of platelets to the VWF strings is shown. The bottom channel shows an injured area with platelets and the immediate, contiguous uninjured downstream area resembling in appearance the uninjured top channel. (B) Same as in panel A, but with the inclusion of hPF4 (25 µg/mL) and the indicated monoclonal antibody (10 µg/mL) and studied at the time points indicated. Representative binding of labeled KKO and platelets are shown. Platelet accumulation at 10 and 40 minutes is shown. (C) Same as in panel B, but for N ≥ 5 separate studies. Means ± 1 SEM are shown. P values were determined using a 1-way ANOVA.
hPF4-VWF-HIT antibody complexes are prothrombotic in vitro. (A) Representative fields showing (top) uninjured and (bottom) injured endothelium as in Figure 1B, but after exposure to the flow of recalcified whole human blood for 10 minutes. Binding of platelets to the VWF strings is shown. The bottom channel shows an injured area with platelets and the immediate, contiguous uninjured downstream area resembling in appearance the uninjured top channel. (B) Same as in panel A, but with the inclusion of hPF4 (25 µg/mL) and the indicated monoclonal antibody (10 µg/mL) and studied at the time points indicated. Representative binding of labeled KKO and platelets are shown. Platelet accumulation at 10 and 40 minutes is shown. (C) Same as in panel B, but for N ≥ 5 separate studies. Means ± 1 SEM are shown. P values were determined using a 1-way ANOVA.
We then sought to identify the platelet components that contribute to adhesion to VWF strings in the presence of PF4+KKO. Platelets bind to shear-activated VWF via the GPIb-IX complex, a pathway that can be blocked by the anti-GPIbα monoclonal antibody AK2.22 The hPF4-VWF-KKO complexes might also enhance platelet adherence by activating the signal transducing receptor FcγRIIA, a process that can be blocked by the anti-FcγRIIA monoclonal antibody IV.3.21 Each antibody added individually blocked HIT antibody-induced adherence of platelets in whole blood to the VWF strings in the presence of PF4+KKO (Figure 5A-B; supplemental Figure 5). By 40 minutes postinfusion, blocking the GPIb-IX-VWF axis appears to be more effective. Addition of IV.3 did not increase the inhibitory effect of AK2. This suggests that the GPIb-IX-VWF axis continues to have a primary role in the binding of platelets, but that the process is amplified in HIT by the presence of hPF4-VWF-HIT antibody complexes on the endothelium.

Binding of platelets to hPF4-VWF-HIT antibody complexes is dependent on both their FcγRIIA and GPIb/IX receptors. (A) As in Figure 4B, representative fields showing platelet accumulation on injured endothelium after exposure to the flow of recalcified human blood in the presence or absence of PF4 and KKO and the GPIb-IX-blocking antibody AK2 or FcγRIIA-blocking antibody IV.3 or both. (B) In each case, binding of platelets is shown relative to injured endothelium with added KKO only as the comparator. Means ± 1 SEM are shown for N ≥ 5 separate injured studies. P values were determined using a 1-way ANOVA. See supplemental Figure 5 for additional controls.
Binding of platelets to hPF4-VWF-HIT antibody complexes is dependent on both their FcγRIIA and GPIb/IX receptors. (A) As in Figure 4B, representative fields showing platelet accumulation on injured endothelium after exposure to the flow of recalcified human blood in the presence or absence of PF4 and KKO and the GPIb-IX-blocking antibody AK2 or FcγRIIA-blocking antibody IV.3 or both. (B) In each case, binding of platelets is shown relative to injured endothelium with added KKO only as the comparator. Means ± 1 SEM are shown for N ≥ 5 separate injured studies. P values were determined using a 1-way ANOVA. See supplemental Figure 5 for additional controls.
Drug interactions with hPF4-VWF-HIT antibody complexes in vitro
Patients with HIT may develop thrombi while on prophylactic or therapeutic doses of heparin (plasma levels 0.1 and 0.3-0.7 U/mL, respectively38 ). Heparin binds to hPF4 with high affinity,30 which can disassociate PF4 from cell surface GAGs.2 Therefore, we asked whether hPF4-VWF-HIT antibody complexes are stable and would persist in the presence of clinically relevant levels of heparin. Heparin at ≥0.4 U/mL reduced hPF4 and subsequent KKO binding to VWF released from injured endothelium within 10 minutes (supplemental Figure 6A). However, PF4 is persistently released from platelets that are continuously being activated on the surfaces and trapped within developing thrombi, and PF4 levels in the vicinity of platelet-rich thrombi have been estimated at >100 µg/mL.39 To recapitulate this setting, we perfused solutions containing varying concentrations of hPF4 (0-100 µg/mL), either without or with 0.4 U/mL heparin added along with a fixed amount of KKO in the microfluidic photochemical injury system. As PF4 levels increased, binding of both PF4 and KKO to VWF strings increased even in the presence of heparin (supplemental Figure 6B). This suggests that hPF4 binding to VWF strings is preserved and hPF4-VWF-HIT antibody complexes can assemble in affected individuals even in the presence of continued heparin anticoagulation and may thereby be pathobiologically relevant even in the presence of ongoing heparin exposure as well as those who had their heparin stopped.
Therapeutic strategies to prevent VWF-related thrombosis have included metalloproteinase ADAMTS1328 and NAC27 by digesting high-molecular-weight VWF strings or preventing their formation, respectively. We asked whether PF4 and or HIT antibody would cause thrombi to develop resistance to ADAMTS13 or NAC assessed by the number and length of extruded VWF strings after endothelial injury in the microfluidic system. PF4 alone did not protect VWF strings from digestion by plasma concentrations of ADAMTS13 (0.7 µg/mL).40 PF4 plus KKO showed a trend toward resistance to ADAMTS13, but the effect did not reach statistical significance within the 10-minute timeframe examined (Figure 6A-B). Neither PF4 or PF4 plus KKO fully protect VWF from the effects of NAC. These studies suggest that approaches used to prevent or disrupt VWF-mediated thrombosis might be effective in HIT.

Inhibition of hPF4-VWF-HIT antibody complexes in vitro. Effect of ADAMTS13 or NAC on hPF4-VWF and hPF4-VWF-HIT antibody complexes. (A) Representative images from studies in Figure 3A after infusion of PF4 (25 µg/mL) into each lane. In some studies, KKO (10 µg/mL), ADAMTS13 (0.7 µg/mL), and/or NAC (10 mg/mL) was added as indicated. (B) The number of VWF strings present in the channel after exposure to the indicated solution is expressed relative to those present in channels following injury and exposure to hPF4 + KKO alone. Means ± 1 SEM are shown for N ≥ 5 studies. P values were determined by 1-way ANOVA.
Inhibition of hPF4-VWF-HIT antibody complexes in vitro. Effect of ADAMTS13 or NAC on hPF4-VWF and hPF4-VWF-HIT antibody complexes. (A) Representative images from studies in Figure 3A after infusion of PF4 (25 µg/mL) into each lane. In some studies, KKO (10 µg/mL), ADAMTS13 (0.7 µg/mL), and/or NAC (10 mg/mL) was added as indicated. (B) The number of VWF strings present in the channel after exposure to the indicated solution is expressed relative to those present in channels following injury and exposure to hPF4 + KKO alone. Means ± 1 SEM are shown for N ≥ 5 studies. P values were determined by 1-way ANOVA.
The role of VWF in the murine model of HIT
We then extended these studies to ask whether VWF-targeted therapeutics might be beneficial in vivo in part by preventing/dissociating intravascular immune complexes. We were unable to visualize multimeric VWF within thrombi with or without KKO induction in the murine HIT model, consistent with previous observations in other settings.41-43 Consequently, we used a functional evaluation to assess the role of VWF in HIT. As previously reported, injection of KKO into “HIT mice” reduced the time to occlusion after laser injury from ∼60 minutes to ∼30 minutes using the rose bengal photochemical carotid arterial injury model.3 Infusion of 0.8 mg/kg NAC, a well-tolerated dose44 shown to reduce thrombus size and stability in vitro,11 markedly prolonged the time to complete occlusion induced by KKO and increased blood flow through the injured carotid (Figure 7A-B). ADAMTS13, at a dose shown previously to attenuate microthrombi in a murine model of subarachnoid hemorrhage,28 significantly reduced the time to occlusion to that seen after infusion of TRA, although total blood flow did not improve significantly (Figure 7A-B).

The role of VWF in arterial thrombosis in murine models of HIT. (A-B) Rose bengal photochemical injury studies of the carotid artery under the indicated interventions. (C-D) Cremaster laser arteriole injury studies. (A) Time to occlusion (<10% of initial blood flow volume) and (B) AUC of each individual sample relative to the average AUC displaced by mice infused with TRA. AUC indicates the total flow through the vessel in HIT mice induced with KKO with or without exposure to NAC (0.8 mg/g) or ADAMTS13 (0.1 µg/g). Mice infused with TRA served as a negative control. Individual data points are shown. P values were determined by a 2-sided Mann-Whitney t test. (C) Fibrin accumulation and (D) platelet accumulation 15 minutes after antibody infusion ± ADAMTS13 compared with measurements before intervention. N = 3 mice (15 injuries) per arm. Mean and 1 SEM are shown. P values were determined by 1-way ANOVA.
The role of VWF in arterial thrombosis in murine models of HIT. (A-B) Rose bengal photochemical injury studies of the carotid artery under the indicated interventions. (C-D) Cremaster laser arteriole injury studies. (A) Time to occlusion (<10% of initial blood flow volume) and (B) AUC of each individual sample relative to the average AUC displaced by mice infused with TRA. AUC indicates the total flow through the vessel in HIT mice induced with KKO with or without exposure to NAC (0.8 mg/g) or ADAMTS13 (0.1 µg/g). Mice infused with TRA served as a negative control. Individual data points are shown. P values were determined by a 2-sided Mann-Whitney t test. (C) Fibrin accumulation and (D) platelet accumulation 15 minutes after antibody infusion ± ADAMTS13 compared with measurements before intervention. N = 3 mice (15 injuries) per arm. Mean and 1 SEM are shown. P values were determined by 1-way ANOVA.
We also examined the effects of ADAMTS13 on clot size using a cremaster laser arteriole injury model in HIT that we previously described in which an initial thrombus is allowed to form before antibody injection, and the growth of the thrombus 15 minutes after antibody infusion is noted.10 As previously observed, KKO, but not TRA infusions resulted in further growth of the thrombus (Figure 7C-D). Coinfusion of ADAMTS13 markedly decreased both the amount of fibrin and platelets incorporated into the clots after KKO, but not after TRA infusions (Figure 7C-D), further supporting the role of VWF in the prothrombotic nature of HIT.
Discussion
HIT is an intensely prothrombotic disorder that can have devastating clinical consequences even after withdrawal of heparin and institution of antithrombotic agents.9 One potential explanation for these findings is the involvement of antigenic complexes between PF4 and non-heparin haptens that activate prothrombotic pathways relatively insusceptible to inhibition of thrombin or FXa. Based on the timing of disease onset in previously unexposed individuals, the induction of HIT requires exposure to heparin, which leads to the formation of immune complexes composed of antibodies and PF4/heparin complexes. However, PF4 also forms antigenic complexes on cell surfaces with GAGs expressed by platelets, monocytes, endothelial cells, and neutrophils,2-5 leading to the elaboration of tissue factor3 and other prothrombotic sequelae.45 Antigenic complexes can also form between PF4 and diverse polyanions that may promote thrombus extension and propagate the disorder even when heparin has been stopped, including polyphosphates released from platelets7 and DNA from neutrophil-derived extracellular traps (NETs).4,45
Vascular injury is a known risk factor for HIT-associated arterial and venous thrombosis,46,47 and we have shown that HIT antibodies bind to the endothelial lining peri-thrombus in a murine model of HIT.10 We now show that part of the bound PF4 is to extended strings of VWF polymers released from injured cells in addition to binding to the endothelial surface GAGs. Like PF4 bound to various other polymeric molecules, including heparin, heparans, NETS, and polyphosphates, PF4-VWF strings are recognized by HIT antibodies. We, therefore, expect that the same epitope is being exposed on PF4 in each case.
For VWF, 3-dimensional reconstructions of events in a microfluidic channel suggest that PF4 binds along the surface of the elongated strings of VWF polymers where a specific binding site is exposed by shear and that are physically close to allow PF4 to cross aggregate these sites. It remains to be determined whether the PF4-binding sites are exposed as a result of shear forces within VWF monomeric units or whether shear brings preexisting binding sites into proximity and thereby promotes oligomerization of PF4 as reported for heparin.31,32 In fact, VWF is a glycoprotein decorated with a substantial number of O- and N-carbohydrates,48 many of which are modified by sialic acid that are important for its function and clearance.49 It is possible that VWF self-association brings these sialylated regions closer together. We previously showed that PF4 bound to thrombomodulin’s chondroitin sulfate side-chain exposes a HIT antigenic site.33 Binding of HIT antibody decreases the ability of thrombomodulin to activate protein C and contribute to the prothrombotic nature of HIT. Finally, the exact molar ratio of these shear-induced sites on the VWF strings to added PF4 is unclear because only a subset of acceptable sites may allow PF4 oligomerization and exposure of HIT antigenic sites.
The net result is the generation of regularly spaced antigenic sites that bind HIT antibodies and potentially form an IgG Fc-rich network that can initiate complement activation50 and that recruits and activates platelets, monocytes, and neutrophils expressing signal-transducing FcγRIIA.2-4 It is likely that as the response to injury spreads downstream, additional PF4 released from activated platelets encounters additional VWF released from activated endothelial cells, expanding the vascular surface expressing HIT antigen, binding of HIT antibody, and perpetuating prothrombotic pathways that are independent of heparin. Binding of PF4 to GAGs or to other components on the injured endothelium may precede or contribute to this feed-forward process, which can eventuate in formation of the large platelet-rich thrombi sometimes seen in HIT that first earned it the name of “white clot syndrome.”51
The involvement of VWF in HIT thrombus propagation as described in these studies may also contribute to the limitations of current antithrombotic therapy directed at thrombin and FXa52 while offering potential new targets for intervention. Platelet adhesion to the injured vasculature in the presence of PF4 and HIT antibody was blocked by inhibition of FcγRIIA and GPIb-IX. Both NAC, which reduces the disulfide bonds linking of individual monomers of VWF27 and ADAMTS13, which cleaves VWF multimers under shear,28 attenuated photochemical injury-induced carotid arterial occlusion induced by HIT antibody in a murine model. However, NAC had a much more pronounced effect on carotid occlusion in the murine model. This could be because of other hemostatic effect of NAC, a relatively nonspecific agent, or because ADAMTS13 has been rendered relatively ineffective, for example by oxidation of its target cleavage site on VWF.11 Whether similar approaches will prevent HIT thrombi and prove to be effective in the clinical setting remains to be established.
In summary, PF4 binds to sites interspersed along strings of VWF released from injured endothelium, allowing formation of HIT antigenic complexes. The antigenic targets are distributed periodically along the VWF strings under flow conditions that may enhance antibody avidity as well. PF4-VWF-HIT antibody complexes bind platelets through a pathway that involves both FcγRIIA and GPIb-IX. We propose that the release of VWF strings exposes a high-density epitope that fosters binding of PF4 and HIT antibodies and promotes binding and activation of platelets through their surface FcγRIIA and/or by activating complement. VWF cleavage by ADAMTS13 or depolymerization of VWF strings by NAC attenuates thrombotic arterial occlusion induced by HIT antibodies in a mouse model. These findings suggest that VWF may play a significant role in the propagation of HIT in space and in time. VWF may thereby play a role on the arterial side of the circulation analogous to the effect of NETs in the propagation of venular injuries.4 Drugs targeting VWF biology may be an effective new therapeutic approach in HIT, especially for arteriolar thrombi.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
For original data, please contact the corresponding author.
The online version of this article contains a data supplement.
Acknowledgments
The authors thank Adam Cuker at the Perelman School of Medicine at the University of Pennsylvania for providing the HIT and control plasma samples.
This work was supported by grants from the National Institutes of Health National Heart, Lung, and Blood Institute T32HL007971 (I.J.); P01 HL110860, R01 HL139448, and R01 HL142122 (M.P., L.R., and D.B.C.); P01 HL110860 and R01 HL136512 (G.M.A.); R01 HL112633 (J.A.L.); and R01 HL137991 (D.W.C.).
Authorship
Contribution: I.J. and A.S. performed the described studies and interpreted data; I.J. wrote the first draft of the paper and contributed to its editing; A.S. prepared the revised manuscript; V.H., G.T.K., and J.C. contributed to the studies; G.M.A., D.W.C., J.A.L., D.B.C., and L.R. provided experimental design, data interpretation, and assistance in editing the manuscript; and M.P. provided overall study guidance, interpretation, and manuscript preparation.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Lubica Rauova, Children’s Hospital of Philadelphia, 3615 Civic Center Blvd, ARC, Room 316F, Philadelphia, PA 19104; e-mail: lubica@email.chop.edu.
