

Key Points
ASA prevents infection-induced coagulopathy, improves microvascular perfusion, and reduces tissue damage while preserving immune response.

Abstract
Antiplatelet therapies have been proposed for the treatment of sepsis, a syndrome resulting from a dysregulated immune response and inappropriate activation of coagulation. Acetylsalicylic acid (ASA) may serve as a potential therapeutic strategy to prevent infection-induced coagulopathy and associated tissue damage. Using intravital microscopy, we found that Staphylococcus aureus infection induced neutrophil recruitment, platelet aggregation, and neutrophil extracellular trap (NET) release in the liver. Mice pretreated with ASA, or animals receiving ASA 3 hours postinfection, had significantly reduced platelet aggregation and NET release. Additionally, ASA-treated mice had reduced intravascular thrombin activity and microvascular occlusion as compared with untreated S aureus–infected mice. This inhibition of coagulation was accompanied by decreased levels of alanine aminotransferase and aspartate aminotransferase in the plasma, indicating less liver damage. Finally, bacterial loads (colony-forming units per milliliter) in liver, lung, and spleen were not different between groups, and the phagocytic capacity of Kupffer cells was preserved following ASA treatment. These results suggest that ASA may serve as a therapeutic approach to sepsis through its ability to reduce the deleterious action of immunothrombi while maintaining innate immune functions.
Introduction
Sepsis is a devastating condition resulting from a dysregulated immune response to infection, organ damage, shock, and death in 15% to 25% of cases.1,2 During this response, coagulation factors interact with immune cells and platelets, resulting in the formation of immunothrombi, complex structures of fibrin, platelets, and leukocytes.3 Immunothrombi are associated with critical complications of sepsis, including disseminated intravascular coagulation4 and aberrant coagulation resulting in microthrombi, organ dysfunction, and patient mortality.5,6
In addition to clotting, platelets also respond directly to infection,7-9 demonstrating a binary role of hemostasis and immunity. Platelets modulate host immunity, including the release of neutrophil extracellular traps (NETs).3,10-12 Although primarily involved in pathogen capture and clearance, NETs also activate thrombin, amplifying clot formation.13-15
Given the central role of platelets in inflammation and thrombosis, we assessed the impact of an antiplatelet drug on infection-induced coagulopathy. Acetylsalicylic acid (ASA; aspirin) is the most widely used drug globally; 20% of American adults and 49% of those older than 65 years of age take daily ASA prophylactically for thrombotic disorders.16,17 The current study utilizes intravital microscopy (IVM) to assess the impact of ASA on immunothrombosis in sepsis.
Study design
Mice
C57Bl/6 mice were purchased from The Jackson Laboratory. Experiments were approved by University of Calgary Animal Care Committee in compliance with the Canadian Council for Animal Care.
Experimental protocols
Staphylococcus aureus (USA300-2406) was cultured to mid-log phase in brain-heart infusion–chloramphenicol broth, washed, and resuspended in saline.15 A total of 2 × 107 colony-forming units (CFUs) were administered IV or intraperitoneally 6 hours prior to analysis. For treatment, 12.5 μg/g (low dose in mice18 ) of ASA (Sigma-Aldrich) was administered intraperitoneally 1 hour preinfection or 3 hours postinfection.
Antibodies and reagents
Fluorophore-labeled anti-Ly6G (clone 1A8), anti-CD49b, anti-CD11b (clone M1/70), and goat–anti-mouse neutrophil elastase (M18) were used. Other markers were Sytox Green (Life Technologies), 5-FAM/QXL 520 fluorescence resonance energy transfer substrate for thrombin (SensoLyte 520 Thrombin Activity Assay; AnaSpec, Inc), 1.00-µm Fluoresbrite YG microspheres (Polysciences, Inc), dihydroethidium (ThermoFisher), and fluorescein isothiocyanate–dextran.
IVM
Following anesthesia (10 mg/kg xylazine/ketamine hydrochloride), the liver was prepared as previously described15 and imaging was performed on an inverted Leica SP8 microscope (Leica Microsystems).
Image analysis
Analysis of NET staining, thrombin cleavage, and vessel occlusion was performed as described.15 Platelet aggregates were categorized by aggregate size. Neutrophil counts, bead capture, and reactive oxygen species (ROS) generation were manually counted.
Cell counts
Counting beads were added to red blood cell–lysed whole blood, and neutrophils were enumerated by flow cytometry. Platelet-neutrophil aggregates were identified as CD41+ neutrophils (Ly6G+).
Plasma proteins
Plasma alanine aminotransferase (ALT) and aspartate aminotransferase (AST) levels were measured by Calgary Laboratory Services. Thrombin-antithrombin (TAT) complexes were measured in plasma by enzyme-linked immunosorbent assay (Abcam).
CFUs
Organs were harvested, weighed, homogenized (GentleMACS Tissue Dissociator), filtered through a 100-μm strainer, centrifuged (5 minutes, 4°C, 2500 rpm), and resuspended in 1 mL of phosphate-buffered saline. Ten-fold dilutions were performed in chloramphenicol (1 mg/mL); 10 μL of each was plated on brain-heart-infusion agar plates and incubated at 37°C overnight.
Cytokine analysis
Plasma cytokine levels were determined using a MILLIPLEX MAP Mouse Cytokine/Chemokine Panel as per the manufacturer’s instructions (Millipore).
Statistical analysis
Data are presented as mean plus or minus standard error of the mean (SEM). One-way analysis of variance (ANOVA) with a post hoc Tukey test, the Student t test, or 2-way ANOVA with a post hoc Bonferroni test were used for statistical analysis as appropriate. Significance was set at P < .05.
Results and discussion
Control and ASA-treated mice were challenged with S aureus IV and imaged by IVM 6 hours postinfection. Infection significantly increased neutrophil recruitment and platelet aggregation in the liver. Neutrophil recruitment was significantly inhibited in ASA-pretreated mice whereas platelet aggregation was attenuated by either pretreatment or, to a lesser extent, by administration of ASA 3 hours postinfection (Figure 1A-C). These results are in line with previous studies that suggest neutrophil recruitment to the liver in response to bacterial infection is platelet independent.19
![ASA treatment reduces the host inflammatory response in the liver following systemic bacteremia. (A) Representative IVM images from an uninfected control animal (i) and at 6 hours after IV infection with 2 × 107 CFU of S aureus (ii). (B) Representative images of neutrophil recruitment to the liver of S aureus–infected control mice (i) and S aureus–infected mice pretreated with ASA (ii) or mice treated with ASA 3 hours after infection (iii). (iv) Quantification of the number of neutrophils per field of view (FOV; N = 4-9). (C) Representative images of platelet aggregation in the liver of control mice infected with S aureus (i), ASA-pretreated mice infected with S aureus (ii), or S aureus–infected mice treated with ASA 3 hours after infection (iii). (iv) Quantification of platelet aggregates of indicated sizes in animals from various treatment groups (N = 3-6). (D) Representative images of NET production (extracellular neutrophil elastase [NE] staining) in the liver of control mice infected with S aureus (i), ASA-treated mice infected with S aureus (ii), and mice treated with ASA 3 hours after infection (iii). (iv) Quantification of the area of NE staining (as a percentage of an FOV) within the liver of mice from various treatment groups (N = 3-6). (A-D) Labelling of cellsin vivo was achieved by IV injection of fluorescently-conjugated antibodies against CD49b (platelets; red), Ly6G (neutrophils; yellow), and neutrophil elastase (NE; cyan). Hepatocytes appear as dark green due to autofluorescence. Scale bar, 50 μm. (E) Quantification of the area of extracellular DNA staining (as a percentage of an FOV) within the liver of mice from various treatment groups (N = 3-5). Peripheral blood neutrophil (F) and platelet (G) counts and neutrophil-platelet aggregates (Ly6G+CD41+ cells) (H) from the different treatment groups (N = 4-5). Values represent the mean obtained from at least 5 FOVs in each animal. Data are represented as mean plus or minus SEM. Statistical analysis was conduced using a 1-way ANOVA with a post hoc Tukey test for panels Biv, Div, E-H and a 2-way ANOVA with a post hoc Bonferroni test for panel Civ. *P < .05, **P < .01, ***P < .001 vs control; #P < .05, ##P < .01, ###P < .001 vs S aureus.](https://ash.silverchair-cdn.com/ash/content_public/journal/blood/135/15/10.1182_blood.2019002783/1/m_bloodbld2019002783f1.png?Expires=1765003101&Signature=MyS0U3eb7ma0xS7CNszDjGK1hSyz49AaPzBolk4nflgUwg5V~sjMbC4sYeOorY7ycCVY5WKgsVeEPwp2NBdCJjO6HrBD4cX2qBVVVlfLpRCK1dhJRXcizxd0GAr65Td5JG~ZRhmNLvHkhVB70UVSvi9BTSXvao7iLFohsHQgdoUyTp2PsPL8y7ViTaihvUSEnJHceR9LU-UyMyYaaS3aDZSjjo3~2OmBHN0vmcrsrgO-Be9cBUUIOlRv8m-bNYByWjugJnpsLIId6exdn1kULYL-pOGX01cDWKOGrcnJ-w~xkP-uoebNhP6HTUepxl8TzruQzv-7~geltdleNMq84g__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
ASA treatment reduces the host inflammatory response in the liver following systemic bacteremia. (A) Representative IVM images from an uninfected control animal (i) and at 6 hours after IV infection with 2 × 107 CFU of S aureus (ii). (B) Representative images of neutrophil recruitment to the liver of S aureus–infected control mice (i) and S aureus–infected mice pretreated with ASA (ii) or mice treated with ASA 3 hours after infection (iii). (iv) Quantification of the number of neutrophils per field of view (FOV; N = 4-9). (C) Representative images of platelet aggregation in the liver of control mice infected with S aureus (i), ASA-pretreated mice infected with S aureus (ii), or S aureus–infected mice treated with ASA 3 hours after infection (iii). (iv) Quantification of platelet aggregates of indicated sizes in animals from various treatment groups (N = 3-6). (D) Representative images of NET production (extracellular neutrophil elastase [NE] staining) in the liver of control mice infected with S aureus (i), ASA-treated mice infected with S aureus (ii), and mice treated with ASA 3 hours after infection (iii). (iv) Quantification of the area of NE staining (as a percentage of an FOV) within the liver of mice from various treatment groups (N = 3-6). (A-D) Labelling of cellsin vivo was achieved by IV injection of fluorescently-conjugated antibodies against CD49b (platelets; red), Ly6G (neutrophils; yellow), and neutrophil elastase (NE; cyan). Hepatocytes appear as dark green due to autofluorescence. Scale bar, 50 μm. (E) Quantification of the area of extracellular DNA staining (as a percentage of an FOV) within the liver of mice from various treatment groups (N = 3-5). Peripheral blood neutrophil (F) and platelet (G) counts and neutrophil-platelet aggregates (Ly6G+CD41+ cells) (H) from the different treatment groups (N = 4-5). Values represent the mean obtained from at least 5 FOVs in each animal. Data are represented as mean plus or minus SEM. Statistical analysis was conduced using a 1-way ANOVA with a post hoc Tukey test for panels Biv, Div, E-H and a 2-way ANOVA with a post hoc Bonferroni test for panel Civ. *P < .05, **P < .01, ***P < .001 vs control; #P < .05, ##P < .01, ###P < .001 vs S aureus.
ASA treatment reduces the host inflammatory response in the liver following systemic bacteremia. (A) Representative IVM images from an uninfected control animal (i) and at 6 hours after IV infection with 2 × 107 CFU of S aureus (ii). (B) Representative images of neutrophil recruitment to the liver of S aureus–infected control mice (i) and S aureus–infected mice pretreated with ASA (ii) or mice treated with ASA 3 hours after infection (iii). (iv) Quantification of the number of neutrophils per field of view (FOV; N = 4-9). (C) Representative images of platelet aggregation in the liver of control mice infected with S aureus (i), ASA-pretreated mice infected with S aureus (ii), or S aureus–infected mice treated with ASA 3 hours after infection (iii). (iv) Quantification of platelet aggregates of indicated sizes in animals from various treatment groups (N = 3-6). (D) Representative images of NET production (extracellular neutrophil elastase [NE] staining) in the liver of control mice infected with S aureus (i), ASA-treated mice infected with S aureus (ii), and mice treated with ASA 3 hours after infection (iii). (iv) Quantification of the area of NE staining (as a percentage of an FOV) within the liver of mice from various treatment groups (N = 3-6). (A-D) Labelling of cellsin vivo was achieved by IV injection of fluorescently-conjugated antibodies against CD49b (platelets; red), Ly6G (neutrophils; yellow), and neutrophil elastase (NE; cyan). Hepatocytes appear as dark green due to autofluorescence. Scale bar, 50 μm. (E) Quantification of the area of extracellular DNA staining (as a percentage of an FOV) within the liver of mice from various treatment groups (N = 3-5). Peripheral blood neutrophil (F) and platelet (G) counts and neutrophil-platelet aggregates (Ly6G+CD41+ cells) (H) from the different treatment groups (N = 4-5). Values represent the mean obtained from at least 5 FOVs in each animal. Data are represented as mean plus or minus SEM. Statistical analysis was conduced using a 1-way ANOVA with a post hoc Tukey test for panels Biv, Div, E-H and a 2-way ANOVA with a post hoc Bonferroni test for panel Civ. *P < .05, **P < .01, ***P < .001 vs control; #P < .05, ##P < .01, ###P < .001 vs S aureus.
Given that neutrophil-platelet interactions stimulate NET production,11,15 and studies have indicated that ASA-treated platelets have a reduced capacity to induce NETs from human neutrophils,12 we examined the effect of ASA treatment on NETs in vivo. NETs were visualized by either anti–neutrophil elastase (NE) antibody or Sytox Green labeling of extracellular DNA. Infected mice had significantly increased NE and Sytox Green staining (Figure 1D-E) compared with controls. Treatment with ASA significantly reduced staining for NETs.
ASA effects were not limited to liver. Although the number of circulating neutrophils in blood was not altered (Figure 1F), ASA treatment attenuated the observed decrease in circulating platelets following infection (Figure 1G). Platelet consumption in infected mice was correlated with increased platelet-neutrophil aggregates in blood; these aggregates were also reduced in ASA-treated animals (Figure 1H). Plasma levels of tumor necrosis factor α, MIP 1β, MCP-1, interleukin 10, MIP2, and IP-10 were elevated at 6 hours after infection, returning toward baseline by 24 hours. Levels of MIP-1α increased after 24 hours whereas RANTES elevated early and remained high through 24 hours. Administration of ASA 3 hours postinfection reduced plasma levels of tumor necrosis factor α and IP-10 (supplemental Figure 1, available on the Blood Web site).
NETs potentiate thrombin generation and drive intravascular coagulation.15 To understand this process in ASA-treated animals, thrombin generation was visualized using an activatable fluorescent peptide as previously described.15 IV-infected mice generated significantly more thrombin than controls (Figure 2A), and ASA pretreatment significantly reduced thrombin generation. These observations were supported by measuring plasma TAT complexes where ASA treatment significantly reduced TAT levels (Figure 2B).
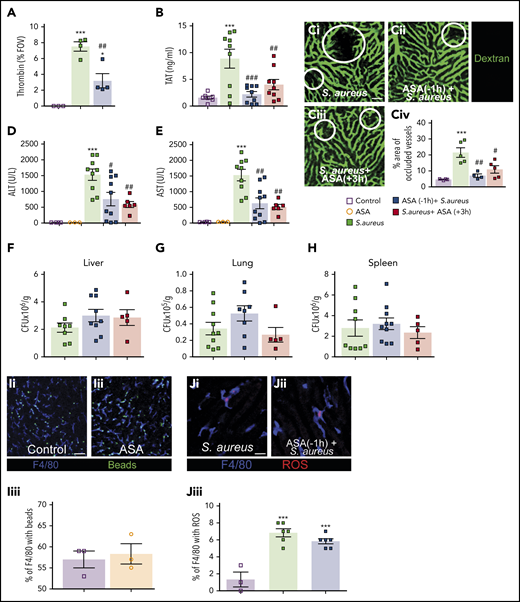
ASA treatment reduces infection-induced coagulopathy but did not affect pathogen clearance. (A) Quantitative analysis of thrombin probe fluorescence within the liver microcirculation of control mice, S aureus–treated mice, or S aureus–treated mice that received ASA 1 hour prior infection (N = 3-4). Values represent the percentage of an FOV occupied by the fluorescent probe. (B) TAT complex plasma levels in control mice, S aureus–infected mice, and mice treated with ASA 1 hour prior to, or 3 hours after, infection (N = 7-9). (C) Mice were injected with fluorescein isothiocyanate–dextran as a contrast agent to identify perfused liver vasculature in S aureus–infected mice (i), in S aureus–infected mice pretreated with ASA (ii), or mice treated with ASA 3 hours after infection (iii). (iv) Quantification of the percentage of an FOV occupied by occluded vessels (N = 4-5). Liver vasculature was stained by IV injection of fluorescently-conjugated dextran (green); scale bar, 50 μm. (D-H) ALT (D) and AST (E) were quantified in plasma of control and S aureus–infected mice with and without ASA treatment (N = 3-10). After 6 hours of infection, the liver (F), lungs (G), and spleen (H) were collected and bacterial CFUs were measured in infected and ASA-treated mice (N = 5-10). Values are presented as the number of CFUs per gram of tissue. (I) Representative images of sterile bead capture from the circulation by KCs in control (i) and ASA-treated mice (ii) (N = 3). Quantification of bead capture by KCs in control and ASA-treated mice (iii). (J) Representative images of ROS production by KCs in control infected (i) and ASA-pretreated mice infected with S aureus (ii). Quantification of ROS+ KCs in infected control and ASA-treated mice (iii) (N = 3-6). (I-J) Labelling of cells in vivo was achieved by IV injection of fluorescently-conjugated antibodies against F4/80 (Kupffer cells; blue). Flurescent polystyrene beads were injected IV (green) in panel I whereas dihydroethidium was injected IV in panel J. Dihydroethidium is converted to a precipitant in the presence of ROS (red); scale bar, 50 μm. Values represent the percentage of KCs per FOV that stain positive for ROS production. Values represent the mean obtained from at least 5 FOVs in each animal. Data are represented as mean plus or minus SEM. Statistical analysis was conduced using a 1-way ANOVA with a post hoc Tukey test for panels A-B, Civ, D-H, and Jiii and a Student t test for Iiii. ***P < .001 vs control; #P < .05, ##P < .01, ###P < .001 vs S aureus.
ASA treatment reduces infection-induced coagulopathy but did not affect pathogen clearance. (A) Quantitative analysis of thrombin probe fluorescence within the liver microcirculation of control mice, S aureus–treated mice, or S aureus–treated mice that received ASA 1 hour prior infection (N = 3-4). Values represent the percentage of an FOV occupied by the fluorescent probe. (B) TAT complex plasma levels in control mice, S aureus–infected mice, and mice treated with ASA 1 hour prior to, or 3 hours after, infection (N = 7-9). (C) Mice were injected with fluorescein isothiocyanate–dextran as a contrast agent to identify perfused liver vasculature in S aureus–infected mice (i), in S aureus–infected mice pretreated with ASA (ii), or mice treated with ASA 3 hours after infection (iii). (iv) Quantification of the percentage of an FOV occupied by occluded vessels (N = 4-5). Liver vasculature was stained by IV injection of fluorescently-conjugated dextran (green); scale bar, 50 μm. (D-H) ALT (D) and AST (E) were quantified in plasma of control and S aureus–infected mice with and without ASA treatment (N = 3-10). After 6 hours of infection, the liver (F), lungs (G), and spleen (H) were collected and bacterial CFUs were measured in infected and ASA-treated mice (N = 5-10). Values are presented as the number of CFUs per gram of tissue. (I) Representative images of sterile bead capture from the circulation by KCs in control (i) and ASA-treated mice (ii) (N = 3). Quantification of bead capture by KCs in control and ASA-treated mice (iii). (J) Representative images of ROS production by KCs in control infected (i) and ASA-pretreated mice infected with S aureus (ii). Quantification of ROS+ KCs in infected control and ASA-treated mice (iii) (N = 3-6). (I-J) Labelling of cells in vivo was achieved by IV injection of fluorescently-conjugated antibodies against F4/80 (Kupffer cells; blue). Flurescent polystyrene beads were injected IV (green) in panel I whereas dihydroethidium was injected IV in panel J. Dihydroethidium is converted to a precipitant in the presence of ROS (red); scale bar, 50 μm. Values represent the percentage of KCs per FOV that stain positive for ROS production. Values represent the mean obtained from at least 5 FOVs in each animal. Data are represented as mean plus or minus SEM. Statistical analysis was conduced using a 1-way ANOVA with a post hoc Tukey test for panels A-B, Civ, D-H, and Jiii and a Student t test for Iiii. ***P < .001 vs control; #P < .05, ##P < .01, ###P < .001 vs S aureus.
Intravascular coagulation contributes to vessel occlusion and organ damage.15 Vascular occlusion was identified by introducing a fluorescent contrast agent (labeled dextran) into circulation. The percentage of occluded vessels in infected mice (IV) was significantly higher than control mice (Figure 2C); however, ASA treatment prevented this vascular occlusion. Moreover, ASA treatment decreased both ALT and AST (markers of organ dysfunction/damage) in the plasma of infected mice (Figure 2D-E). Histological examination of the liver identifies small, focal, necrotic patches that are prevented in ASA-treated animals (supplemental Figure 2). These results suggest that targeting the NET-platelet-thrombin axis can diminish intravascular coagulation and associated end-organ damage.
As platelets also contribute to the immune response, we next determined the impact of ASA on host immunity. Critically, ASA treatment did not alter bacterial clearance in the liver, lungs, or spleen (Figure 2F-H). This finding is in agreement with a recent report that NET formation and immunothrombi are dispensable for limiting dissemination of bloodstream bacterial infections.20 It is known that platelets and Kupffer cells (KCs) work in concert to clear pathogens from the blood21 ; importantly, ASA did not impact the capacity of KCs to capture circulating microspheres from the bloodstream (Figure 2I). Production of ROS by phagocytes also contributes to pathogen destruction. Using dihydroethidium for direct assessment of ROS production in vivo,22 we determined that ASA treatment did not alter ROS generation by KCs following phagocytosis of S aureus (Figure 2J). These results indicate that bacterial clearance from the bloodstream is not negatively impacted by ASA treatment.
As sepsis is often caused by an initially localized infection, such as peritonitis, we repeated experiments by injecting bacteria intraperitoneally. As with IV infection, ASA reduced neutrophil recruitment, platelet aggregation, and NET generation following intraperitoneal S aureus infection (supplemental Figure 3A-D). Additionally, ASA did not alter blood neutrophil counts, but partially reverted platelet depletion, reduced platelet-neutrophil aggregate formation, TAT complex generation, and vessel occlusion induced by intraperitoneal infection (supplemental Figure 3E-I). Most importantly, ASA did not alter bacterial clearance in liver, lungs, or spleen of infected animals (supplemental Figure 3J-K). More studies considering other models of localized infection, such as pneumonia, and with different bacterial species are needed to support the potential efficacy of ASA in clinical sepsis.
ASA is the most widely used drug in the world. Millions of patients take a low dose of ASA daily as a prophylactic therapy for thrombotic disorders. Our findings suggest that the presence of a low dose of ASA prior to bacterial infection may offer some protection to the patient without compromising the host immune response. Moreover, our findings support the potential use of ASA as a therapeutic for coagulopathy in sepsis and provide unique insights into the specific cellular mechanisms altered by ASA administration in the context of infection. As ASA targets both cyclooxygenase 1 (COX-1) and COX-2, the current study does not positively identify platelet COX-1 as the specific therapeutic target, though we believe this model is reflective of the overall physiological response of patients treated with ASA. Importantly, our results are in agreement with clinical studies showing that low-dose ASA usage reduces hospital mortality in sepsis,23-25 potentially providing a therapeutic avenue to improve patient outcomes in this critical area.
For original data, please e-mail the corresponding author.
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Acknowledgments
The authors thank Braedon McDonald for helpful insights and comments regarding model development.
This work was supported by grants from the Canadian Foundation for Innovation, Alberta Innovates and Advanced Education, the Heart and Stroke Foundation of Canada (HSFC), and the Natural Sciences and Engineering Research Council (NSERC) (C.N.J.). C.N.J. was supported by the Canada Research Chairs Program. A.C. was supported by the Beverley Phillips Rising Star Postdoctoral Fellowship and the University of Calgary, Cumming School of Medicine Postdoctoral Scholar Program. R.P.D. was funded by the Canadian Liver Foundation and the University of Calgary Faculty of Graduate Studies Indigenous Graduate Award.
Authorship
Contribution: A.C. performed experiments, analyzed the data, prepared figures, and wrote the manuscript; R.P.D., H.G., and M.W.L. performed experiments and contributed to the generation of the manuscript; and C.N.J. conceived, designed, and supervised the research, analyzed data, and wrote the manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Craig N. Jenne, Department of Microbiology, Immunology, and Infectious Diseases, University of Calgary, 3330 Hospital Dr NW, Calgary, AB T2N 4N1, Canada; e-mail: cnjenne@ucalgary.ca.
