

In this issue of Blood, report that acetylsalicylic acid (ASA) helps prevent Staphylococcus aureus sepsis-induced intravascular coagulation and liver dysfunction by limiting neutrophil-mediated microvascular thrombosis.1 The authors show that ASA reduces neutrophil and platelet accumulation as well as the formation of procoagulant neutrophil extracellular traps (NETs) in the liver microvasculature of infected mice. Thus, ASA inhibits thrombin formation and platelet consumption in this model. Remarkably, the beneficial effects of ASA occur without impairing bacterial clearance when ASA was administered 1 hour before or 3 hours after infection. Importantly, no bleeding complications were reported in ASA-treated mice.
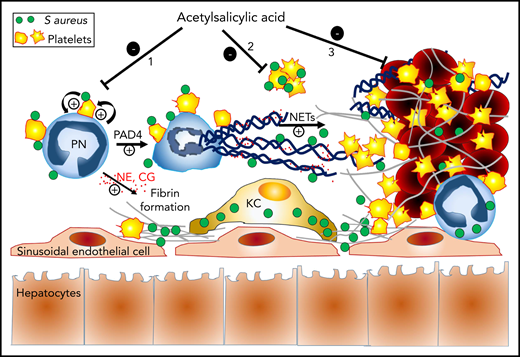
Proposed mechanisms of the protective action of ASA against S aureus sepsis-induced hepatic thrombosis. Once in the blood, circulating S aureus triggers an immunothrombotic cascade resulting in intravascular coagulation and occlusion of liver sinusoids. Circulating S aureus activates neutrophils and platelets through direct physical interactions (eg, via toll-like receptors and other pattern-recognition receptors) and secretion of immunomodulatory proteins, thus stimulating neutrophil-platelet reciprocal activating interactions. These interactions lead to the release of neutrophil elastase (NE) and cathepsin-G (CG) by neutrophils (PN), as well as to PAD4-dependent formation of procoagulant NETs. Together with nucleosomes, free and NETs-associated NE and CG enhance tissue factor- and factor XII-dependent coagulation, by degrading the tissue factor pathway inhibitor. Intravascular fibrin deposits and NETs help to capture and kill circulating S aureus, while at the same time, they promote liver injury and dysfunction by occluding sinusoids. Depending on the timing of administration, treatment with ASA can prevent thrombosis in sinusoids and associated liver injury by acting on at least 3 possible levels: (1) prevention of NETosis through inhibition of neutrophil-platelet interactions and reciprocal activation, (2) inhibition of S aureus–induced platelet aggregation, and (3) inhibition of platelet incorporation and aggregation on preformed prothrombotic NETs. The antibacterial action of “non-NETotic” neutrophils and other immune cells like Kupffer cells (KC) helps minimize the impact of ASA treatment on bacterial elimination.
Proposed mechanisms of the protective action of ASA against S aureus sepsis-induced hepatic thrombosis. Once in the blood, circulating S aureus triggers an immunothrombotic cascade resulting in intravascular coagulation and occlusion of liver sinusoids. Circulating S aureus activates neutrophils and platelets through direct physical interactions (eg, via toll-like receptors and other pattern-recognition receptors) and secretion of immunomodulatory proteins, thus stimulating neutrophil-platelet reciprocal activating interactions. These interactions lead to the release of neutrophil elastase (NE) and cathepsin-G (CG) by neutrophils (PN), as well as to PAD4-dependent formation of procoagulant NETs. Together with nucleosomes, free and NETs-associated NE and CG enhance tissue factor- and factor XII-dependent coagulation, by degrading the tissue factor pathway inhibitor. Intravascular fibrin deposits and NETs help to capture and kill circulating S aureus, while at the same time, they promote liver injury and dysfunction by occluding sinusoids. Depending on the timing of administration, treatment with ASA can prevent thrombosis in sinusoids and associated liver injury by acting on at least 3 possible levels: (1) prevention of NETosis through inhibition of neutrophil-platelet interactions and reciprocal activation, (2) inhibition of S aureus–induced platelet aggregation, and (3) inhibition of platelet incorporation and aggregation on preformed prothrombotic NETs. The antibacterial action of “non-NETotic” neutrophils and other immune cells like Kupffer cells (KC) helps minimize the impact of ASA treatment on bacterial elimination.
The current study by Carestia et al is one of several studies implicating platelet/neutrophil interactions in sepsis-induced intravascular coagulation and organ dysfunction. In 2007, 3 years after the discovery of NETs and of their antibacterial activity,2 Kubes’ group showed that sepsis-induced platelet/neutrophil interactions stimulated the formation of NETs, and provided the first in vitro evidence that NETs could form and trap bacteria under flow conditions.3 The ability of neutrophils and NETs to capture circulating bacteria was later confirmed in vivo in mouse models of sepsis.4,5 Results by Massberg et al further suggested that the procoagulant activity of neutrophils could also help limit bacterial dissemination by immobilization of bacteria onto intravascular fibrin deposits in liver sinusoids.4 Although these milestone studies pointed out the potential protective action of neutrophils and NETs during sepsis, several important studies that followed stressed the duality of NETs in this thromboinflammatory setting by demonstrating that NETs also contributed to sepsis-induced tissue injury and organ dysfunction.5-7 In fact, peptidylarginine deiminase-4-deficient mice (PAD4−/− mice), which are defective for NETosis, as well as mice treated with DNAse to eliminate intravascular NETs, showed reduced sepsis-induced thrombin formation and platelet aggregation, thus leading to improved microvascular perfusion and reduced organ damage.5-7 Strikingly, neither PAD4 deficiency nor DNase treatment affected bacteremia. Thus, although NETs can capture circulating bacteria in experimental sepsis, specific targeting of this component of neutrophil antimicrobial activity can be overcome, possibly due to redundancy with and/or compensation by other antibacterial host defense mechanisms. These experimental findings showing that targeting NETs can prevent thrombotic complications of sepsis support the hypothesis forwarded in 2006 by Brown et al, who, noting that accumulation and activation of neutrophils in the microvasculature were key features of sepsis-induced multiple organ failure, proposed that “targeting neutrophils and their interactions with blood vessel walls could be a worthwhile therapeutic strategy for sepsis.”8 Moreover, the deleterious role of NETs may contribute to the life-threatening syndrome of sepsis, with organ dysfunction caused by an exaggerated and dysregulated immune response to infection (see figure).
By targeting platelets with ASA, Carestia et al considered the central role of platelets in inflammation and thrombosis and thus chose to tackle the immunothrombotic cascade upstream of NETs. This strategy offers a prophylactic action to prevent intravascular coagulation, before NETs are formed. It is also worth noting that, despite clinical evidence that ASA, or antiplatelet therapy in general, could improve sepsis outcome,9 there is a dearth of preclinical and mechanistic data that explore and validate this beneficial effect. The results of Carestia et al provide important insights into the possible mechanisms underlying the protective action of ASA in sepsis.
Given the high mortality rate of sepsis and increasing antimicrobial resistance, there is a great need for new therapeutic options for sepsis. The work of Carestia et al could have clinical implications. Future studies may determine whether the efficacy and safety of ASA the authors describe here in models of sepsis induced by IV or intraperitoneal injection of S aureus also apply to other models and with different bacterial species. Platelets have indeed been shown to continuously prevent bleeding in various inflammatory situations, where they intervene to both inhibit bacterial growth and prevent bleeding at the primary site of infection.10
Conflict-of-interest disclosure: The author declares no competing financial interests.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal