

Key Points
AMPK deficiency decreases acetyl-CoA, histone acetylation, and BRD4 recruitment to chromatin in AML stem cells.
Inhibition of AMPK synergizes with BET inhibition to suppress AML.

Abstract
Altered metabolism fuels 2 hallmark properties of cancer cells: unlimited proliferation and differentiation blockade. Adenosine monophosphate–activated protein kinase (AMPK) is a master regulator of bioenergetics crucial for glucose metabolism in acute myeloid leukemia (AML), and its inhibition delays leukemogenesis, but whether the metabolic function of AMPK alters the AML epigenome remains unknown. Here, we demonstrate that AMPK maintains the epigenome of MLL-rearranged AML by linking acetyl-coenzyme A (CoA) homeostasis to Bromodomain and Extra-Terminal domain (BET) protein recruitment to chromatin. AMPK deletion reduced acetyl-CoA and histone acetylation, displacing BET proteins from chromatin in leukemia-initiating cells. In both mouse and patient-derived xenograft AML models, treating with AMPK and BET inhibitors synergistically suppressed AML. Our results provide a therapeutic rationale to target AMPK and BET for AML therapy.
Introduction
Emerging evidence has illuminated links between metabolism and epigenetics. S-adenosylmethionine, a cofactor of DNA and histone methyltransferases, is produced via the 1-carbon metabolism pathway,1 whereas α-ketoglutarate and acetyl-coenzyme A (CoA), cofactors for demethylases and acetyltransferases, respectively, are produced mainly by glucose metabolism.2,3 These epigenetic regulators are frequently found mutated or dysregulated in cancers, particularly in acute myeloid leukemia (AML), in which DNMT3A, TET2, and polycomb group proteins are among the most frequently mutated genes.4 The metabolic pathways that feed the epigenetic regulators are also frequently dysregulated in cancer.5,6 These metabolic and epigenetic alterations in cancers are being exploited for therapies; however, resistance to cancer monotherapies is almost inevitable.7 Mechanistic insights into how metabolism feeds epigenetics in cancer may reveal new strategies that target both processes to achieve durable therapeutic responses.
Adenosine monophosphate–activated protein kinase (AMPK) is a master regulator of cellular energetics that activates catabolism (such as glycolysis and mitochondrial biogenesis) and inhibits anabolism upon metabolic stress.8 AMPK has a context-dependent role in tumorigenesis, suppressing or promoting tumorigenesis in mouse models of cancers.9-13 We and others have shown in several leukemia models that AMPK promotes leukemogenesis by maintaining metabolic homeostasis, highlighting the importance of the metabolic pathways governed by AMPK in leukemia.9,12-14 Whether the metabolic homeostasis governed by AMPK modulates AML epigenome remains unclear, and understanding this interaction may broaden the potential of targeting AMPK for therapies.
Histone acetylation governed by acetyl-CoA homeostasis is tightly linked to cell proliferation. Nutrients increase acetyl-CoA and histone acetylation, thereby inducing growth-promoting genes and cell division.15-17 Acetylated histones are recognized by histone acetylation readers, including the Bromodomain and Extra-Terminal domain (BET) or the Yaf9, ENL, AF9, Taf14, and Sas5 (YEATS) domain proteins.18,19 BET and YEATS then recruit histone modifiers, chromatin remodelers, P-TEFb, or the Mediator complex to activate gene expression.20,21 Because BET proteins are critical for transcriptional regulation in AML and are being pursued as a potential therapy,22,23 deeper understanding of how BET recruitment is regulated may lead to combination therapies designed to enhance the efficacy of BET inhibitors.
Here, we identified AMPK as a link between acetyl-CoA homeostasis and BET protein recruitment to chromatin in MLL-rearranged AML. AMPK deletion reduced acetyl-CoA and histone acetylation levels; however, extracellular supply of acetate restored levels of both. AMPK deletion depleted polyacetylated histones, which have a high affinity for BET binding. As a consequence, AMPK deletion reduced the occupancy of the BET protein BRD4 to superenhancers regulating leukemogenic genes, thus reducing their expression. In murine and human patient-derived xenograft models, genetic and pharmacological approaches to inhibit both AMPK and BRD4 synergistically suppressed AML. Our results unveil a unique vulnerability of AML, safeguarded by metabolic regulator AMPK and epigenetic reader BRD4, that can be therapeutically targeted.
Methods
Mice
C57/BL6 mice were used for murine AML transplantation. Mx1-Cre; Prkaa1fl/fl; Prkaa2fl/fl mice were described previously.13 Brd4fl/fl mice were obtained from EUCOMM and backcrossed to C57BL/6 mice. A total of 0.5 μg/g body mass of polyinosinic:polycytidylic acid (poly[I:C]) was injected every other day for 14 days to induce Mx1-Cre expression. NSG-SGM324 mice were irradiated at 250 cGy before transplantation of human AML cells. All procedures regarding the use of mice were approved by Baylor College of Medicine Institutional Animal Care and Use Committees and the Institutional Review Board committee. Human t(9;11)-rearranged AML25 cells were collected under LAB01-473 protocol approved by the MD Anderson Institutional Review Board committee.
Retroviral transduction
MLL-AF9 or MOZ-TIF2 oncogenes in pMIG (MSCV-IRES-GFP) retroviral vectors were used for transduction. Thirty days after deleting AMPK with poly(I:C) administration, lineage−c-kit+Sca-1+ (LSK) cells were spin infected, as previously described.13
CRISPR/Cas9-mediated gene editing in murine AML cells
sgRNA against Rosa26, Acss1, Acss2, and Acss3 were designed according to CRISPR DESIGN (http://crispr.mit.edu/). Cells were electroporated as previously described.26 Sequences of the sgRNA and detailed methods are provided in supplemental Methods, available on the Blood Web site.
RNA-seq
cDNA libraries from sorted cells were generated using the Smart-seq2 protocol27 and sequenced on a NextSeq platform. Raw sequencing data were converted to fastq files and aligned to the reference genome (mm10), using STAR. DESeq2 was used for data processing, normalization, and differential expression analysis.
ChIP-seq
Native chromatin immunoprecipitation (ChIP)-seq was performed as described before.28 In brief, 50 000 leukemic-granulocyte monocyte progenitors (L-GMPs) were sorted, and native chromatin obtained by MNase treatment was incubated with antibodies listed in supplemental Table 3. Library preparation was performed with a NEB Ultra II kit following the manufacturer’s protocols (New England Biolabs, Ipswich, MA) and sequenced with a NextSeq platform. Reads were mapped to the mm10 genome, using Bowtie2. Homer was used to identify peaks. Superenhancers were mapped using the ROSE2 software package.29
Statistics
Statistics were determined with a paired Student t test, analysis of variance, or log-rank test with Prism software (GraphPad). Group data always represent mean ± standard deviation.
Results
A metabolite screen identifies acetate to rescue defective proliferation of AMPK-deficient AML
We previously generated a MLL-AF9 driven murine AML model in which the 2 genes encoding the kinase subunits of AMPK, Prkaa1 and Prkaa2, are conditionally deleted before retroviral transduction.13 Mx1-Cre; Prkaa1fl/fl; Prkaa2fl/fl (AMPK KO) and Prkaa1fl/fl; Prkaa2fl/fl (AMPK WT) mice were treated with poly(I:C) to induce Cre, and lineage−c-kit+Sca-1+ progenitor cells were transduced with MLL-AF9 retrovirus and transplanted. Metabolomic profiling of WT and AMPK KO AML cells isolated from recipient mice showed that AMPK deletion significantly decreased the amounts of relevant metabolites, including 2-phosphoglycerate (2-PG)/3-PG, lactate, citrate, succinate, acetyl-CoA, and ATP (Figure 1A), consistent with its known role in promoting glycolysis and the tricarboxylic acid cycle.
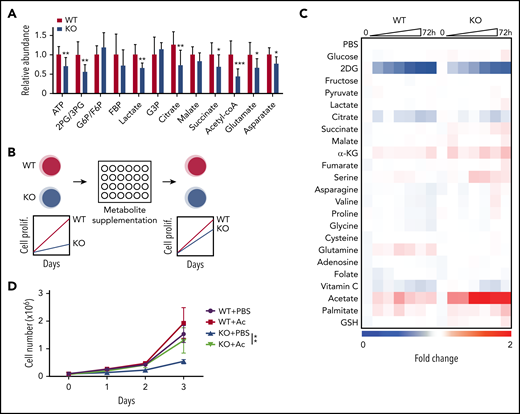
A metabolite screen identifies acetate to promote proliferation of AMPK-deficient AML cells. (A) Metabolomics analysis with mass spectrometry revealed that AMPK-deficient AML cells had decreased metabolites in glycolysis and tricarboxylic acid cycle, as well as amino acids and nucleotides pathways (n = 9). (B) A schematic of a screening strategy using metabolite supplementation to rescue the defective proliferation of AMPK-deficient AML cells. (C) Results from the metabolite screening identified acetate to rescue AMPK-deficient AML. Wild-type (WT) or AMPK-deficient (KO) AML cells were cultured in the presence of the indicated compound for 72 hours, and cell proliferation was monitored by a 3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium (MTS) assay. 2-deoxyglucose (2DG), an inhibitor of glycolysis, was included as a control. Fold changes in signals were normalized against those from wells supplemented with phosphate-buffered saline. (D) WT or AMPK KO AML cells were cultured with (Ac) or without (phosphate-buffered saline) 2 mM acetate supplementation, and cell proliferation was monitored (n = 3). All data represent mean ± standard deviation; *P < .05; **P < .01; ***P < .001 by Student’s t-test.
A metabolite screen identifies acetate to promote proliferation of AMPK-deficient AML cells. (A) Metabolomics analysis with mass spectrometry revealed that AMPK-deficient AML cells had decreased metabolites in glycolysis and tricarboxylic acid cycle, as well as amino acids and nucleotides pathways (n = 9). (B) A schematic of a screening strategy using metabolite supplementation to rescue the defective proliferation of AMPK-deficient AML cells. (C) Results from the metabolite screening identified acetate to rescue AMPK-deficient AML. Wild-type (WT) or AMPK-deficient (KO) AML cells were cultured in the presence of the indicated compound for 72 hours, and cell proliferation was monitored by a 3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium (MTS) assay. 2-deoxyglucose (2DG), an inhibitor of glycolysis, was included as a control. Fold changes in signals were normalized against those from wells supplemented with phosphate-buffered saline. (D) WT or AMPK KO AML cells were cultured with (Ac) or without (phosphate-buffered saline) 2 mM acetate supplementation, and cell proliferation was monitored (n = 3). All data represent mean ± standard deviation; *P < .05; **P < .01; ***P < .001 by Student’s t-test.
As metabolites reduced by AMPK deletion may be at least partially responsible for the defective leukemogenic potential, we next tested whether exogenous supply of metabolites that feed into the attenuated metabolic pathways could restore proliferation in AMPK KO AML cells (Figure 1B). WT and AMPK KO AML cells were cultured in the presence of 22 selected metabolites involved in glycolysis, tricarboxylic acid cycle, amino acids, and redox regulators, and cell proliferation was monitored by a 3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium (MTS) assay (Figure 1C). Acetate significantly rescued AMPK KO cells in this screen (Figure 1C), which was independently confirmed by quantifying cell proliferation (Figure 1D). Before acetate treatment, WT AML cells exhibited blast-like morphologies with large nucleus, whereas AMPK KO AML cells exhibited differentiated morphologies, increased expression of a myeloid marker Gr-1, and decreased expression of Myc, Meis1, and Hoxa9, suggesting that they are undergoing myeloid differentiation (Figure 2A-B). Interestingly, acetate treatment of AMPK KO AML cells reduced Gr-1 expression and induced leukemia-associated genes, and slightly increased the frequency of blast-like cells (Figures 2A-B). These results suggest that acetate metabolism prevents myeloid differentiation of AMPK KO AML cells.
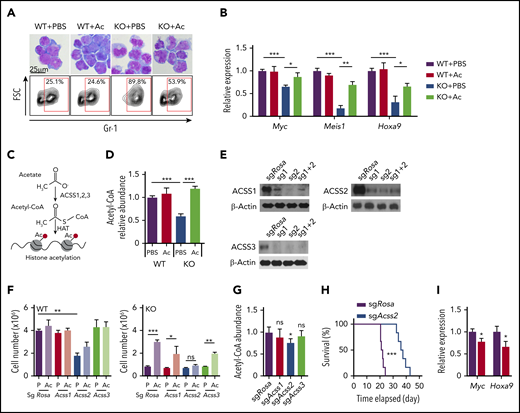
Acetate prevents myeloid differentiation of AMPK-deficient AML cells through Acss2-mediated production of acetyl-CoA. (A) Giemsa staining (upper panels) and flow cytometry to detect Gr-1 expression (lower panels) of AML cells cultured with or without acetate (n = 3). (B) Quantitative PCR to detect Myc, Meis1, and Hoxa9 expression in AML cells cultured with or without acetate (n = 3). (C) A schematic illustration of acetate conversion to acetyl-CoA by Acss. (D) Measurement of intracellular acetyl-CoA in AMPK KO AML cells by acetate supplementation (n = 3). (E) Immunoblotting of ACSS1, ACSS2, and ACSS3 after deleting the corresponding genes in MLL-AF9-induced AML cells with CRISPR/Cas9. Two sgRNA were used individually or in combination. (F) WT and AMPK KO AML cells were ablated for Rosa26 (control), Acss1, Acss2, or Acss3 and cultured in the presence (Ac) or absence (P: phosphate-buffered saline) of acetate, and cell proliferation was monitored (n = 3). (G) Acetyl-coA levels of MLL-AF9-induced AML cells after deleting Rosa26, Acss1, Acss2, or Acss3 (n = 3). (H) Survival of mice after transplanting Rosa26- or Acss2-deleted AML cells (n = 6). (I) Quantitative PCR of Myc and Hoxa9 transcripts in Acss2-deleted AML cells (red bars) compared with Rosa26-deleted cells (black bars) (n = 3). All data represent mean ± standard deviation. *P < .05; **P < .01; ***P < .001 by Student’s t-test or 1-way ANOVA, except for comparison of the survival curves, in which the significance was accessed by a log-rank test.
Acetate prevents myeloid differentiation of AMPK-deficient AML cells through Acss2-mediated production of acetyl-CoA. (A) Giemsa staining (upper panels) and flow cytometry to detect Gr-1 expression (lower panels) of AML cells cultured with or without acetate (n = 3). (B) Quantitative PCR to detect Myc, Meis1, and Hoxa9 expression in AML cells cultured with or without acetate (n = 3). (C) A schematic illustration of acetate conversion to acetyl-CoA by Acss. (D) Measurement of intracellular acetyl-CoA in AMPK KO AML cells by acetate supplementation (n = 3). (E) Immunoblotting of ACSS1, ACSS2, and ACSS3 after deleting the corresponding genes in MLL-AF9-induced AML cells with CRISPR/Cas9. Two sgRNA were used individually or in combination. (F) WT and AMPK KO AML cells were ablated for Rosa26 (control), Acss1, Acss2, or Acss3 and cultured in the presence (Ac) or absence (P: phosphate-buffered saline) of acetate, and cell proliferation was monitored (n = 3). (G) Acetyl-coA levels of MLL-AF9-induced AML cells after deleting Rosa26, Acss1, Acss2, or Acss3 (n = 3). (H) Survival of mice after transplanting Rosa26- or Acss2-deleted AML cells (n = 6). (I) Quantitative PCR of Myc and Hoxa9 transcripts in Acss2-deleted AML cells (red bars) compared with Rosa26-deleted cells (black bars) (n = 3). All data represent mean ± standard deviation. *P < .05; **P < .01; ***P < .001 by Student’s t-test or 1-way ANOVA, except for comparison of the survival curves, in which the significance was accessed by a log-rank test.
Cells convert acetate to acetyl-CoA via acyl-CoA synthetase short chain family member 1 (ACSS1) and ACSS3, localized to mitochondrion, or ACSS2, localized to the cytoplasm and nucleus (Figure 2C).30 We postulated that AMPK KO AML cells use acetate to produce acetyl-CoA and promote proliferation. Acetyl-CoA levels in AMPK KO cells, both in the bulk of AML cells as well as in the leukemia-initiating cells (LICs), termed L-GMPs in this model (supplemental Figure 1A),31-33 were reduced, and acetate supplementation increased the acetyl-CoA pool of AMPK KO AML cells (Figure 2D). We then deleted Acss1, Acss2, or Acss3, using a CRISPR/Cas9-based method,26,34 and confirmed deletion by immunoblotting, T7 endonuclease assay, and sequencing (Figure 2E; supplemental Figure 1B-G). Deletion of Acss1 or Acss3 had negligible effects on cell proliferation in both WT and AMPK KO AML cells, regardless of the presence of acetate (Figure 2F). In contrast, Acss2 deletion significantly reduced proliferation of WT AML cells and reduced acetyl-CoA levels (Figure 2F-G). Moreover, Acss2 deletion rendered AMPK KO cells insensitive to acetate (Figure 2F). Thus, Acss2 mediates the effects of acetate in promoting AML cell proliferation.
We then tested whether Acss2 regulates leukemogenesis in vivo. We deleted Acss2 by CRISPR from freshly isolated AML cells and transplanted the cells into irradiated recipient mice. Mice transplanted with Acss2-deleted AML cells had a significantly delayed onset of leukemogenesis compared with control-edited AML (21 vs 38 days; Figure 2H) and reduced the expression of Myc and Hoxa9 (Figure 2I). Together, these results establish that acetyl-CoA homeostasis governed by Acss2 regulates leukemic gene expression and leukemogenesis.
AMPK maintains histone acetylation in LICs
Recent studies have established a link between cellular acetyl-CoA levels and histone acetylation.15-17,35-38 Immunoblotting with an acetyl-lysine antibody revealed that AMPK deletion reduced acetylation of nuclear polypeptides of approximately 10 to 20 kDa in size, suggesting that AMPK deletion decreases histone acetylation in AML (supplemental Figure 2A). Indeed, immunoblotting with pan-acetyl-H3 and pan-acetyl-H4 antibodies showed reduced acetylation of H3 and H4 in AMPK KO AML cells (Figure 3A). Further, we found significant reduction of H3K9ac, H3K27ac, H4K5ac, H4K8ac, and H4K12ac in AMPK KO AML cells, whereas acetylation of H3K14ac, H3K18ac, p53, α-tubulin, or CBP was not appreciably affected by AMPK deletion (Figures 3A-B). L-GMPs exhibited a similar reduction in histone acetylation (Figure 3A). Acss2-deleted cells also showed decreased histone acetylation (supplemental Figure 2B). In addition, acetate supplementation restored the acetylation levels of histones in AMPK KO AML cells (supplemental Figure 2C). These results establish that AMPK is critical to maintain histone acetylation levels in LICs.
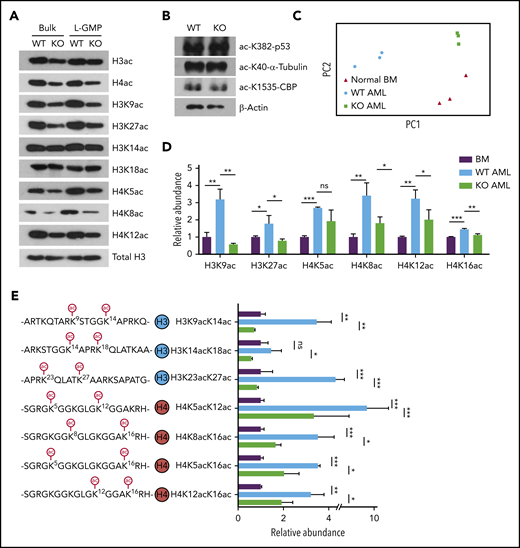
AMPK KO AML cells markedly downregulate histones that are acetylated on multiple lysine residues. (A-B) Immunoblotting with WT or AMPK KO bulk (GFP+) AML cells or the L-GMP population to detect acetylated histones (A) or acetylated p53, α-tubulin, or CBP (B). (C) Principal component analysis of the histone proteoform data set obtained from normal bone marrow (BM) cells, WT AML cells, and AMPK KO AML cells. (D) Acetylation status of individual lysine residues from the histone proteoform analysis, normalized against normal BM cells (n = 3). (E) The abundance of histone proteoforms that were acetylated on 2 lysine residues (n = 3). All data represent mean ± standard deviation. *P < .05; **P < .01; ***P < .001 by 1-way ANOVA.
AMPK KO AML cells markedly downregulate histones that are acetylated on multiple lysine residues. (A-B) Immunoblotting with WT or AMPK KO bulk (GFP+) AML cells or the L-GMP population to detect acetylated histones (A) or acetylated p53, α-tubulin, or CBP (B). (C) Principal component analysis of the histone proteoform data set obtained from normal bone marrow (BM) cells, WT AML cells, and AMPK KO AML cells. (D) Acetylation status of individual lysine residues from the histone proteoform analysis, normalized against normal BM cells (n = 3). (E) The abundance of histone proteoforms that were acetylated on 2 lysine residues (n = 3). All data represent mean ± standard deviation. *P < .05; **P < .01; ***P < .001 by 1-way ANOVA.
AMPK is required to recruit BET to chromatin
Our data revealed that loss of AMPK reduced histone acetylation in AML cells; however, whether this reduction specifically affects BET recruitment to chromatin remains unknown. The BET bromodomains (BDs) have a binding preference toward histone H3 and H4 peptides that are acetylated on multiple sites. BD1 of BRD2 or BRDT binds diacetylated H4K5acK12ac or H4K5acK8ac with higher affinity than monoacetylated H4, respectively.39-41 BD1 and BD2 of BRD4 bind preferentially to polyacetylated histone H4 and H3, respectively.42 We hypothesized that AMPK deletion reduces the amount of polyacetylated histone H3 and H4 that recruit BET proteins to chromatin.
We took a top-down mass spectrometric approach to determine the level of polyacetylated histones in WT and AMPK KO AML.43,44 Histones from AML cells were analyzed by mass spectrometer without protease digestion to preserve the information of the combinations of modifications within a single polypeptide, referred to as proteoforms.45 Principal component analysis showed that AMPK deletion has profound effects on histone proteoforms. The histone proteoforms between normal bone marrow cells and primary AML cells were significantly different, and the proteoform profiles of AMPK KO AML cells clustered separately from WT AML cells (Figures 3C; supplemental Figure 2E). The overall acetylation levels of H3K9ac, H3K27ac, H4K5ac, H4K8ac, and H4K12ac were all significantly reduced in AMPK KO AML cells, confirming the immunoblotting results (Figure 3D; supplemental Figure 2D). Importantly, histones that are polyacetylated were significantly reduced by AMPK deletion, such as diacetylated histone H4 proteoforms H4K5acK12ac, H4K8acK16ac, H4K5acK16ac, and H4K12acK16ac (Figure 3E; supplemental Table 1). We also found almost all combinations of diacetylation on histone H3 were reduced in AMPK KO cells, including H3K9acK14ac and H3K14acK18ac, which bind strongly to BRD4-BD2 (Figure 3E).42 These diacetylated histone H3 and H4 proteoforms were more abundant in AML cells compared with normal bone marrow cells, suggesting that leukemogenesis is associated with an overall increase in diacetylated histone proteoforms (supplemental Figure 2F). Thus, AMPK may regulate gene expression in AML by maintaining the polyacetylated histone proteoforms that have high affinity for BET proteins.
We then examined the gene expression and genome-wide histone acetylation profiles of WT and AMPK KO L-GMPs by RNA-seq and ChIP-seq. Gene set enrichment analysis of WT and AMPK KO L-GMPs revealed that the MLL-AF9, Myc, and BET target genes (sensitive to a BET inhibitor JQ146 ) were all downregulated in L-GMPs after AMPK deletion (Figure 4A). Specifically, Hoxa9, Meis1, and Myc were significantly reduced in AMPK KO L-GMPs (Figure 4B). Motif analysis of the gene expression profile revealed decreased expression of genes with Myc or ETS motifs in AMPK KO L-GMPs compared with WT (Figure 4C). ChIP-seq showed a widespread reduction in H3K9ac, H3K27ac, and H4K8ac signals around the transcriptional start sites in AMPK KO L-GMPs (Figure 5A). Because H3K27ac and H4K8ac also mark active enhancers, some of which are bound by BRD4,46 we examined the enrichment of H3K27ac and H4K8ac on enhancer elements defined by the enrichment of H3K27ac or BRD4. We found that H3K27ac and H4K8ac signals were enriched on enhancer elements of L-GMPs and were significantly reduced by AMPK deletion, suggesting that AMPK positively regulates enhancer activity in L-GMPs (Figure 5B-C; supplemental Figure 3A).
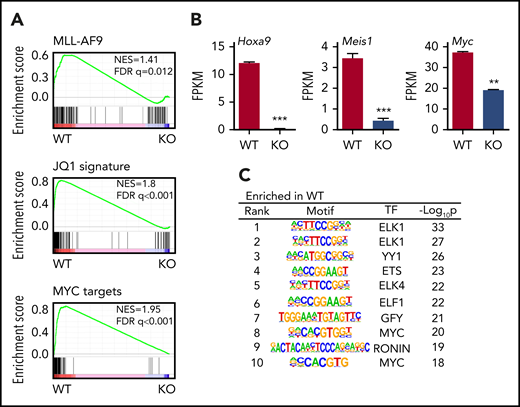
AMPK regulates leukemic gene networks in LICs. (A) Gene set enrichment analysis revealed reduced expression of MLL-AF9 target genes, JQ1 suppressed genes, and Myc target genes in AMPK KO L-GMPs. (B) Expression of Hoxa9, Meis1, and Myc as shown by the fragments per kilobase of transcript per million mapped reads (FPKM). (n = 3). (C) Gene set enrichment analysis, using the motif gene sets identified Myc motif as the most enriched motif in WT L-GMPs gene expression profile. All data represent mean ± standard deviation. *P < .05; **P < .01; ***P < .001 by Student’s t-test.
AMPK regulates leukemic gene networks in LICs. (A) Gene set enrichment analysis revealed reduced expression of MLL-AF9 target genes, JQ1 suppressed genes, and Myc target genes in AMPK KO L-GMPs. (B) Expression of Hoxa9, Meis1, and Myc as shown by the fragments per kilobase of transcript per million mapped reads (FPKM). (n = 3). (C) Gene set enrichment analysis, using the motif gene sets identified Myc motif as the most enriched motif in WT L-GMPs gene expression profile. All data represent mean ± standard deviation. *P < .05; **P < .01; ***P < .001 by Student’s t-test.
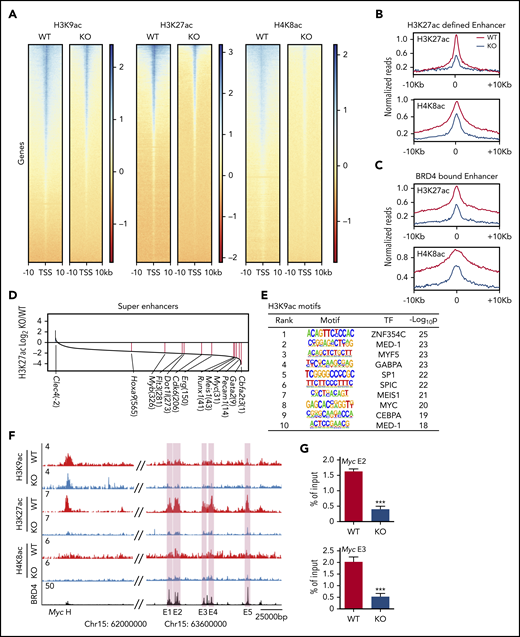
AMPK maintains histone acetylation at Brd4 binding sites. (A) Density plots of H3K9ac, H3K27ac, and H4K8ac ChIP-seq signals centered around the transcription start site. (B-C) Meta profiles of the indicated ChIP-seq dataset centered around the peak apex of 10 615 enhancer elements defined by H3K27ac signal (B) and 6522 enhancer elements defined by BRD4 occupancy based on Roe et al46 (C). (D) A waterfall plot depicting the differences in H3K27ac signals at superenhancers between WT and AMPK KO L-GMPs. The numbers in parentheses indicate the rankings of each gene. (E) Motif analysis of H3K9ac ChIP-seq data set showing the transcription factor motifs enriched in WT L-GMPs compared with AMPK KO L-GMPs. (F) ChIP-seq occupancy profiles of H3K9ac, H3K27ac, and H4K8ac of both AMPK-proficient and AMPK-deficient L-GMPs, as well as the BRD4 profile46 at the Myc locus. Myc enhancer elements E1-E5 are shown in pink shades. (G) ChIP-quantitative polymerase chain reaction to detect BRD4 occupancy at 2 Myc enhancers (E2 and E3) with WT and AMPK KO AML (n = 3). All data represent mean ± standard deviation. *P < .05; **P < .01; ***P < .001 by 1-way ANOVA.
AMPK maintains histone acetylation at Brd4 binding sites. (A) Density plots of H3K9ac, H3K27ac, and H4K8ac ChIP-seq signals centered around the transcription start site. (B-C) Meta profiles of the indicated ChIP-seq dataset centered around the peak apex of 10 615 enhancer elements defined by H3K27ac signal (B) and 6522 enhancer elements defined by BRD4 occupancy based on Roe et al46 (C). (D) A waterfall plot depicting the differences in H3K27ac signals at superenhancers between WT and AMPK KO L-GMPs. The numbers in parentheses indicate the rankings of each gene. (E) Motif analysis of H3K9ac ChIP-seq data set showing the transcription factor motifs enriched in WT L-GMPs compared with AMPK KO L-GMPs. (F) ChIP-seq occupancy profiles of H3K9ac, H3K27ac, and H4K8ac of both AMPK-proficient and AMPK-deficient L-GMPs, as well as the BRD4 profile46 at the Myc locus. Myc enhancer elements E1-E5 are shown in pink shades. (G) ChIP-quantitative polymerase chain reaction to detect BRD4 occupancy at 2 Myc enhancers (E2 and E3) with WT and AMPK KO AML (n = 3). All data represent mean ± standard deviation. *P < .05; **P < .01; ***P < .001 by 1-way ANOVA.
Because BRD4 binds superenhancers with high levels of H3K27ac and activates the expression of proto-oncogenes,46,47 we compared the levels of H3K27ac modification in superenhancers using the ROSE2 (Rank Ordering of Super-Enhancers 2) algorithm.29 Most (771 of 781) of the superenhancers had higher levels of H3K27ac enrichment in WT L-GMPs (Figure 5D). The top superenhancers with enriched H3K27ac in WT L-GMPs were those in close proximity to genes encoding leukemogenic transcription factors such as Cbfa2t3, Gata2, Myc, Meis1, and Runx1. Other upregulated superenhancers found in WT L-GMPs were associated with Erg, Cdk6, Dot1l, Myb, and Hoxa9, all of which are known to promote leukemogenesis (Figure 5D). Motifs analysis of the H3K9ac-, H3K27ac-, and H4K8ac-enriched peaks identified enrichment of these transcription factors in WT compared with AMPK KO L-GMPs (Figure 5E; supplemental Figure 3B). Accordingly, acetylated histones, particularly H3K27ac, were enriched at the enhancer elements of Myc in L-GMPs, which were reduced by deleting AMPK (Figure 5F). In contrast, the few superenhancers enriched in AMPK KO L-GMPs included those associated with the Clec4 cluster involved in inflammation and highly expressed in mature myeloid cells (supplemental Figure 3E). Thus, AMPK deletion reduces histone acetylation on promoters and superenhancers within L-GMPs, reducing the expression of key leukemogenic transcription factors.
We then tested whether reduced histone acetylation on gene regulatory elements attenuates BRD4 recruitment. Deletion of AMPK reduced recruitment of BRD4 to Myc, Meis1, and Erg loci (Figure 5G; supplemental Figure 3C-D), which was partially rescued by acetate supplementation (supplemental Figure 4A). Deletion of Acss2 by CRISPR also decreased histone H3K27ac and BRD4 recruitment at these loci (supplemental Figure 4B). Collectively, these results demonstrate that deletion of AMPK or Acss2 reduces polyacetylated histones, reduces histone acetylation on the regulatory elements of leukemogenic genes, and attenuates BRD4 recruitment to these elements.
Because MYC is a critical player in leukemogenesis regulated by BRD4, we tested whether overexpression of Myc would rescue the defects of AMPK KO AML. Retroviral expression of Myc partially rescued the defective cell expansion observed by deleting AMPK or after compound C treatment, and accelerated the onset of leukemogenesis by AMPK KO AML cell in vivo (supplemental Figure 4C-F). These data highlight that attenuation of the MYC leukemogenic gene program is at least partly responsible for the impaired AML progression after AMPK deletion.
AMPK inhibition sensitizes AML to BET inhibition
To test whether AMPK and Brd4 genetically interact to promote leukemogenesis, we generated compound heterozygous mutant mice of AMPK and Brd4. Mx1-Cre; Prkaa1+/fl; Prkaa2+/fl (AMPK heterozygous), Mx1-Cre; Brd4+/fl mice (Brd4 heterozygous), and Mx1-Cre; Prkaa1+/fl; Prkaa2+/fl; Brd4+/fl (AMPK/Brd4 double heterozygous) mice were treated with poly(I:C) to induce Cre, lineage−c-kit+Sca-1+ progenitor cells isolated, and transduced with MLL-AF9 before transplantation into irradiated recipient mice. Wild-type, AMPK heterozygous, and Brd4 heterozygous cells caused AML in recipient mice with median survival of between 54 and 57 days, demonstrating that heterozygosity of AMPK or Brd4 does not affect leukemogenesis (Figure 6A). In contrast, AMPK/Brd4 double-heterozygous cells infected with MLL-AF9 had significantly delayed onset of leukemogenesis (median survival, 125 days). Recipients of AMPK/Brd4 double heterozygous AML also had significantly fewer GFP+ AML cells in the blood and L-GMPs in the bone marrow compared with other groups (Figure 6B-C; supplemental Figure 5A). These results establish the genetic interaction between AMPK and Brd4 in AML, and further support the possibility that AMPK KO AMLs are sensitive to BET inhibition.
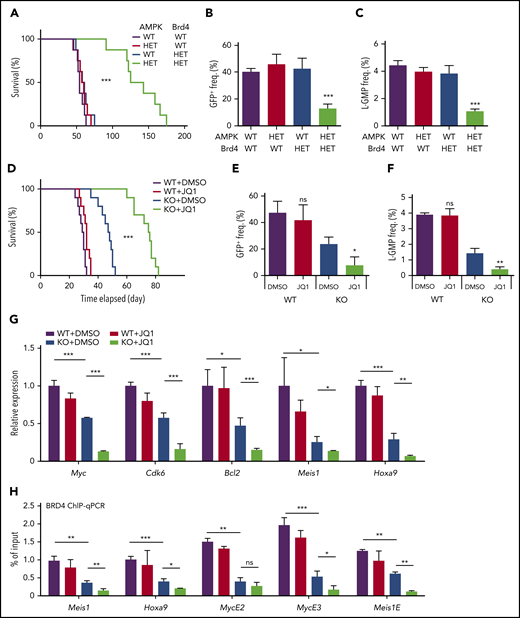
Inhibition of AMPK and BET synergistically suppresses leukemogenesis, BET recruitment, and leukemic gene expression. (A) A survival curve of mice transplanted with MLL-AF9 transformed AML cells with the indicated genotypes (n = 8). (B-C) Flow cytometry analysis of moribund mice with the indicated genotypes to detect GFP+ AML cells in the peripheral blood (B) and L-GMPs in the bone marrow (C) (n = 3). (D) A survival curve of mice transplanted with WT or AMPK KO AML cells that were then treated with or without JQ1 (n = 10). (E-F) Flow cytometry analysis of moribund mice with the indicated genotypes and treatments to detect GFP+ AML cells in the peripheral blood (E) and L-GMPs (F) in the bone marrow (n = 3). (G) Quantitative PCR with L-GMPs isolated from recipient mice transplanted with WT or AMPK KO AML then treated with or without JQ1 (n = 3). (H) ChIP-quantitative polymerase chain reaction analysis using AML cells treated with or without JQ1 to detect BRD4 occupancy at the promoter regions of Meis1 and Hoxa9, and the enhancer elements of Myc and Meis1 (n = 3). All data represent mean ± standard deviation. *P < .05; **P < .01; ***P < .001 by 1-way ANOVA, except for comparison of the survival curves in which the significance was accessed by a log-rank test.
Inhibition of AMPK and BET synergistically suppresses leukemogenesis, BET recruitment, and leukemic gene expression. (A) A survival curve of mice transplanted with MLL-AF9 transformed AML cells with the indicated genotypes (n = 8). (B-C) Flow cytometry analysis of moribund mice with the indicated genotypes to detect GFP+ AML cells in the peripheral blood (B) and L-GMPs in the bone marrow (C) (n = 3). (D) A survival curve of mice transplanted with WT or AMPK KO AML cells that were then treated with or without JQ1 (n = 10). (E-F) Flow cytometry analysis of moribund mice with the indicated genotypes and treatments to detect GFP+ AML cells in the peripheral blood (E) and L-GMPs (F) in the bone marrow (n = 3). (G) Quantitative PCR with L-GMPs isolated from recipient mice transplanted with WT or AMPK KO AML then treated with or without JQ1 (n = 3). (H) ChIP-quantitative polymerase chain reaction analysis using AML cells treated with or without JQ1 to detect BRD4 occupancy at the promoter regions of Meis1 and Hoxa9, and the enhancer elements of Myc and Meis1 (n = 3). All data represent mean ± standard deviation. *P < .05; **P < .01; ***P < .001 by 1-way ANOVA, except for comparison of the survival curves in which the significance was accessed by a log-rank test.
We then treated both wild-type and AMPK KO AML cells with JQ1, a prototypical BET inhibitor. Treatment of WT AML cells with JQ1 reduced cell proliferation in a dose-dependent manner in vitro (supplemental Figure 5B).23 AMPK KO AML cells were significantly more sensitive to JQ1 than WT AML cells, exhibiting no expansion in the presence of 0.05 ng/μL JQ1, which only mildly suppressed AMPK-proficient cells (supplemental Figure 5C). Thus, AMPK deletion sensitizes AML cells to BET inhibition.
Next, we treated AML recipient mice with JQ1. Recipient mice of MLL-AF9-induced AML cells were treated with varying doses of JQ1 for 10 days starting 10 days after transplantation. JQ1 treatment significantly extended the survival of WT recipient mice at 10 mg/kg/d, whereas 2.5 mg/kg/d had no discernible effects (supplemental Figure 5D). We previously showed that AMPK KO AML mice survive longer than controls,13 and here we found that JQ1 further extended survival, even at a dose (2.5 mg/kg/d) that had no therapeutic effect in mice transplanted with WT AML (Figure 6D). Deletion of Acss2 by CRISPR also rendered AML more sensitive to JQ1 (supplemental Figure 5E). Low-dose JQ1 treatment significantly reduced leukemic burden in mice with AMPK KO AML, as determined by the reduced percentage of leukemic GFP+ cells in blood, suppression of splenomegaly, and increased frequency of Gr-1+ mature myeloid cells in the bone marrow (Figure 6E; supplemental Figure 5F-G). Low-dose JQ1treatment further depleted the L-GMPs that were already reduced by deleting AMPK, indicating that AMPK deletion sensitizes L-GMPs to BET inhibition (Figure 6F). We also tested the effects of AMPK deletion and JQ1 in another murine AML model driven by a MOZ-TIF2 oncogene. Deletion of AMPK from MOZ-TIF2-induced AML cells reduced histone acetylation and rendered them sensitive to low dose of JQ1, similar to the MLL-AF9-induced model (supplemental Figure 5H-I).
BET inhibition decreased the occupancy of Brd4, and thus the expression of key leukemic genes in AMPK KO L-GMPs. The expression of Myc, Cdk6, Bcl2, Meis1, and Hoxa9 in L-GMPs isolated from recipient mice was the lowest in mice with AMPK deletion after JQ1 treatment (Figure 6G). ChIP-quantitative polymerase chain reaction of BRD4 showed that a low dose of JQ1 treatment did not affect BRD4 occupancy at the promoters of Meis1 and Hoxa9, nor the enhancer elements of Myc and Meis1, whereas AMPK deletion significantly reduced BRD4 recruitment to these loci (Figure 6H). However, low-dose JQ1 treatment further reduced BRD4 occupancy on these loci in AMPK KO L-GMPs (Figure 6H). Thus, ablation of AMPK makes BRD4 vulnerable to eviction from target genes by JQ1, rendering AML sensitive to BET inhibition.
To test whether JQ1 treatment affects normal hematopoiesis, we treated WT and AMPK KO mice (without AML) with JQ1. We detected no difference in the number of white blood cells, red blood cells, or platelets after JQ1 treatment in either WT or AMPK KO mice (supplemental Figure 5J-K). There were also no differences in the frequencies of hematopoietic stem cells, multipotent progenitors, common myeloid progenitors, megakaryocyte-erythroid progenitors, and GMPs (supplemental Figure 5L). AMPK;Brd4 double-heterozygous mice did not show any changes in hematopoietic progenitor cells or hematopoiesis (supplemental Figure 6). These results demonstrate that suppression of both AMPK and BET proteins blocks AML progression without negatively affecting normal hematopoiesis.
Human AML are sensitive to combinatorial AMPK and BET inhibition
Finally, we tested whether our findings could be extended to human AML. Expansion of MOLM13 cells in culture was suppressed by compound C, which was partially rescued by acetate supplementation (supplemental Figure 7A). The combination of compound C and JQ1 significantly decreased expansion of MOLM13 cells (Figure 7A; supplemental Figure 7B-C) and increased the expression of myeloid differentiation marker CD33 (Figure 7B). To assess whether combinatorial treatment suppressed BRD4 function, we examined the expression of BRD4 target genes and BRD4 occupancy. Expression of HOXA9, MYC, and MEIS1 and BRD4 occupancy on the promoters or enhancers of these genes were significantly reduced on the combinatorial treatment (supplemental Figure 7D-E). These data suggest that AMPK inhibition attenuates BRD4 function in human AML.
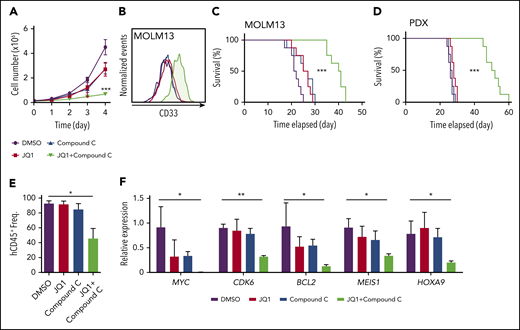
Combinatorial inhibition of AMPK and BET prolongs survival in patient-derived xenograft AML model. (A-B) MOLM13 cells were treated with JQ1 (0.05 ng/μL) and an AMPK inhibitor compound C (0.1 ng/μL) individually or in combination and cell proliferation (A) and expression of a myeloid differentiation marker CD33 (B) were analyzed (n = 3). (C-D) Survival curve of NSG-SGM3 mice treated with JQ1 (2.5 mg/kg/d) and compound C (1 mg/kg/d) individually or in combination after transplantation of MOLM13 cells (C) (n = 8) or patient-derived t(9;11) AML cells (D) (n = 8). (E) Flow cytometry analysis of mice described in (D) to detect human CD45+ cells in the peripheral blood (n = 3). (F) Quantitative PCR using human AML cells isolated from recipient mice as shown in (D) to detect the expression of key leukemogenic genes (n = 3). All data represent mean ± standard deviation. *P < .05; **P < .01; ***P < .001 by 1-way ANOVA, except for comparison of the survival curves in which the significance was accessed by a log-rank test.
Combinatorial inhibition of AMPK and BET prolongs survival in patient-derived xenograft AML model. (A-B) MOLM13 cells were treated with JQ1 (0.05 ng/μL) and an AMPK inhibitor compound C (0.1 ng/μL) individually or in combination and cell proliferation (A) and expression of a myeloid differentiation marker CD33 (B) were analyzed (n = 3). (C-D) Survival curve of NSG-SGM3 mice treated with JQ1 (2.5 mg/kg/d) and compound C (1 mg/kg/d) individually or in combination after transplantation of MOLM13 cells (C) (n = 8) or patient-derived t(9;11) AML cells (D) (n = 8). (E) Flow cytometry analysis of mice described in (D) to detect human CD45+ cells in the peripheral blood (n = 3). (F) Quantitative PCR using human AML cells isolated from recipient mice as shown in (D) to detect the expression of key leukemogenic genes (n = 3). All data represent mean ± standard deviation. *P < .05; **P < .01; ***P < .001 by 1-way ANOVA, except for comparison of the survival curves in which the significance was accessed by a log-rank test.
Next, we transplanted MOLM13 cells into NSG-SGM3 mice24 and treated them with varying doses of JQ1, compound C, or both. Treatment with 10 mg/kg/d JQ1 significantly extended the survival of recipient mice, whereas 2.5 mg/kg/d had no discernible effects (Figure 7C; supplemental Figure 7F). Treatment with compound C also suppressed leukemogenesis, as previously shown13 (supplemental Figure 7G). Strikingly, we found that only 10 days of treatment with the combination of compound C and JQ1, at doses at which single agents had negligible effects, significantly suppressed leukemogenesis (Figure 7C).
Last, we examined whether this combination therapy can suppress leukemogenesis, using a patient-derived xenograft model of AML with a t(9;11) (MLL-AF9) rearrangement.25 This patient-derived xenograft line also responded to JQ1 and compound C single treatments in a dose-dependent manner (supplemental Figure 7H-I). However, low doses of the individual drugs (2.5 mg/kg/d of JQ1; 1 mg/kg/d of compound C) did not affect survival. These low doses significantly extended leukemia-free survival when combined, with a synergistic reduction of human CD45+ AML cells in the bone marrow and expression of MYC, CDK6, BCL2, MEIS1, and HOXA9 (Figure 7D-F). Collectively, these results establish that the combinatorial inhibition of AMPK and BET proteins suppresses human AML by attenuating the expression of key leukemogenic genes.
Discussion
Here, we provide evidence that the master regulator of cellular energetics AMPK links acetyl-CoA homeostasis to BET recruitment, thereby promoting the expression of key leukemogenic genes. Our results establish that chemical inhibition of AMPK and BET proteins synergistically suppresses AML by disabling the LIC gene regulatory networks.
Recent studies have highlighted the importance of acetate metabolism in cancer. Tumors in mice and humans consume large amount of acetate that is oxidized to generate acetyl-CoA for lipid synthesis and histone acetylation.48-51 Consistent with these findings, acetate was able to rescue the defective proliferation of AMPK KO AML cells via ACSS2. Deletion of Acss2 with CRISPR/Cas9 also impaired leukemogenesis in a transplantation model, supporting the notion that acetyl-CoA homeostasis governed by the AMPK/ACSS2 module is critical for AML. Of note, acetate is abundant in mouse plasma, and its turnover rate is one of the fastest among metabolites.52 Future work should shed light on how acetate availability in the leukemia niche affects leukemogenesis.
Recent evidence supports the model in which AMPK protects AML cells under stress. We previously demonstrated that deletion of AMPK depletes LICs in multiple myeloid leukemia models (those initiated by MLL-AF9, MOZ-TIF2, and BCR-ABL oncogenes) and renders them sensitive to metabolic stress caused by dietary restriction.13 Further, a recent report demonstrated that leukemia stem cells in human AML have active mitophagy controlled by the AMPK-FIS1 axis.14 AML stem cells had activated AMPK and increased expression of FIS1 compared with the bulk of AML, and inhibition of the AMPK-FIS1 axis induced differentiation and cell cycle arrest of AML stem cells. Thus, AMPK governs metabolic homeostasis of immature AML cells, and its inhibition has particularly potent suppressive effects on AML.
The results presented here extend the emerging view that AMPK directly or indirectly affects the epigenome and gene expression. AMPK regulates histone modification by phosphorylating histone H2B or H3,53,54 or by phosphorylating EZH2, affecting histone methylation.55 AMPK also regulates DNA demethylation by phosphorylating TET2, affecting the levels of the TET co-factor α-ketoglutarate,56,57 or by phosphorylating DNA methyltransferase 1.58 Our study demonstrates that suppressing AMPK disrupts acetyl-CoA homeostasis and histone acetylation, and profoundly decreases the number of polyacetylated histones that have high affinities for BET binding.42 Because the acetyltransferase p300/CBP is critical for histone acetylation and BRD4 recruitment in MLL-AF9-transformed AML cells,46 AMPK governed acetyl-CoA homeostasis may feed p300/CBP-dependent histone acetylation to recruit BRD4 to chromatin.
BET inhibitors have garnered much interest as therapies for malignancies, exhibiting remarkable antitumor effects against multiple caners including AML.22,23,59-61 However, tumors gain resistance to BET inhibitors by increasing BRD4-Mediator interaction or activating the WNT signaling pathway.61-63 Designing combination therapies that increase antitumor effects without increasing toxicity is necessary to circumvent this issue. In this context, our work proposes a novel combination therapy against AML in which acetyl-CoA homeostasis and acetyl-lysine readers are concurrently inhibited by AMPK and BET inhibitors, respectively. The concept of inhibiting both the source (acetyl-CoA homeostasis) and the genomic readers (BET proteins) of acetylation may be extended beyond AMPK, such as by inhibiting ACSS2 or ATP-citrate lyase, which converts citrate to acetyl-CoA.16,37,48,64
RNA-seq and ChIP-seq data have been deposited in the Gene Expression Omnibus under accession number GSE128001.
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Acknowledgments
The authors thank Catherine Gillespie for critical reading of the manuscript.
This work was supported by the Cancer Prevention and Research Institute of Texas (R1201 and RP160035), the Gabrielle’s Angel Foundation for Cancer Research, and the National Institutes of Health, National Cancer Institute (R01CA193235) and National Institute of Diabetes and Digestive and Kidney Diseases (R01DK107413). D.N. and X.S. are Scholar and Special Fellow of the Leukemia and Lymphoma Society, respectively. Y.J. was supported by training grants from the National Institutes of Health and Cancer Prevention and Research Institute of Texas (T32DK060445 and RP160283). Flow cytometry was partially supported by the National Institutes of Health (National Center for Research Resources grant S10RR024574, National Institute of Allergy and Infectious Diseases AI036211 and National Cancer Institute P30CA125123) for the Baylor College of Medicine Cytometry and Cell Sorting Core. Sequencing was partially supported by the National Institutes of Health, National Institute of Diabetes and Digestive and Kidney Diseases (P30DK56338) and National Cancer Institute (P30CA125123). Metabolomics Core Services was supported by grant U24 DK097153 of National Institutes of Health Common Funds Project to the University of Michigan.
Authorship
Contribution: Y.J. performed experiments with help from T.H., X.S., and A.K.; T.W. performed histone proteoform analysis under the supervision of N.L.Y.; K.E. performed the enhancer analysis under the supervision of C.Y.L.; K.A.H. performed bioinformatics analysis of sequencing data; M.Y.K. provided human AML samples; and Y.J. and D.N. designed the experiments, analyzed the results, and wrote the manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Daisuke Nakada, Department of Molecular and Human Genetics, Baylor College of Medicine, Houston, TX 77030; e-mail: nakada@bcm.edu.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal