

Key Points
IL-7R expression is a functional biomarker of T-ALL cells with leukemia-initiating potential and plays a crucial role in T-ALL pathogenesis.
Targeting IL-7R–mediated signaling hampers leukemia-initiating activity and progression of human T-ALL.

Abstract
T-cell acute lymphoblastic leukemia (T-ALL) is an aggressive hematological malignancy resulting from the dysregulation of signaling pathways that control intrathymic T-cell development. Relapse rates are still significant, and prognosis is particularly bleak for relapsed patients. Therefore, development of novel therapies specifically targeting pathways controlling leukemia-initiating cell (LIC) activity is mandatory for fighting refractory T-ALL. The interleukin-7 receptor (IL-7R) is a crucial T-cell developmental pathway that is commonly expressed in T-ALL and has been implicated in leukemia progression; however, the significance of IL-7R/IL-7 signaling in T-ALL pathogenesis and its contribution to disease relapse remain unknown. To directly explore whether IL-7R targeting may be therapeutically efficient against T-ALL relapse, we focused on a known Notch1-induced T-ALL model, because a majority of T-ALL patients harbor activating mutations in NOTCH1, which is a transcriptional regulator of IL-7R expression. Using loss-of-function approaches, we show that Il7r-deficient, but not wild-type, mouse hematopoietic progenitors transduced with constitutively active Notch1 failed to generate leukemia upon transplantation into immunodeficient mice, thus providing formal evidence that IL-7R function is essential for Notch1-induced T-cell leukemogenesis. Moreover, we demonstrate that IL-7R expression is an early functional biomarker of T-ALL cells with LIC potential and report that impaired IL-7R signaling hampers engraftment and progression of patient-derived T-ALL xenografts. Notably, we show that IL-7R–dependent LIC activity and leukemia progression can be extended to human B-cell acute lymphoblastic leukemia (B-ALL). These results have important therapeutic implications, highlighting the relevance that targeting normal IL-7R signaling may have in future therapeutic interventions, particularly for preventing T-ALL (and B-ALL) relapse.
Introduction
T-cell acute lymphoblastic leukemia (T-ALL) is an aggressive acute lymphoblastic leukemia (ALL) subtype resulting from the transformation of T-cell precursors developing in the thymus.1,2 Although intensive chemotherapy led to improved outcomes in recent years, relapses are still frequent and are associated with poor prognosis.1,3 Therefore, current research efforts focus on developing novel targeted therapies against specific signaling pathways involved in disease pathogenesis and relapse. The interleukin-7 receptor (IL-7R) has recently emerged as a promising therapeutic target for ∼10% of T-ALL (and ∼2% of B-cell acute lymphoblastic leukemia [B-ALL]) cases harboring gain-of-function oncogenic mutations in IL7R, the gene encoding the IL-7Rα subunit.4-7 IL-7R is a heterodimer formed by IL-7Rα and a common γ chain (γc), shared between the receptors for IL-2, IL-4, IL-9, IL-15, and IL-21.8,9 Normal IL-7R signaling is induced by IL-7–mediated heterodimerization of IL-7Rα and γc,8 and it plays an essential role in human T-cell development.10 However, mutant IL-7R can signal independently of IL-7 and γc, owing to disulfide bond formation and homodimerization of 2 mutant IL-7Rα chains, which leads to cell transformation and leukemogenesis.4-6,11 Therefore, targeting mutant IL-7R itself6 or IL-7R downstream signaling pathways, including JAK/STAT and PI3K/Akt/mTOR,12-14 has been proposed as a useful therapeutic approach for treating patients with IL7R-activating mutations.
In addition to the oncogenic role of mutant IL7R, normal IL-7R/IL-7 signaling is involved in a major proportion of T-ALL cases expressing conventional IL-7Rs that mediate leukemia survival and in vitro proliferation in response to IL-7.15-21 IL-7R/IL-7 signaling also contributes to T-ALL progression in vivo in patient-derived xenografts (PDXs),22 and this function can be extended to some B-ALL cases that express functional IL-7Rs,23,24 indicating that IL-7R plays a general role in ALL progression. However, whether normal IL-7R function could also modulate leukemogenesis remains an open question. Supporting this view, IL-7–transgenic mice and AKR/J mice, which naturally overexpress IL-7Rα, spontaneously develop B-cell and/or T-cell lymphomas.25-28 Also, increased membrane IL-7R density leading to enhanced IL-7 signaling cooperates with T-cell oncogenes in mouse T-ALL.29 Moreover, IL7R is a transcriptional NOTCH1 target in human T-cell development and T-ALL.30,31 Considering that oncogenic NOTCH1 mutations occur in >65% of T-ALL patients,32 normal IL-7R/IL-7 signaling may critically impact T-ALL pathogenesis and relapse in a major proportion of T-ALL cases expressing oncogenic NOTCH1, not only in those harboring IL-7R–related genetic aberrations. However, formal proof that normal IL-7R expression participates in T-ALL pathogenesis is still lacking and whether induction of the leukemogenic process requires IL-7R signaling remains unknown. In this study, we used conclusive loss-of-function approaches to demonstrate that IL-7R signaling is essential for T-ALL pathogenesis induced by Notch. We also show that IL-7R expression is a functional biomarker of human T-ALL cells with leukemia-initiating potential and extend this finding to B-ALL, providing direct evidence that targeting normal IL-7R itself offers a feasible and effective therapeutic strategy for a large proportion of ALL patients.
Methods
Cell lines, primary ALL samples, and OP9 cultures
HPB-ALL and Jurkat cell lines (American Type Culture Collection) were cultured in RPMI 1640 with 10% fetal bovine serum (FBS; Gibco). Human samples were obtained according to the Declaration of Helsinki principles and to the procedures approved by the Spanish National Research Council Bioethics Committee after informed consent. Primary T-ALL and B-ALL cells were obtained by centrifugation, on Lymphoprep (STEMCELL Technologies), of peripheral blood (PB) or bone marrow (BM) samples from patients at diagnosis (supplemental Tables 1 and 2, available on the Blood Web site). For in vitro cultures, human T-ALL or B-ALL cells or mouse T-ALL cells were cultured onto OP9 cells expressing GFP (OP9-GFP)33 or DL4 Notch ligand (OP9-DL4)34 in α-MEM with 20% FBS and recombinant human (rh)IL-7 (200 IU/mL; National Institute of Biological Standards and Controls). When indicated, cultures were supplemented with 100 nM γ-secretase inhibitor (GSI) Compound E (Enzo Life Sciences) or dimethyl sulfoxide (DMSO) as vehicle. For IL-7R blocking, T-ALL cells were cultured onto OP9-DL4 cells with IL-7 (200 IU/mL) and an anti–IL-7Rα neutralizing monoclonal antibody (mAb; 10 μg/mL; Dendritics) or a mouse immunoglobulin G1 (IgG1) control.
Flow cytometry
Mouse anti-human mAbs used included anti-CD5–PE–Cy5 (Beckman Coulter), anti-CD7–PE (Life Technologies), anti-CD45–V450, anti-CD127–biotin, anti-HLA-DR–PE (BD Biosciences), and anti-CD10–PerCPCy5.5 (BioLegend). Anti-mouse mAbs were anti-CD8–FITC (Invitrogen); anti-CD44–PE, anti-CD3ε–PE, anti-CD4–PerCP, anti-CD11b–FITC, anti-Gr1–PE, anti-H2-Kb–PE, anti-H2-Kb–biotin, anti-TCRβ–FITC (all from BD Biosciences); and anti-IL7Rα–biotin and anti-CD25–APC (eBioscience). Biotinylated antibodies (Abs) were developed using Streptavidin-APC (eBioscience). Background fluorescence was determined with irrelevant isotype-matched Abs (BD Biosciences). For cell cycle studies, cells were incubated with 10 µg/mL Hoestch 33342 (Sigma-Aldrich) before culture. Cell proliferation was assessed after incubation with CellTrace Violet (Thermo Fisher Scientific) and cultured for the indicated times. Flow cytometry was performed using a FACSCalibur or a FACSCanto II (BD Biosciences).
Western blotting
Activation of signaling pathways downstream of IL-7R was analyzed by western blotting of cells incubated with 200 IU/mL rhIL-7 at 37°C for the indicated times. Whole-cell lysates (RIPA buffer) separated on 10% sodium dodecyl sulfate–polyacrylamide gel electrophoresis (Bio-Rad) were transferred to polyvinylidene difluoride membranes, as described,30 and membranes were incubated with Abs against STAT5, phospho-STAT5–Tyr694, AKT, phospho-AKT–Ser473, phospho-ERK, ERK, BCL2, and intracellular Notch1 (ICN1) (Cell Signaling). α-Tubulin expression (Sigma-Aldrich) was analyzed as loading control. Washed membranes were incubated with horseradish peroxidase–conjugated anti-mouse or anti-rabbit Abs for 1 hour and developed using Lumi-LightPLUS (Roche).
ChIP assays
Total DNA was extracted from thymocytes from embryonic day 14.5 Swiss mouse embryos. Chromatin immunoprecipitation (ChIP) assays were performed using a rat IgG1 anti-mouse RBP-Jk Ab (Cosmo Bio) or an irrelevant rat IgG1 Ab (BD Biosciences).30 Unbound chromatin (input) and immunoprecipitated DNA samples were analyzed by semiquantitative polymerase chain reaction (PCR), using primers recognizing the RBP-Jk binding site in mouse Il7r promoter (located at −1937 bp from the ATG translation initiation codon of Il7r) or the RBP-Jk site in Il2ra promoter35 (supplemental Table 3).
Luciferase reporter assays
A 2235-bp fragment containing the 5′ upstream regulatory region of mouse Il7r (from −58 bp to −2293 bp upstream of the ATG translation initiation codon; Ensembl, ENSMUSG00000003882) was PCR amplified using Hot Start DNA polymerase (QIAGEN) and cloned into the BglII and MluI sites of pGL3Basic firefly luciferase reporter vector (Promega). Site-directed mutagenesis of the Il7r RBP-Jk binding site was performed using conventional PCR. The mutated sequence was confirmed by sequencing and cloned into pGL3. Specific primers used are listed in supplemental Table 3. Jurkat cells were cotransfected by electroporation (264 V, 975 μF) with the Il7r luciferase reporter vector containing wild-type (wt) or mutated RBP-Jk binding sites, together with the MigR1 retroviral vector encoding ICN1 and GFP or only GFP,36 and/or with MigR1 encoding a dominant-negative mutant form of the Notch coactivator mastermind-like1 (dnMAML1) fused to GFP,37 plus the constitutively active Renilla reniformis luciferase-producing vector prL-CMV (Promega). Luciferase activities were determined in triplicates after 48 hours using the Dual Luciferase Reporter Assay (Promega) and expressed as fold induction relative to transfection with control plasmids.
Real-time quantitative PCR
Short hairpin RNA (shRNA)-transduced cells were analyzed for IL7R transcription by quantitative PCR using TaqMan probes (Applied Biosystems), as described.30 Glyceraldehyde-3-phosphate dehydrogenase was used as endogenous control.
Isolation of Lin− c-kit+ hematopoietic progenitors from mouse BM
Lineage-negative cells (Lin−) were isolated by immunomagnetic sorting from BM samples of 6- to 10-week-old C57BL/6 mice (The Jackson Laboratory), IL-7Rα–transgenic mice,38 or IL-7Rα–deficient B6.129S7-Il7rtm1lmx mice.39,40 Briefly, cells were sequentially incubated with biotin-coupled Abs against B220/CD45R, Gr1.1, CD11b, CD3, or Ter-119 (BD Biosciences) and streptavidin-coupled magnetic spheres (Miltenyi Biotec). Lin− c-kit+ hematopoietic progenitor cells (HPCs) were isolated by cell sorting using a FACSAria (BD Biosciences) after labeling with APC-coupled anti-CD117 (c-kit) (BD Biosciences).
Xenotransplantation assays
Animal studies were performed in accordance with guidelines approved by the Animal Experimentation Ethics Committee of the Comunidad de Madrid Animal Research Review Board. Sublethally irradiated (1.5 Gy) 6- to 10-week-old RAG-2−/− × γc−/−41 or NOD.Cg-Prkdcscidγc−/− (NSG; The Jackson Laboratory) immunodeficient mice were subjected to IV injection with T-ALL or B-ALL patient samples (first grafts) or with xenografts from host mice (second grafts) (106 cells per mice).
Generation of mouse T-ALL
T-ALL was generated from mouse BM Lin− c-kit+ HPCs transduced with ICN1 and GFP, following a modification of the original model.42 ICN1 was PCR amplified from the MigR1-ICN1 retroviral vector36 and cloned into the BamHI and XhoI sites of pSIN-BX42 containing IRES-eGFP. ICN1-IRES-eGFP was cloned into the BamHI and NotI sites of the pHRSIN (pHRSIN-ICN1) lentiviral vector.43 Lentiviral particles were generated by lipofection (jetPEI; Polyplus) of 293T cells using VSV-G (pMD2G), viral structural proteins gag/pol (psPAX2), and pHRSIN-ICN1 or pHRSIN control vector. Viral supernatants were collected at 48 hours and 72 hours after transfection. Murine HPCs were transduced for 16 to 24 hours (10-30 multiplicity of infection) in fibronectin-coated plates (50 μg/mL, RetroNectin; Takara) in RPMI 1640 plus 10% FBS containing mouse SCF (50 IU/mL), rhFLT3L (50 IU/mL), rhTPO (50 ng/mL) (all from PeproTech), and rhIL-6 (25 IU/mL; National Institute of Biological Standards and Controls). After transduction, cells were injected IV into sublethally irradiated NSG mice (1-2 × 105 cells per mouse) rather than isogenic mice, as originally described,42 because the former would be expected to develop leukemia with reduced latency and higher penetrance compared with the latter (11-40 weeks after transplant, 50% penetrance).42
shRNA lentiviral constructs and transductions
For silencing endogenous IL-7R expression, 5 shRNAs directed against human IL7R (shIL7R) (Sigma-Aldrich Mission TCRN shRNA Target set, TRCN0000058228, TCRN0000058229, TCRN0000058230, TCRN0000058231, TRCN0000058232) were assayed by transfection of HPB-ALL cells and puromycin selection (1 μg/mL; Sigma-Aldrich). Selected shIL7Rs were then cloned under the U6 promoter into the pHRSIN-GFP lentiviral vector. For shIL7R transduction, T-ALL and B-ALL cells were cultured for 48 hours onto OP9-DL4 stromal cells or OP9-GFP cells, respectively, in the presence of rhIL-7 (200 IU/mL) and transduced for 24 hours with lentiviral vectors encoding shIL7R or scrambled shRNA (shsc) as control.
Leukemia-initiating cell assays
T-ALL leukemia-initiating cell (LIC) activity was analyzed by transplantation into NSG mice under limiting dilution conditions (80 to 8 × 104 cells per mouse, ≥ 4 mice per dose). Transplanted mice were analyzed by flow cytometry for the presence of human CD7+CD45+ blasts in PB at 5 and 7 weeks posttransplant, and the frequency of leukemia-free mice was calculated using ELDA software (http://bioinf.wehi.edu.au/software/elda/).44
Statistics
Statistical significance was determined using a 2-tailed Student t test, and Kaplan-Meyer survival curves were compared using the log-rank test, using GraphPad software.
Results
IL-7R expression is a biomarker of human T-ALL cells with leukemia-initiating potential
We analyzed IL-7R expression in a panel of primary T-ALL samples representative of distinct T-cell developmental stages according to the European Group for Immunological Characterization of Leukemias classification, which were obtained from pediatric or adult patients at the time of diagnosis (supplemental Table 1). Most T-ALL samples expressed surface IL-7R (93.3%, n = 14) at variable levels (supplemental Figure 1A) and responded to IL-7 in vitro, as indicated by the induction of STAT5 phosphorylation. T-ALL cells also showed constitutive expression of the antiapoptotic IL-7R target BCL245,46 (supplemental Figure 1B), suggesting that IL-7R is also functional in vivo. Sequencing analysis revealed that most IL-7R+ T-ALLs (14/16) expressed the normal IL7R gene, whereas only 2 of them displayed IL7R-activating mutations (supplemental Table 1). Therefore, expression of normal IL-7R is a common feature of human T-ALLs arrested at distinct developmental stages. According to the fact that IL7R is a transcriptional Notch1 target,30,31 ICN1 expression, which is indicative of constitutive Notch1 activation, was observed in all T-ALL samples analyzed by western blotting (supplemental Figure 1B; and data not shown), and NOTCH1-activating mutations were confirmed in 4 of 6 T-ALL patient samples analyzed by NGS (66%, close to reported data32 ) (supplemental Table 1). Consequently, treatment of ICN1+ T-ALL cells in IL-7–supplemented OP9-GFP cell cocultures with an inhibitor of Notch1 activation (GSI) resulted in specific IL-7R downregulation (Figure 1A). Conversely, T-ALL cells incubated with DMSO vehicle maintained IL-7R expression (Figure 1A) and progressively outcompeted cells with lower IL-7R expression levels (Figure 1B). In addition, T-ALL cell numbers decreased in OP9-DL4 cocultures supplemented with a blocking anti–IL-7Rα mAb (Figure 1C), supporting that Notch1-induced IL-7R expression allows for IL-7–mediated proliferation and expansion of primary T-ALL cells in vitro. Confirming this view, CellTrace Violet labeling of primary human T-ALL cells with heterogeneous levels of IL-7R revealed that proportions of cells undergoing division in response to IL-7 were significantly increased in the subset with higher IL-7R expression levels (Figure 1D-E). Moreover, cell cycle studies showed increased proportions of S+G2/M cycling cells in the IL-7R+ T-ALL subset compared with the IL-7R−/lo T-ALL subset in response to several IL-7 concentrations (Figure 1F); however, no differences in cell apoptosis were observed (data not shown), confirming that IL-7R expression provides a proliferation advantage to T-ALL cells in vitro.
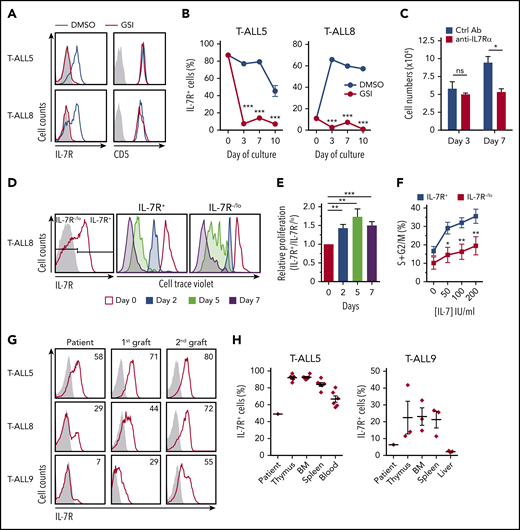
In vitro and in vivo proliferation advantage of primary T-ALL cells expressing IL-7R induced by activated Notch1. (A) IL-7R and CD5 flow cytometry analysis of primary T-ALL cells (T-ALL5 and T-ALL8, from T-ALL patients 5 and 8, respectively) cocultured for 3 days onto OP9-GFP stromal cells supplemented with rhIL-7 and either a Notch activation inhibitor (red) or DMSO as vehicle (blue). Representative results from 3 experiments are shown. Shaded graphs represent background staining. (B) Percentages of primary human T-ALL cells expressing IL-7R cultured as in (A) for the indicated days. Mean ± standard error of the mean (SEM) of triplicates from a representative experiment are shown (n = 3). (C) Total numbers of primary T-ALL5 cells recovered at the indicated days from cocultures set up onto OP9-DL4 cells in the presence of rhIL-7 and either a neutralizing anti–IL-7Rα or an isotype-matched control antibody. Data are mean ± SEM (n = 3). (D) Flow cytometry of IL-7R+ (middle panel) and IL-7R−/lo (right panel) human primary T-ALL8 cells electronically gated as shown (left panel) upon labeling with CellTrace Violet and culture onto OP9-GFP cells supplemented with rhIL-7 for the indicated times. Data are representative of triplicate cultures from 2 independent experiments. (E) Relative proliferation of IL-7R+ vs IL-7R-/lo cells in (D), calculated as CellTrace Violet dilution ratio. (F) Proliferation potential of IL-7R+ and IL-7R−/lo T-ALL8 cells cultured onto OP9-GFP stromal cells supplemented with increasing concentrations of rhIL-7. Percentages of cells in the S+G2/M phases of the cell cycle were determined by Hoestch labeling and flow cytometry of cells electronically gated as in (D) after 3 days of culture. Data are mean ± SEM of triplicate cultures from 2 independent experiments. (G) IL-7R expression levels displayed by primary T-ALL cells from 3 patients analyzed by flow cytometry at the time of diagnosis (Patient) or after serial transplantation and thymus engraftment into consecutive Rag2−/−γc−/− immunodeficient mice (1st graft and 2nd graft). Numbers show percentages of positive cells. Shaded graphs show background staining with irrelevant isotype-matched antibodies. (H) Percentages of IL-7R–expressing T-ALL cells engrafting the indicated organs 10 to 12 weeks after transplant into immunodeficient mice in (G). Data are mean ± SEM of 3 to 7 mice. Percentages at the time of diagnosis (Patient) are shown for comparison. *P < .05, **P < .01, ***P < .001. ns, not significant.
In vitro and in vivo proliferation advantage of primary T-ALL cells expressing IL-7R induced by activated Notch1. (A) IL-7R and CD5 flow cytometry analysis of primary T-ALL cells (T-ALL5 and T-ALL8, from T-ALL patients 5 and 8, respectively) cocultured for 3 days onto OP9-GFP stromal cells supplemented with rhIL-7 and either a Notch activation inhibitor (red) or DMSO as vehicle (blue). Representative results from 3 experiments are shown. Shaded graphs represent background staining. (B) Percentages of primary human T-ALL cells expressing IL-7R cultured as in (A) for the indicated days. Mean ± standard error of the mean (SEM) of triplicates from a representative experiment are shown (n = 3). (C) Total numbers of primary T-ALL5 cells recovered at the indicated days from cocultures set up onto OP9-DL4 cells in the presence of rhIL-7 and either a neutralizing anti–IL-7Rα or an isotype-matched control antibody. Data are mean ± SEM (n = 3). (D) Flow cytometry of IL-7R+ (middle panel) and IL-7R−/lo (right panel) human primary T-ALL8 cells electronically gated as shown (left panel) upon labeling with CellTrace Violet and culture onto OP9-GFP cells supplemented with rhIL-7 for the indicated times. Data are representative of triplicate cultures from 2 independent experiments. (E) Relative proliferation of IL-7R+ vs IL-7R-/lo cells in (D), calculated as CellTrace Violet dilution ratio. (F) Proliferation potential of IL-7R+ and IL-7R−/lo T-ALL8 cells cultured onto OP9-GFP stromal cells supplemented with increasing concentrations of rhIL-7. Percentages of cells in the S+G2/M phases of the cell cycle were determined by Hoestch labeling and flow cytometry of cells electronically gated as in (D) after 3 days of culture. Data are mean ± SEM of triplicate cultures from 2 independent experiments. (G) IL-7R expression levels displayed by primary T-ALL cells from 3 patients analyzed by flow cytometry at the time of diagnosis (Patient) or after serial transplantation and thymus engraftment into consecutive Rag2−/−γc−/− immunodeficient mice (1st graft and 2nd graft). Numbers show percentages of positive cells. Shaded graphs show background staining with irrelevant isotype-matched antibodies. (H) Percentages of IL-7R–expressing T-ALL cells engrafting the indicated organs 10 to 12 weeks after transplant into immunodeficient mice in (G). Data are mean ± SEM of 3 to 7 mice. Percentages at the time of diagnosis (Patient) are shown for comparison. *P < .05, **P < .01, ***P < .001. ns, not significant.
To next assess the contribution of IL-7R signaling to T-ALL progression in vivo, we analyzed IL-7R expression in patient-derived T-ALL xenografts serially transplanted into immunodeficient mice. This model revealed a progressive increase in IL-7R expression levels in leukemic cells infiltrating distinct organs of serially transplanted mice (Figure 1G-H), indicating that IL-7R confers a selective growth advantage in response to endogenous mouse IL-7,22,47 which is responsible for leukemia progression in vivo. Therefore, IL-7R/IL-7 signaling could impact T-ALL LIC function. To directly investigate this possibility, T-ALL patient cells were sorted using FACS, based on surface IL-7R expression levels, into IL-7R−/lo and IL-7Rhi cells (supplemental Figure 2), and LIC potential was assessed in a limiting dilution xenograft assay. Kinetic analysis of PB T-ALL engraftment revealed a fivefold to sixfold enriched LIC frequency of IL-7Rhi cells compared with IL-7R−/lo cells at distinct times posttransplant (Figure 2A), and the frequency of disease-free mice was significantly higher in mice transplanted with IL-7R−/lo cells (Figure 2B). We next scored for in vivo engraftment and progression of T-ALL patient cells in which IL7R encoding the IL-7Rα subunit was silenced by specific shIL7R compared with control T-ALL cells expressing shsc. Designed shIL7Rs (supplemental Figure 3A) were validated by their ability to reduce IL7R messenger RNA and surface IL-7Rα expression in T-ALL cell lines, leading to impaired IL-7R signaling in response to IL-7, as measured by STAT5 phosphorylation (supplemental Figure 3B-C). Primary T-ALL samples transduced with the selected shIL7R (shIL7R5) or shsc were transplanted into NSG mice. GFP expression analyses showed similar transduction efficiencies for shsc- and shIL7R-transduced cells (Figure 2C,E), but leukemia burden analysis of transplanted mice revealed a drastic reduction in shIL7R-transduced cell numbers infiltrating all tissues analyzed compared with shsc-transduced T-ALL cells, indicating that IL-7R silencing strikingly impairs T-ALL engraftment and progression in host mice (Figure 2D,F). In fact, reduced IL-7R expression in shIL7R-transduced T-ALL cells resulted in decreased proportions of cycling cells (Figure 2G-I). Collectively, these results show that IL-7R expression is crucial for human T-ALL LIC function. Therefore, LIC activity is enriched in IL-7R+ T-ALL cells; thus, IL-7R expression can be considered a useful biomarker of human T-ALL cells with LIC potential.
![IL-7R signaling is essential for LIC function and progression of human T-ALL in vivo. (A) LIC potential of IL-7Rhi (blue) and IL-7R−/lo (red) primary human T-ALL8 cells (from T-ALL patient 8) that were sorted by fluorescence-activated cell sorting based on IL-7R expression levels (high or negative-to-low, respectively), as shown in supplemental Figure 2, and transplanted into NSG mice under limiting dilution conditions (from 80 to 8 × 104 cells per mouse, ≥4 mice per dose). LIC frequency was calculated using ELDA software by recording numbers of leukemia-free mice in the PB at 5 and 7 weeks posttransplant (p.t.). (B) Kaplan-Meier curves of leukemia-free mice transplanted in (A) with 80 FACS-sorted IL-7Rhi or IL-7R−/lo T-ALL8 cells. Flow cytometry analysis of the efficiency of transduction of human primary T-ALL5 (from T-ALL patient 5) (C) and T-ALL8 (E) cells with lentiviral vectors encoding an shRNA against IL-7R (shIL7R) or an shsc, together with GFP. Numbers indicate the percentages of transduced (GFP+) cells. Relative numbers of transduced (GFP+) T-ALL5 (D) or T-ALL8 (F) cells engrafting the indicated organs of NSG mice euthanized when they presented advanced symptoms of disease (7-8 weeks posttransplant) and analyzed by flow cytometry. Data are percentages (mean ± standard error of the mean [SEM]) of GFP+ engrafted cells normalized to percentages of GFP+ injected cells (C,E) (n = 5). (G) Flow cytometry of IL-7R surface expression on primary human T-ALL5 cells upon transduction with shIL7R or shsc (GFP+) and culture for 3 days onto OP9-GFP cells plus rhIL-7 (left panels). Line graphs show percentages of IL-7R+ cells on electronically gated GFP+ transduced cells from a representative experiment (right panels). Shaded graphs show background staining with irrelevant isotype-matched antibodies. (H) Surface IL-7R expression levels on primary T-ALL5 cells transduced and cultured as in (G). Data are mean ± SEM of mean fluorescence intensity (MFI) values of GFP+ transduced cells (n = 3). (I) Percentages of T-ALL5 cycling cells transduced and cultured as in (G). Data are percentages (mean ± SEM) of GFP+ transduced cells in the S+G2/M phases of the cell cycle, as determined by Hoestch labeling and flow cytometry (n = 3). *P < .05, **P < .01.](https://ash.silverchair-cdn.com/ash/content_public/journal/blood/134/24/10.1182_blood.2019000982/4/m_bloodbld2019000982f2.png?Expires=1769206809&Signature=i6Dn~wYZTe-Gf6L2d9x3NzMp24b1YqNFWLI1~p~yNjLKw55mF-0WINPs8sCpCJZuorCtM90AwFThFqdgNDJN98LukfC~SVBeCEBfOW0uO7pZzxz3ETiTshlI0v2hNV6eQlR20XVkEho6fmfEBHwqlhPebafGyam8NsmRG3HQKYdftLbZmkP2o3pnAzMYiCnkrEJSHYtbNeTrKi-wRL4ylaO~RiQRlCOiCi8UTOmtWl4C-sKeZ0Am78NDaSe7I9Yvc9l3xdzZ7F-pLNWsjYFuMnP0pknAY90I4mprg6L9KgStCFUptgK7hPZFBGPp1Nt1C1BGnojZygwjfUnDPZSP8Q__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
IL-7R signaling is essential for LIC function and progression of human T-ALL in vivo. (A) LIC potential of IL-7Rhi (blue) and IL-7R−/lo (red) primary human T-ALL8 cells (from T-ALL patient 8) that were sorted by fluorescence-activated cell sorting based on IL-7R expression levels (high or negative-to-low, respectively), as shown in supplemental Figure 2, and transplanted into NSG mice under limiting dilution conditions (from 80 to 8 × 104 cells per mouse, ≥4 mice per dose). LIC frequency was calculated using ELDA software by recording numbers of leukemia-free mice in the PB at 5 and 7 weeks posttransplant (p.t.). (B) Kaplan-Meier curves of leukemia-free mice transplanted in (A) with 80 FACS-sorted IL-7Rhi or IL-7R−/lo T-ALL8 cells. Flow cytometry analysis of the efficiency of transduction of human primary T-ALL5 (from T-ALL patient 5) (C) and T-ALL8 (E) cells with lentiviral vectors encoding an shRNA against IL-7R (shIL7R) or an shsc, together with GFP. Numbers indicate the percentages of transduced (GFP+) cells. Relative numbers of transduced (GFP+) T-ALL5 (D) or T-ALL8 (F) cells engrafting the indicated organs of NSG mice euthanized when they presented advanced symptoms of disease (7-8 weeks posttransplant) and analyzed by flow cytometry. Data are percentages (mean ± standard error of the mean [SEM]) of GFP+ engrafted cells normalized to percentages of GFP+ injected cells (C,E) (n = 5). (G) Flow cytometry of IL-7R surface expression on primary human T-ALL5 cells upon transduction with shIL7R or shsc (GFP+) and culture for 3 days onto OP9-GFP cells plus rhIL-7 (left panels). Line graphs show percentages of IL-7R+ cells on electronically gated GFP+ transduced cells from a representative experiment (right panels). Shaded graphs show background staining with irrelevant isotype-matched antibodies. (H) Surface IL-7R expression levels on primary T-ALL5 cells transduced and cultured as in (G). Data are mean ± SEM of mean fluorescence intensity (MFI) values of GFP+ transduced cells (n = 3). (I) Percentages of T-ALL5 cycling cells transduced and cultured as in (G). Data are percentages (mean ± SEM) of GFP+ transduced cells in the S+G2/M phases of the cell cycle, as determined by Hoestch labeling and flow cytometry (n = 3). *P < .05, **P < .01.
IL-7R signaling is essential for LIC function and progression of human T-ALL in vivo. (A) LIC potential of IL-7Rhi (blue) and IL-7R−/lo (red) primary human T-ALL8 cells (from T-ALL patient 8) that were sorted by fluorescence-activated cell sorting based on IL-7R expression levels (high or negative-to-low, respectively), as shown in supplemental Figure 2, and transplanted into NSG mice under limiting dilution conditions (from 80 to 8 × 104 cells per mouse, ≥4 mice per dose). LIC frequency was calculated using ELDA software by recording numbers of leukemia-free mice in the PB at 5 and 7 weeks posttransplant (p.t.). (B) Kaplan-Meier curves of leukemia-free mice transplanted in (A) with 80 FACS-sorted IL-7Rhi or IL-7R−/lo T-ALL8 cells. Flow cytometry analysis of the efficiency of transduction of human primary T-ALL5 (from T-ALL patient 5) (C) and T-ALL8 (E) cells with lentiviral vectors encoding an shRNA against IL-7R (shIL7R) or an shsc, together with GFP. Numbers indicate the percentages of transduced (GFP+) cells. Relative numbers of transduced (GFP+) T-ALL5 (D) or T-ALL8 (F) cells engrafting the indicated organs of NSG mice euthanized when they presented advanced symptoms of disease (7-8 weeks posttransplant) and analyzed by flow cytometry. Data are percentages (mean ± standard error of the mean [SEM]) of GFP+ engrafted cells normalized to percentages of GFP+ injected cells (C,E) (n = 5). (G) Flow cytometry of IL-7R surface expression on primary human T-ALL5 cells upon transduction with shIL7R or shsc (GFP+) and culture for 3 days onto OP9-GFP cells plus rhIL-7 (left panels). Line graphs show percentages of IL-7R+ cells on electronically gated GFP+ transduced cells from a representative experiment (right panels). Shaded graphs show background staining with irrelevant isotype-matched antibodies. (H) Surface IL-7R expression levels on primary T-ALL5 cells transduced and cultured as in (G). Data are mean ± SEM of mean fluorescence intensity (MFI) values of GFP+ transduced cells (n = 3). (I) Percentages of T-ALL5 cycling cells transduced and cultured as in (G). Data are percentages (mean ± SEM) of GFP+ transduced cells in the S+G2/M phases of the cell cycle, as determined by Hoestch labeling and flow cytometry (n = 3). *P < .05, **P < .01.
Il7r is transcriptionally induced by oncogenic Notch1 at early stages of T-ALL pathogenesis
The addiction of human T-ALL cells with LIC activity to IL-7R function suggests that IL-7R signaling could contribute to T-ALL pathogenesis. To investigate this possibility, we used a modification of the reported murine model of T-ALL induced by oncogenic Notch1,42 in which irradiated immunodeficient NSG (H2Kd) mice, rather than isogenic mice, were transplanted with C57BL/6 (H2Kb) BM Lin− c-kit+ HPCs transduced with ICN1 and GFP, which efficiently engrafted the BM of host mice (Figure 3A) and generated ectopic CD4+CD8+ double-positive (DP) CD3/TCRαβ+ aberrant cells (Figure 3B). NSG hosts allowed for a rapid expansion of ICN1-transduced cells, which finally overgrew nontransduced HPCs and led to an aggressive leukemia with a short latency (≤5 weeks) and a 100% penetrance that resulted in a short mouse survival (≤40 days; Figure 3C-D). Importantly, essentially all leukemic cells developing from murine ICN1-transduced HPCs in the BM of distinct host mice (moT-ALL1 and moT-ALL2) acquired a DP phenotype and high IL-7R expression levels, whereas conventional DP thymocytes derived from nontransduced progenitors displayed low IL-7R expression levels (Figure 3E). IL-7R expression was likewise observed in most murine leukemic cells infiltrating spleen, thymus, and liver (Figure 3F), indicating that Notch1-dependent oncogenesis parallels IL-7R induction. Notably, levels of IL-7R expression correlated with those of ICN1, as shown in a representative (moT-ALL3) leukemia (Figure 3G), suggesting a direct regulation of IL-7R expression by Notch1.

Notch1-dependent T-ALL pathogenesis parallels the induction of IL-7R expression. (A) Schematic diagram of mouse T-ALL generation. BM Lin− c-kit+ HPCs from C57BL/6 (H2-Kb+) mice were transduced with a lentiviral vector encoding the active form of Notch1 plus GFP (ICN1+) and then transplanted into NSG (H2-Kd+) immunodeficient mice. Mice were euthanized and analyzed when advanced symptoms of disease were evident. (B) CD4, CD8, CD3, and TCRαβ expression analyzed by flow cytometry (right panels) on electronically gated (left panel) ICN1-transduced (ICN1+) or nontransduced (ICN1−) H2-Kb+ donor cells engrafting the BM of recipient mice at 2 weeks posttransplant. (C) Percentages of ICN1+ and ICN1− H2-Kb+ donor cells engrafting the BM of mice transplanted as in (A) at the indicated days posttransplant. Percentages at the day of transplant (t=0) are shown for comparison. Mean ± standard error of the mean (SEM) of 3 to 7 mice per group are shown. (D) Kaplan-Meier survival curves of mice transplanted with BM HPCs transduced with ICN1 and GFP (ICN1) or GFP alone (Ctrl), as in (A). (E) Flow cytometry of IL-7R (right panels) and CD4/CD8 (left panels) expression displayed by conventional CD4+CD8+ DP cells from a normal C57BL/6 thymus (top row) or by moT-ALL1 and moT-ALL2 primary mouse leukemias (middle and bottom rows, respectively) generated in 2 independent mice transplanted as in (A). Shaded graphs show background staining with irrelevant isotype-matched antibodies. (F) Percentages of IL-7R–expressing cells within normal DP thymocytes from C57BL/6 mice (normal thymus) or ICN1+ DP moT-ALL2 cells engrafting the indicated organs of mice transplanted as in (A). Mean ± SEM values of ≥7 mice per group are shown. (G) IL-7R expression levels (right panel) displayed by primary T-ALL cells (moT-ALL3) generated as in (A) and electronically gated for high (ICN1hi) or intermediate (ICN1int) ICN1 expression (left panel). ICN1 expression was determined by GFP flow cytometry. **P < .01, ***P < .001.
Notch1-dependent T-ALL pathogenesis parallels the induction of IL-7R expression. (A) Schematic diagram of mouse T-ALL generation. BM Lin− c-kit+ HPCs from C57BL/6 (H2-Kb+) mice were transduced with a lentiviral vector encoding the active form of Notch1 plus GFP (ICN1+) and then transplanted into NSG (H2-Kd+) immunodeficient mice. Mice were euthanized and analyzed when advanced symptoms of disease were evident. (B) CD4, CD8, CD3, and TCRαβ expression analyzed by flow cytometry (right panels) on electronically gated (left panel) ICN1-transduced (ICN1+) or nontransduced (ICN1−) H2-Kb+ donor cells engrafting the BM of recipient mice at 2 weeks posttransplant. (C) Percentages of ICN1+ and ICN1− H2-Kb+ donor cells engrafting the BM of mice transplanted as in (A) at the indicated days posttransplant. Percentages at the day of transplant (t=0) are shown for comparison. Mean ± standard error of the mean (SEM) of 3 to 7 mice per group are shown. (D) Kaplan-Meier survival curves of mice transplanted with BM HPCs transduced with ICN1 and GFP (ICN1) or GFP alone (Ctrl), as in (A). (E) Flow cytometry of IL-7R (right panels) and CD4/CD8 (left panels) expression displayed by conventional CD4+CD8+ DP cells from a normal C57BL/6 thymus (top row) or by moT-ALL1 and moT-ALL2 primary mouse leukemias (middle and bottom rows, respectively) generated in 2 independent mice transplanted as in (A). Shaded graphs show background staining with irrelevant isotype-matched antibodies. (F) Percentages of IL-7R–expressing cells within normal DP thymocytes from C57BL/6 mice (normal thymus) or ICN1+ DP moT-ALL2 cells engrafting the indicated organs of mice transplanted as in (A). Mean ± SEM values of ≥7 mice per group are shown. (G) IL-7R expression levels (right panel) displayed by primary T-ALL cells (moT-ALL3) generated as in (A) and electronically gated for high (ICN1hi) or intermediate (ICN1int) ICN1 expression (left panel). ICN1 expression was determined by GFP flow cytometry. **P < .01, ***P < .001.
Functional assays showed that IL-7R expressed by ICN1-induced murine T-ALL cells responded to IL-7 and promoted STAT5 and AKT activation, leading to T-ALL cell expansion in culture (Figure 4A-B). Supporting the contribution of IL-7R signaling to murine T-ALL progression also in vivo, serial transplantation of ICN1-induced murine T-ALL cells into immunodeficient mice revealed increased IL-7R expression levels in sequential grafts (Figure 4C), which correlated with increased leukemia aggressiveness and decreased median survival of host mice (Figure 4D). Therefore, as shown in humans, IL-7R is a biomarker of T-ALL cells with LIC potential in mice.
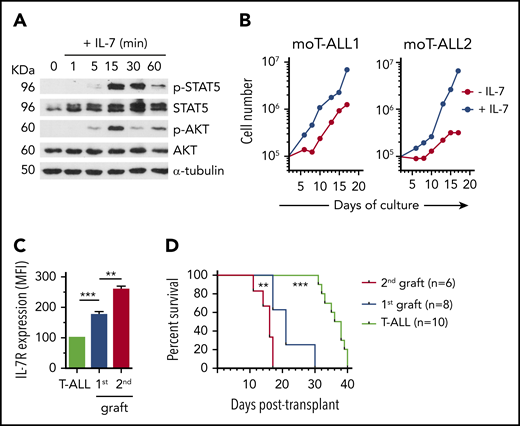
IL-7R expression is a biomarker of murine T-ALL cells with LIC potential. (A) Immunoblot analysis of STAT5 and AKT activation in a representative primary murine T-ALL (moT-ALL1), generated as in Figure 3A, and stimulated with IL-7 for the indicated times. α-Tubulin expression is shown as loading control. Molecular weight (KDa) is indicated on the left. (B) Total numbers of primary murine T-ALL cells (moT-ALL1 and moT-ALL2), generated as in Figure 3A, and cultured for the indicated days onto OP9-GFP cells in the presence or absence of IL-7. Data from a representative experiment of 3 are shown. (C) IL-7R expression levels of primary murine T-ALL cells (moT-ALL4) analyzed by flow cytometry immediately after isolation from the spleen of diseased mice in Figure 3A or after serial transplantation and spleen engraftment into consecutive NSG-immunodeficient mice (1st and 2nd grafts). Mean fluorescence intensity (MFI) values (mean ± SEM) from 3 to 12 mice are shown. (D) Kaplan-Meier survival curves of NSG mice serially transplanted with primary murine moT-ALL4 cells as in (C). **P < .01, ***P < .001.
IL-7R expression is a biomarker of murine T-ALL cells with LIC potential. (A) Immunoblot analysis of STAT5 and AKT activation in a representative primary murine T-ALL (moT-ALL1), generated as in Figure 3A, and stimulated with IL-7 for the indicated times. α-Tubulin expression is shown as loading control. Molecular weight (KDa) is indicated on the left. (B) Total numbers of primary murine T-ALL cells (moT-ALL1 and moT-ALL2), generated as in Figure 3A, and cultured for the indicated days onto OP9-GFP cells in the presence or absence of IL-7. Data from a representative experiment of 3 are shown. (C) IL-7R expression levels of primary murine T-ALL cells (moT-ALL4) analyzed by flow cytometry immediately after isolation from the spleen of diseased mice in Figure 3A or after serial transplantation and spleen engraftment into consecutive NSG-immunodeficient mice (1st and 2nd grafts). Mean fluorescence intensity (MFI) values (mean ± SEM) from 3 to 12 mice are shown. (D) Kaplan-Meier survival curves of NSG mice serially transplanted with primary murine moT-ALL4 cells as in (C). **P < .01, ***P < .001.
IL7R is a transcriptional NOTCH1 target in humans,30 but this functional link has not been demonstrated in mice, despite the fact that human and mouse IL7R/Il7r promoters share a conserved CSL/RBP-Jk binding site, located at −1937 bp from the ATG initiation codon of murine Il7r30 (Figure 5A). ChIP assays of CD4−CD8−IL-7R+ mouse embryonic thymocytes (Figure 5B) revealed that the reported site of murine Il7r promoter could bind endogenous RBP-Jk as efficiently as the Notch1 target Il2ra (Figure 5C-D), and luciferase reporter assays confirmed that active Notch1 induced Il7r gene transcription through RBP-Jk binding to the Il7r promoter (Figure 5E). Moreover, reporter activity due to active Notch1 constitutively expressed in Jurkat cells was significantly inhibited by ectopic expression of mutant Notch coactivator dnMAML1,37 and site-directed mutagenesis at the RBP-Jk binding site likewise resulted in impaired transcription from the Il7r reporter construct (Figure 5F), supporting functional involvement of this site in Notch1-induced murine IL-7R expression. Therefore, Il7r transcription and IL-7Rα protein expression are part of the oncogenic program induced by aberrant Notch1 signaling during murine T-ALL pathogenesis.
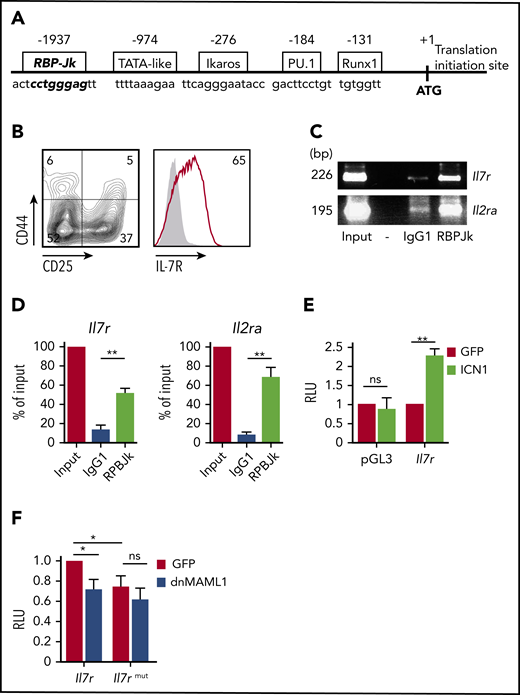
Il7r is a transcriptional Notch1 target in murine T-ALL pathogenesis. (A) Schematic representation of the 5′ regulatory region of murine Il7r. Numbers represent base pairs from the translation initiation site (+1, atg, initiation codon). The sequence of a putative RBP-Jκ binding site (bold type) was identified at −1937 bp. Binding sequences for other regulators are also shown. (B) Flow cytometry analysis of IL-7R (right panel) and CD44 and CD25 (left panel) expression on thymocytes from embryonic day 14.5 murine fetal thymus. Numbers indicate percentages of positive cells. Shaded graph shows background staining with irrelevant isotype-matched antibodies. (C) Representative ChIP assay of fetal murine thymocytes in (B), using anti–RBP-Jκ or isotype-matched control antibody (IgG1). PCR amplification of input and immunoprecipitated DNA was performed using primer pairs spanning the RBP-Jκ site identified in Il7r or the reported Il2ra RBP-Jκ site. Size in base pairs (bp) is indicated on the left (n = 3); -, empty lane. (D) Quantitative analysis of ChIP assays in (C). Results are shown as percentage of enrichment compared with input. Bars represent mean ± standard error of the mean (SEM) of 3 independent experiments. Luciferase reporter assays of Jurkat cells cotransfected with the reporter vector pGL3 (empty or containing the Il7r RBP-Jκ binding site) along with a retrovirus encoding ICN1 and GFP (ICN1) or GFP alone (GFP) as control (n = 3) (E) or pGL3 containing the wt or a mutated RBP-Jκ binding site in Il7r, along with retrovirus encoding dnMAML1 fused to GFP (dnMAML1) or GFP alone (GFP) (n = 5) (F). Data are fold induction of relative luciferase activity (RLU) ± SEM. *P < .05, **P < .01. ns, not significant.
Il7r is a transcriptional Notch1 target in murine T-ALL pathogenesis. (A) Schematic representation of the 5′ regulatory region of murine Il7r. Numbers represent base pairs from the translation initiation site (+1, atg, initiation codon). The sequence of a putative RBP-Jκ binding site (bold type) was identified at −1937 bp. Binding sequences for other regulators are also shown. (B) Flow cytometry analysis of IL-7R (right panel) and CD44 and CD25 (left panel) expression on thymocytes from embryonic day 14.5 murine fetal thymus. Numbers indicate percentages of positive cells. Shaded graph shows background staining with irrelevant isotype-matched antibodies. (C) Representative ChIP assay of fetal murine thymocytes in (B), using anti–RBP-Jκ or isotype-matched control antibody (IgG1). PCR amplification of input and immunoprecipitated DNA was performed using primer pairs spanning the RBP-Jκ site identified in Il7r or the reported Il2ra RBP-Jκ site. Size in base pairs (bp) is indicated on the left (n = 3); -, empty lane. (D) Quantitative analysis of ChIP assays in (C). Results are shown as percentage of enrichment compared with input. Bars represent mean ± standard error of the mean (SEM) of 3 independent experiments. Luciferase reporter assays of Jurkat cells cotransfected with the reporter vector pGL3 (empty or containing the Il7r RBP-Jκ binding site) along with a retrovirus encoding ICN1 and GFP (ICN1) or GFP alone (GFP) as control (n = 3) (E) or pGL3 containing the wt or a mutated RBP-Jκ binding site in Il7r, along with retrovirus encoding dnMAML1 fused to GFP (dnMAML1) or GFP alone (GFP) (n = 5) (F). Data are fold induction of relative luciferase activity (RLU) ± SEM. *P < .05, **P < .01. ns, not significant.
IL-7R signaling is essential for Notch1-mediated T-ALL pathogenesis
To investigate the contribution of IL-7R to T-ALL pathogenesis, we compared the impact of active Notch1 on HPCs from Il7r transgenic mice38 or wt C57BL/6 mice transplanted into immunodeficient NSG mice. We found that both ICN1-transduced HPCs developed ectopic DP CD3+TCRαβ+ aberrant cells with equivalent kinetics (supplemental Figure 4A-B) and generated an aggressive T-cell leukemia that infiltrated peripheral organs with similar efficiencies, despite the fact that IL-7R expression levels were significantly higher in Il7r-transgenic mice than in wt leukemic cells (supplemental Figure 4C-D). Accordingly, median survival was identical in both settings (supplemental Figure 4E). These data suggest that IL-7R expression levels induced by oncogenic Notch1 in normal HPCs are sufficient for ICN1-dependent leukemogenesis, but they did not provide formal evidence of IL-7R involvement in the process.
To conclusively establish whether IL-7R signaling collaborates with oncogenic Notch1 in T-ALL pathogenesis, we next analyzed the impact of oncogenic Notch1 in Il7r-deficient (Il7r−/−) HPCs.39,40 Strikingly, Il7r deletion prevented ectopic development of ICN1+ aberrant cells in the BM of transplanted mice, whereas nontransduced Il7r−/− HPCs were capable of engrafting the host BM as efficiently as wt HPCs, generating normal myeloid cells that reached the periphery (Figure 6A-D); however, lymphoid generation was impaired as expected.39,40 More importantly, Il7r deletion completely abrogated Notch1-induced leukemogenesis in transplanted mice, which showed a disease-free survival period > 20 weeks, with no signs of engraftment or expansion of ICN1+ cells in thymus, spleen, liver, or PB (Figure 6E-F). In contrast, ICN1+ wt HPCs generated ectopic DP aberrant T cells, which infiltrated peripheral organs and produced an overt leukemia with a median survival < 40 days (Figure 6B,D-E). Therefore, oncogenic Notch1 is not sufficient for cell transformation in the absence of IL-7R expression. These data demonstrate that IL-7R signaling is crucial for Notch1-induced T-ALL pathogenesis.
![IL-7R signaling is essential for Notch1-induced T-ALL pathogenesis. BM Lin− c-kit+ HPCs isolated from wt or Il7r−/− H2-Kb+ mice were transduced with lentiviral vectors encoding ICN1 and GFP and transplanted into H2-Kb− immunodeficient mice. (A) CD4 and CD8 expression of electronically gated transduced (ICN1+) and nontransduced (ICN1−) H2-Kb+Il7r wt or Il7r−/− donor cells engrafting the BM of transplanted mice at 4 weeks posttransplant. Results are representative of 1 of ≥18 mice per group (n = 3). Engraftment potential of ICN1-transduced (B) and total H2-Kb+ (C) donor cells. Data are percentages (mean ± standard error of the mean [SEM]) of donor cells recovered from the peripheral blood of ≥18 mice per group at 4 weeks posttransplant (n = 3). (D) Percentages of myeloid-lineage (CD11b+ or Gr1.1+), B-lineage (B220+), T-lineage (DP; CD4+CD8+ and/or CD8+CD4−) or Lin− (CD11b−, Gr1.1−, B220−, CD4−, CD8−) cells derived from transduced (ICN1+) and nontransduced (ICN1−) donor cells engrafting the spleen of transplanted mice at 5 weeks posttransplant. Mean ± SEM values from ≥18 mice per group are shown (n = 3). (E) Kaplan-Meier survival curves of immunodeficient mice transplanted with wt or Il7r−/− BM HPCs transduced with ICN1. (F) Percentages of ICN1+ transduced cells recovered at 5 weeks posttransplant from the indicated organs of immunodeficient mice transplanted with wt or Il7r−/− BM HPCs. Mean ± SEM percentages of ≥14 mice per group are shown (n = 3). **P < .01, ***P < .001. ns, not significant.](https://ash.silverchair-cdn.com/ash/content_public/journal/blood/134/24/10.1182_blood.2019000982/4/m_bloodbld2019000982f6.png?Expires=1769206809&Signature=SQo9SP24Xjl0pAeQj2hD0RR95ht31r2gaEsj7zXxTAnW7HPlRwf3U~Hq8UXCMbzCgYZZxtztyjNtolYKsBgL6OV-mMIlSMULkude-8W4kOXpjx7lFMxUcZ~GMTI2~Js7zh1Pbi0z6rqb4u33F1TKtilsGG0ydw1NUg1O~-LDh88uxrX2F36xBuLwMiAm-HkTisy4pETcWCSzbXjeg8MG6NuN9MZu73NAeW6TCoN3~UNB6-4SSisQbFdILo8ylt~Gbvg1rkza2hw3Dp6qApdcbvOcIys34N-xwrkWO8jFEJb~LZbeObO7H1UfYI5Gb0vzcmyJzsa69i~b4uuyYc8SYg__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
IL-7R signaling is essential for Notch1-induced T-ALL pathogenesis. BM Lin− c-kit+ HPCs isolated from wt or Il7r−/− H2-Kb+ mice were transduced with lentiviral vectors encoding ICN1 and GFP and transplanted into H2-Kb− immunodeficient mice. (A) CD4 and CD8 expression of electronically gated transduced (ICN1+) and nontransduced (ICN1−) H2-Kb+Il7r wt or Il7r−/− donor cells engrafting the BM of transplanted mice at 4 weeks posttransplant. Results are representative of 1 of ≥18 mice per group (n = 3). Engraftment potential of ICN1-transduced (B) and total H2-Kb+ (C) donor cells. Data are percentages (mean ± standard error of the mean [SEM]) of donor cells recovered from the peripheral blood of ≥18 mice per group at 4 weeks posttransplant (n = 3). (D) Percentages of myeloid-lineage (CD11b+ or Gr1.1+), B-lineage (B220+), T-lineage (DP; CD4+CD8+ and/or CD8+CD4−) or Lin− (CD11b−, Gr1.1−, B220−, CD4−, CD8−) cells derived from transduced (ICN1+) and nontransduced (ICN1−) donor cells engrafting the spleen of transplanted mice at 5 weeks posttransplant. Mean ± SEM values from ≥18 mice per group are shown (n = 3). (E) Kaplan-Meier survival curves of immunodeficient mice transplanted with wt or Il7r−/− BM HPCs transduced with ICN1. (F) Percentages of ICN1+ transduced cells recovered at 5 weeks posttransplant from the indicated organs of immunodeficient mice transplanted with wt or Il7r−/− BM HPCs. Mean ± SEM percentages of ≥14 mice per group are shown (n = 3). **P < .01, ***P < .001. ns, not significant.
IL-7R signaling is essential for Notch1-induced T-ALL pathogenesis. BM Lin− c-kit+ HPCs isolated from wt or Il7r−/− H2-Kb+ mice were transduced with lentiviral vectors encoding ICN1 and GFP and transplanted into H2-Kb− immunodeficient mice. (A) CD4 and CD8 expression of electronically gated transduced (ICN1+) and nontransduced (ICN1−) H2-Kb+Il7r wt or Il7r−/− donor cells engrafting the BM of transplanted mice at 4 weeks posttransplant. Results are representative of 1 of ≥18 mice per group (n = 3). Engraftment potential of ICN1-transduced (B) and total H2-Kb+ (C) donor cells. Data are percentages (mean ± standard error of the mean [SEM]) of donor cells recovered from the peripheral blood of ≥18 mice per group at 4 weeks posttransplant (n = 3). (D) Percentages of myeloid-lineage (CD11b+ or Gr1.1+), B-lineage (B220+), T-lineage (DP; CD4+CD8+ and/or CD8+CD4−) or Lin− (CD11b−, Gr1.1−, B220−, CD4−, CD8−) cells derived from transduced (ICN1+) and nontransduced (ICN1−) donor cells engrafting the spleen of transplanted mice at 5 weeks posttransplant. Mean ± SEM values from ≥18 mice per group are shown (n = 3). (E) Kaplan-Meier survival curves of immunodeficient mice transplanted with wt or Il7r−/− BM HPCs transduced with ICN1. (F) Percentages of ICN1+ transduced cells recovered at 5 weeks posttransplant from the indicated organs of immunodeficient mice transplanted with wt or Il7r−/− BM HPCs. Mean ± SEM percentages of ≥14 mice per group are shown (n = 3). **P < .01, ***P < .001. ns, not significant.
Addiction to IL-7R signaling of primary B-ALL cells
Because some B-ALL cases express functional IL-7R and respond to IL-7, leading to heterogeneous proliferative responses,17-19,23,48 we sought to investigate whether IL-7R signaling can mediate B-ALL progression (supplemental Table 2; supplemental Figure 5). Supporting this possibility, PDX assays revealed that IL-7R expression increased progressively in serially transplanted B-ALL xenografts, which activate IL-7R signaling in response to IL-7 (supplemental Figure 5). Moreover, shRNA-mediated IL-7R silencing (Figure 7A,C) dramatically impaired B-ALL PDX engraftment into immunodeficient mice, as indicated by a marked inhibition of leukemia burden in all organs analyzed compared with host mice receiving shsc-transduced B-ALL cells (Figure 7B,D). Likewise, silencing of functional IL-7R expressed by REH and NALM6 B-ALL cell lines impaired tumor growth of subcutaneous implants (supplemental Figure 6). These data suggest that B-ALL cells with LIC activity are addicted to IL-7R signaling, indicating that IL-7R critically cooperates with distinct oncogenes during T-cell or B-cell leukemogenesis.

IL-7R plays a key role in LIC function and progression of human B-ALL. (A) Human primary B-ALL blasts were transduced with lentiviral vectors encoding shIL7R or a scrambled sequence (shsc), together with GFP, and cells were then injected into NSG immunodeficient mice. Mice were euthanized and analyzed for leukemic content when they presented advanced symptoms of disease. Transduction efficiency of transplanted B-ALL8 (from B-ALL patient 8) (A) and B-ALL9 (from B-ALL patient 9) (C) cells. Numbers indicate percentages of GFP+ transduced cells. Percentages of transduced (GFP+) B-ALL8 (B) and B-ALL9 (D) cells engrafting the indicated organs of host mice 10 to 11 weeks posttransplant. Mean (± SEM) percentages are shown (n = 4). **P < .01.
IL-7R plays a key role in LIC function and progression of human B-ALL. (A) Human primary B-ALL blasts were transduced with lentiviral vectors encoding shIL7R or a scrambled sequence (shsc), together with GFP, and cells were then injected into NSG immunodeficient mice. Mice were euthanized and analyzed for leukemic content when they presented advanced symptoms of disease. Transduction efficiency of transplanted B-ALL8 (from B-ALL patient 8) (A) and B-ALL9 (from B-ALL patient 9) (C) cells. Numbers indicate percentages of GFP+ transduced cells. Percentages of transduced (GFP+) B-ALL8 (B) and B-ALL9 (D) cells engrafting the indicated organs of host mice 10 to 11 weeks posttransplant. Mean (± SEM) percentages are shown (n = 4). **P < .01.
Discussion
In this study, we have investigated whether normal IL-7R/IL-7 signaling contributes to leukemia maintenance and progression,15-22 as well as plays a major role in the leukemogenic process, as reported for mutant IL7R signaling.4-7,11,49 This is a key question of great clinical impact, because molecular pathways contributing to cell transformation and LIC activity are prime therapeutic targets for the development of novel strategies aimed at reducing chemotherapy intensity and preventing disease relapse, the major hurdle in T-ALL. We approached this critical issue using a seminal Notch1-induced murine T-ALL model,42 representative of a majority (>60%) of T-ALL patients harboring activating mutations in NOTCH1,32 the transcriptional regulator of IL7R in human T-ALL.30,31 We show that IL-7R overexpression is an early event and a functional marker of Notch1-dependent T-cell leukemogenesis, also involved in T-ALL proliferation and progression. In addition, Il7r loss-of-function approaches provide novel in vivo evidence that normal IL-7R–mediated signaling is essential for Notch1 oncogenicity and T-ALL pathogenesis, and functional assays demonstrate that IL-7R expression contributes to T-ALL LIC function, a finding that can be extended to human B-ALL cases expressing IL-7R. Therefore, IL-7R/IL-7 signaling may cooperate with distinct oncogenes in ALL leukemogenesis and relapse.
The contribution of the IL-7R/IL-7 axis to T-ALL progression has been known for many years17 and might be an expected function for a cytokine pathway that plays a key role in lymphopoiesis.39,40 However, no evidence has yet been provided that normal IL-7R signaling is essential for leukemogenesis. Given that the loss of leukemia induction seen with the Il7r knockout progenitors is similar to what has been reported with a number of other knockout models (ie, Rag2, Slp76)50 with impaired β-selection, our results may simply reflect the fact that the Il7r−/− progenitors cannot differentiate to the β-selection checkpoint that is susceptible to ICN1-induced leukemogenesis. However, the identification of T-ALL cases blocked at the early pre-T/stage I but harboring NOTCH1-activating mutations51 suggests that IL-7R could be required by itself in T-ALL pathogenesis, at least in those T-ALL cases. In addition, the pathological relevance of IL-7R may not be limited to T-ALL cases triggered by oncogenic NOTCH1, as modeled here. Rather, a wider impact can be predicted considering that activation of IL-7R–associated signaling pathways is linked to distinct oncogenic alterations in T-ALL.51 Moreover, there is a high selective pressure for NOTCH1 mutation in distinct T-ALL subtypes, regardless of the driving oncogene involved.51,52 Accordingly, Notch1 activation is an early hallmark of T-cell leukemogenesis and a key regulator of T-ALL LIC activity in a wide range of T-ALL cases,53,54 2 features that could now be extended to the IL-7R pathway.
The proposed contribution of IL-7R to Notch1-induced leukemia is in agreement with the finding that IL-7Rα expression is triggered very early during murine T-ALL generation and is also a hallmark of T-ALL pathogenesis in humans that allows tracking preleukemic and leukemic cell emergence in vivo (data not shown) in a new model of Notch1-induced human T-ALL.55 These data confirm a conserved role for Notch1 as a transcriptional regulator of IL-7Rα expression during leukemia development, as well as suggest the participation of a conserved RBP-Jk binding site.30 In addition, accumulating evidence supports a conserved role for Notch1 in IL-7R regulation via a far 3′ enhancer.31 Collectively, these data suggest that the oncogenic relevance revealed here for the Notch1/IL-7R axis in murine T-ALL pathogenesis might be conserved in humans. Considering that the contribution of the IL-7R/IL-7 axis in leukemia engraftment and progression can be extended to B-ALL, it is possible that mechanisms alternative to oncogenic NOTCH1 might regulate IL7R transcription and, thus, LIC function in ALL pathogenesis. Moreover, there might be a high selective pressure for mutations that induce excessive IL-7Rα–mediated signaling in ALL.29
The general role revealed here for IL-7R as a functional biomarker of ALL cells with LIC potential has significant clinical relevance. New therapeutic developments against ALL now rely on molecular targets involved in LIC activity.56 Therefore, therapeutic approaches targeting LIC activity remain an important challenge for the eradication of residual chemoresistant leukemic blasts in relapsed patients. Thus, our results open a new and major area of research to develop and validate novel IL-7Rα–targeted therapies. Consequently, drugs targeting IL-7Rα itself might be a promising treatment for a great majority of T-ALL patients and some B-ALL patients, regardless of the presence of IL7R-activating mutations,57 as has recently been suggested by anti–IL-7Rα mAb administration in T-ALL and B-cell leukemia xenograft models.58,59 Moreover, the demonstration that IL-7R signaling directly contributes to T-ALL LIC activity and leukemogenesis provides proof of concept that IL-7Rα targeting represents a promising therapeutic approach to eradicate disease relapse. Despite the fact that optimization of such immunotherapeutic strategies is mandatory to prevent potential side effects leading to T-cell aplasia and severe immunodeficiency, we can conclude that therapeutic targeting of IL-7R signaling may lead to improved outcomes in a majority of T-ALL patients, as well as in some B-ALL patients.
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Acknowledgments
The authors thank J. C. Aster, K. Weijer, H. Spits, J. C. Zúñiga-Pflücker, A. Rodríguez, and A. Singer for providing useful reagents and mouse strains and Juan Alcain for technical support.
This work was supported in part by funds from Plan Nacional, Ministerio de Ciencia, Innovación y Universidades SAF2013-44857-R and SAF2016-75442-R (Agencia Estatal de Investigación/European Regional Development Fund, European Union), Fundación Asociación Española Contra el Cáncer (AECC CI13131229, CICPF18030TORI), Fundación Uno Entre Cien Mil, and Fundación Lair. Institutional grants from the Fundación Ramón Areces and Banco de Santander to the Centro de Biología Molecular Severo Ochoa are also acknowledged.
Authorship
Contribution: S.G.-G. and M.L.T. designed experiments and wrote the manuscript; S.G.-G., P.F., M.M., A.E. and T.P. performed experiments; M.R. and A.P.-M. provided human samples and clinical experience; and A.E.C. provided critical animal models and helpful suggestions.
Conflict-of-interest disclosures: The authors declare no competing financial interests.
Correspondence: María L. Toribio, Centro de Biología Molecular Severo Ochoa, CSIC-UAM, C/Nicolás Cabrera, 1, Universidad Autónoma de Madrid, 28049 Madrid, Spain; e-mail: mtoribio@cbm.csic.es.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal