

Abstract
The 2010 and 2017 editions of the European LeukemiaNet (ELN) recommendations for diagnosis and management of acute myeloid leukemia (AML) in adults are widely recognized among physicians and investigators. There have been major advances in our understanding of AML, including new knowledge about the molecular pathogenesis of AML, leading to an update of the disease classification, technological progress in genomic diagnostics and assessment of measurable residual disease, and the successful development of new therapeutic agents, such as FLT3, IDH1, IDH2, and BCL2 inhibitors. These advances have prompted this update that includes a revised ELN genetic risk classification, revised response criteria, and treatment recommendations.
Introduction
Since the 2017 report from the European LeukemiaNet (ELN),1 there has been substantial progress in our knowledge of acute myeloid leukemia (AML). Recent advances significantly influence clinical practice. These advances include insights into the clinical value of genomic abnormalities for diagnosis and prognosis, the clinical significance of inherited predisposition to AML, technological advancements in the quantitative assessment of measurable residual disease (MRD) and their utility for assessing therapeutic response and disease risk, the development of a range of novel therapeutic agents, and developments in allogeneic hematopoietic cell transplantation (HCT), resulting in new disease classification,2 diagnostic and prognostic algorithms, and updated therapeutic practices. The current report highlights these advances and updates their implications for the standard of care and for clinical trials in AML.
Methods
The panel included international members with recognized clinical and research expertise in AML. Literature and relevant abstract review, categorization of evidence, and arrival at consensus recommendations were developed as previously reported.1,3 For diagnosis and management of acute promyelocytic leukemia (APL), readers are referred to the respective recommendations.4
AML classification
Molecular landscape
Somatic mutations drive the development of AML. Although the epigenetic state of leukemia cells, the bone marrow microenvironment, the health of normal hematopoietic cells, and other features are important for leukemia biology, somatic mutations can be assessed readily with current techniques. Leukemia develops from the serial acquisition of somatic mutations in hematopoietic stem and progenitor cells with the capacity to self-renew and propagate the neoplastic clone.5,6 Initiating mutations may lead to an expanded clone of cells that is apparent in the peripheral blood, termed clonal hematopoiesis, a common pre-malignant state that increases in prevalence with age.7 Although some mutations, such as those in DNMT3A, TET2, and ASXL1, are more common in clonal hematopoiesis and appear to be relatively early events in leukemogenesis, others tend to be acquired later in the course of leukemia development, including mutations in FLT3, NRAS, and RUNX1. The combinations of mutations that ultimately drive leukemogenesis are influenced by biological cooperativity and mutual exclusivity between mutated genes.
General classification
The International Consensus Classification of AML2,8 that updated the prior revised fourth edition World Health Organization (WHO) classification of AML9 introduced changes in the blast thresholds and new genetic entities to define AML, further expanding the spectrum of classification identified by cytogenetic and mutational profiles (Table 1). Because of their overriding impact on disease phenotype and disease outcome, genetic aberrations are given priority in defining AML disease classification, with additional predisposing features (therapy-related, prior myelodysplastic syndrome [MDS] or MDS/myeloproliferative neoplasm [MPN], germline predisposition) appended as qualifiers of the primary diagnosis. A summary of the hierarchical classification is depicted in Figure 1.
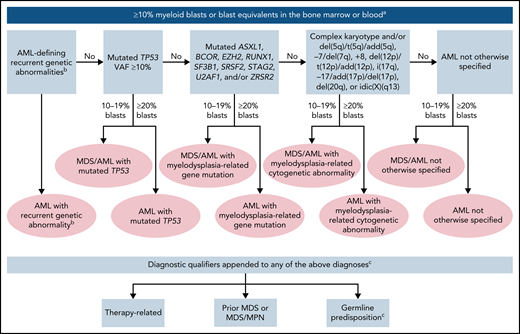
Hierarchical classification of the International Consensus Classification of AML. The classification is hierarchical (ie, AML with recurrent genetic abnormalities takes precedence over all other categories); among the remaining categories, AML with mutated TP53 supersedes AML with myelodysplasia-related gene mutations, and the latter supersedes AML with myelodysplasia-related cytogenetic abnormalities. aMyeloblasts, monoblasts, and megakaryoblasts are included in the blast count. Monoblasts and promonocytes, but not abnormal monocytes, are counted as blast equivalents in AML with monocytic or myelomonocytic differentiation, and promyelocytes in the setting of PML::RARA or variant RARA rearrangement. Cases with prior diagnosis of MPN are excluded and are classified as accelerated (10%-19% blasts) or blast phase (≥20% blasts) MPN. For patients who already have a history of MDS/MPN (eg, CMML), the diagnosis of MDS/MPN should be retained until there are ≥20% blasts/blast equivalents; however, once an AML-defining recurrent genetic abnormality (eg, KMT2A rearrangement or NPM1 mutation) is detected and the blast count is ≥10%, AML-type therapy is recommended. bAML-defining recurrent genetic abnormalities are t(15;17)(q24.1;q21.2)/PML::RARA; t(8;21)(q22;q22.1)/RUNX1::RUNX1T1; inv(16)(p13.1q22) or t(16;16)(p13.1;q22)/CBFB::MYH11; t(9;11)(p21.3;q23.3)/MLLT3::KMT2A; t(6;9)(p22.3;q34.1)/DEK::NUP214; inv(3)(q21.3q26.2) or t(3;3)(q21.3;q26.2)/GATA2, MECOM(EVI1); mutated NPM1; in-frame bZIP mutated CEBPA; t(9;22)(q34.1;q11.2)/BCR::ABL1; other recurrent rearrangements involving RARA, KMT2A, MECOM, and other rare rearrangements as listed in Table 1. The entity is named with the specific genetic abnormality. Cases with BCR::ABL1 rearrangement and 10% to 19% blasts are classified as CML in accelerated phase, and cases with history of CML and ≥20% blasts are classified as CML in myeloid blast phase. cExamples how to append diagnostic qualifiers: AML with myelodysplasia-related cytogenetic abnormality, therapy-related; AML with myelodysplasia-related gene mutation, prior myelodysplastic syndrome; AML with myelodysplasia-related gene mutation, germline RUNX1 mutation (ie, gene or syndrome should be specified).
Hierarchical classification of the International Consensus Classification of AML. The classification is hierarchical (ie, AML with recurrent genetic abnormalities takes precedence over all other categories); among the remaining categories, AML with mutated TP53 supersedes AML with myelodysplasia-related gene mutations, and the latter supersedes AML with myelodysplasia-related cytogenetic abnormalities. aMyeloblasts, monoblasts, and megakaryoblasts are included in the blast count. Monoblasts and promonocytes, but not abnormal monocytes, are counted as blast equivalents in AML with monocytic or myelomonocytic differentiation, and promyelocytes in the setting of PML::RARA or variant RARA rearrangement. Cases with prior diagnosis of MPN are excluded and are classified as accelerated (10%-19% blasts) or blast phase (≥20% blasts) MPN. For patients who already have a history of MDS/MPN (eg, CMML), the diagnosis of MDS/MPN should be retained until there are ≥20% blasts/blast equivalents; however, once an AML-defining recurrent genetic abnormality (eg, KMT2A rearrangement or NPM1 mutation) is detected and the blast count is ≥10%, AML-type therapy is recommended. bAML-defining recurrent genetic abnormalities are t(15;17)(q24.1;q21.2)/PML::RARA; t(8;21)(q22;q22.1)/RUNX1::RUNX1T1; inv(16)(p13.1q22) or t(16;16)(p13.1;q22)/CBFB::MYH11; t(9;11)(p21.3;q23.3)/MLLT3::KMT2A; t(6;9)(p22.3;q34.1)/DEK::NUP214; inv(3)(q21.3q26.2) or t(3;3)(q21.3;q26.2)/GATA2, MECOM(EVI1); mutated NPM1; in-frame bZIP mutated CEBPA; t(9;22)(q34.1;q11.2)/BCR::ABL1; other recurrent rearrangements involving RARA, KMT2A, MECOM, and other rare rearrangements as listed in Table 1. The entity is named with the specific genetic abnormality. Cases with BCR::ABL1 rearrangement and 10% to 19% blasts are classified as CML in accelerated phase, and cases with history of CML and ≥20% blasts are classified as CML in myeloid blast phase. cExamples how to append diagnostic qualifiers: AML with myelodysplasia-related cytogenetic abnormality, therapy-related; AML with myelodysplasia-related gene mutation, prior myelodysplastic syndrome; AML with myelodysplasia-related gene mutation, germline RUNX1 mutation (ie, gene or syndrome should be specified).
Changes to the blast thresholds defining AML
All recurrent genetic abnormalities (Table 1) that define specific subtypes of AML, with the exception of AML with t(9;22)(q34.1;q11.2)/BCR::ABL1, are now considered to establish a diagnosis of AML if there are ≥10% blasts in the bone marrow or blood. The clinical behavior of myeloid neoplasms with these rearrangements reflects the specific genetic abnormality, even for cases presenting with <20% blasts.10-18 This 10% blast threshold aligns with previously AML-defining abnormalities, such as PML::RARA, CBFB::MYH11, and RUNX1::RUNX1T1.19 To avoid potential overlap with chronic myeloid leukemia in accelerated phase, AML with BCR::ABL1 still requires ≥20% blasts.
Although all other AML subtypes require ≥20% blasts for diagnosis, a new category of MDS/AML has been introduced in association with defined genomic abnormalities to include cases with 10% to 19% blasts in the bone marrow or blood to recognize the fact that these cases lie on the border between AML and MDS in terms of their biology and prognosis (Table 1).20-25 Patients diagnosed with MDS/AML should be eligible for either MDS or AML clinical trials and treatment approaches.
Antecedent AML history
An important change to the classification is the removal of the former categories AML with myelodysplasia-related changes (AML-MRC) and therapy-related myeloid neoplasms. Recent data indicate that genetic characteristics, rather than clinical history (de novo, secondary after an antecedent MDS or MDS/MPN, or therapy-related), have most relevance in classifying biologically distinct AML subgroups.6,26 Dysplastic morphology, currently used as a criterion for AML-MRC, lacks independent prognostic significance.27-29 Thus, although a prior history of MDS or MDS/MPN and prior exposure to therapy are still important features to note in the diagnosis, they are now applied as diagnostic qualifiers to the AML-defining category (Table 1; Figure 1).
AML with recurrent genetic abnormalities
This category has been expanded to include additional variant translocations involving RARA, KMT2A, and MECOM, as well as other rare recurring translocations, which are now recognized as AML-defining entities (Table 1).14,30,31 Recent studies show that in-frame mutations affecting the basic leucine zipper (bZIP) region of CEBPA confer a favorable outcome, irrespective of their occurrence as biallelic or monoallelic mutations.32-35 In-frame bZIP variants are found in 90% and 35% of cases with biallelic and monoallelic CEBPA mutations, respectively. Gene expression analyses support a distinct biology associated with CEBPA bZIP mutation in AML. Accordingly, this AML subtype has been redefined to only require an in-frame bZIP CEBPA mutation for classification rather than the previous requirement for biallelic CEBPA abnormalities.
AML with mutated TP53, AML with myelodysplasia-related gene mutations, and AML with myelodysplasia-related cytogenetic abnormalities
Accumulating evidence indicates that from both a clinical and molecular perspective, TP53-mutant AML and MDS represent a distinct disease entity. The vast majority of TP53-mutant cases have complex karyotypes, and in about half, TP53 mutations occur in the absence of other AML-associated gene mutations. Clinically, these myeloid neoplasms are associated with a very poor prognosis.6,36-41 The presence of a pathogenic TP53 mutation (at a variant allele fraction of at least 10%, with or without loss of the wild-type TP53 allele) defines the new entity AML with mutated TP53.
Cases lacking TP53 mutation, but with mutations in ASXL1, BCOR, EZH2, RUNX1, SF3B1, SRSF2, STAG2, U2AF1, and/or ZRSR2, are categorized as AML with myelodysplasia-related gene mutations, irrespective of any prior history of MDS. These mutations are highly associated with AML following prior MDS or MDS/MPN and confer an adverse prognosis even if they occur in de novo AML.6,26,42-45 AML with myelodysplasia-related gene mutations encompasses the prior provisional category of AML with mutated RUNX1.
The new category AML with myelodysplasia-related cytogenetic abnormalities now includes cases previously classified as AML-MRC due to the presence of myelodysplasia-associated cytogenetic findings, but lacking TP53 or myelodysplasia-related gene mutations.46
Of note, the classification is hierarchical (Figure 1); ie, “AML with mutated TP53” takes precendence over “AML with myelodysplasia-related gene mutations,” and the latter supercedes “AML with myelodysplasia-related cytogenetic abnormalities.”
The remaining AML cases are categorized as “AML, not otherwise specified” (irrespective of the presence or absence of multilineage dysplasia). The 4 categories described above are designated as AML/MDS if the bone marrow or blood blast count is 10% to 19% and as AML with ≥20% blasts (Table 1; Figure 1). Cases that have both a specific AML-defining recurrent genetic abnormality and TP53 mutation and/or myelodysplasia-related gene mutations or cytogenetics should be classified according to the defined recurrent genetic abnormality. Although complex karyotypes and certain co-mutation profiles confer adverse prognosis to some genetic AML subtypes, these are captured in the prognostic stratification scheme and do not affect their primary diagnostic classification.
Therapy-related AML
Currently comprising 10% to 15% of all newly diagnosed AML, the incidence of cases showing relatedness to previous therapy for another disease continues to rise due in part to increasing numbers of cancer survivors at risk.47 As mentioned above, “therapy-related AML” is no longer considered a disease entity, but the term “therapy-related” is now used as a diagnostic qualifier to the disease entities that are primarily defined by their genetic profile.
These neoplasms have been thought to be the direct consequence of mutational events induced by cytotoxic therapy and/or selection of chemotherapy-resistant clones.48-50 In general, these AMLs are associated with adverse genetic lesions, and more than 90% show an abnormal karyotype.51,52 The more common subtype, seen in ∼75% of cases, typically presents 5 to 7 years after first exposure to alkylating agents or radiation, is often preceded by MDS, and is frequently accompanied by chromosome 5 and/or 7 abnormalities, complex karyotype, and TP53 mutations.48,49,52,53 Some individuals develop AML after treatment with topoisomerase II inhibitors, with breakage at topoisomerase II sites leading to abnormal recombination and balanced translocations involving KMT2A at 11q23.3, RUNX1 at 21q22.1, or RARA at 17q21.2. In these cases, the latency period is shorter, often it is only 1 to 3 years, and antecedent MDS is rare.
Another pathogenetic pathway is represented by cases with a preexisting myeloid clone that is resistant to chemotherapy.52 Clonal hematopoiesis of indeterminate potential may be the first step in a multi-hit model.54,55 Cases were identified in which the exact TP53 mutation found at diagnosis was already present at low frequency in blood or bone marrow many years before AML development.52 These data suggest a model in which hematopoietic stem cells carrying mutations in TP53 or PPM1D undergo positive selection by cytotoxic therapy, ultimately leading to AML.56,57 Mutations in the RAS/MAPK pathway, alterations in RUNX1 or TP53, and KMT2A rearrangements are also frequent somatic drivers in pediatric AML related to previous therapy, but unlike in adults, most cases appear to represent independent clones arising as a consequence of cytotoxic therapy and not preexisting minor clones.50
Deleterious mutations typical of familial cancer predisposition syndromes in the homologous recombination DNA repair pathway, particularly BRCA1, BRCA2, PALB2, TP53, or CHEK2, are observed in ∼20% of cases.58,59 The identification of such preexisting conditions facilitates screening and counseling of patients prior to treatment of their primary disease, family donor selection for allogeneic HCT, cancer/organ surveillance strategies, and cascade testing within families.60
Germline predisposition
Increasingly, individuals are being recognized as having an inherited germline predisposition to hematopoietic malignancies (Table 2).61,62 Recognition of such hereditary predispositions impacts patient management, especially if there is consideration for an allogeneic HCT and health surveillance strategies for the patient and relatives who share the causative variant. Clinical testing for these syndromes is difficult for most clinicians given their relative lack of experience regarding these conditions, requirement for obtaining germline DNA for testing (Table 3), and a lack of standardization in the field regarding which patients and which genes should be tested.63
Germline predisposition risk should be considered for all patients diagnosed with a hematopoietic malignancy regardless of age, because some germline predisposition alleles, like those in DDX41, can drive hematopoietic malignancies in older age.64,65 When identified, germline predisposing disorders should be applied as diagnostic qualifiers to the specific AML disease category. Key features of the clinical presentation that should prompt consideration of germline testing are given in Table 3. Clinicians should familiarize themselves with academic and commercial testing options, including the culture and sequencing of skin fibroblasts, thereby excluding somatic mutations in hematopoietic cells, and the panel of genes to be analyzed (Table 2).63 Germline variants are categorized as pathogenic, likely pathogenic, variant of uncertain significance, likely benign, or benign; only pathogenic and likely pathogenic variants are considered causative of disease and are followed clinically in families. However, gene variant classification can change over time as additional information regarding gene/allele function and/or segregation data from families becomes available, and variants of uncertain significance in particular are often reclassified as likely pathogenic or pathogenic.
Certain germline disorders are associated with specific characteristics that are important for clinicians to recognize (Table 2), those associated with quantitative and qualitative platelet defects: ANKRD26, ETV6, and RUNX1, and those associated with other organ dysfunction: GATA2 with immunodeficiency; Shwachman Diamond syndrome with exocrine pancreas insufficiency and skeletal dysplasia; Fanconi anemia with facial dysmorphism, squamous cell carcinomas, and liver tumors; and dyskeratosis congenita with pulmonary fibrosis, liver cirrhosis, and vascular anomalies; among others. Some disorders are associated only with myeloid malignancies (eg, CEBPA), whereas others confer risk to a variety of hematopoietic malignancies and solid tumors. The tumor spectrum associated with each disorder may expand over time as more individuals and families are identified. Germline predisposition alleles that confer risk to lymphoid malignancies are emerging and often overlap with the myeloid malignancy risk genes.
Because the treatment plan for many patients with AML includes allogeneic HCT and relatives are the preferred donors, testing for germline risk alleles should be performed as early as possible during clinical management. Use of hematopoietic donor stem cells from carriers of deleterious RUNX1 and CEBPA variants is prohibitive, but we lack data for most predisposition genes and whether any variants are permissive to transplantation.66 Future studies that lead to a comprehensive list of all predisposition genes will advance our ability to provide the best treatments for patients and their families and will facilitate strategies to maintain health for them throughout their lifetimes.
Diagnostic procedures
All tests necessary to establish the diagnosis, risk classification, and the other procedures recommended to be performed at diagnosis are listed in Table 4.
Immunophenotyping
Immunophenotyping by multiparameter flow cytometry (MFC) is required to diagnose AML accurately by identifying cell surface and intracellular markers (Table 5). Because of the heterogeneity of AML, no marker is expressed in all cases. It is also important to identify leukemia-associated immunophenotypes (LAIP) for subsequent MRD monitoring by MFC. In cases where an aspirate is unobtainable and circulating blasts are absent, myeloid phenotype may be confirmed on a core biopsy using immunohistochemistry.
Cytogenetic and molecular studies
Conventional cytogenetic analysis is mandatory in the evaluation of AML. If conventional cytogenetics fails, fluorescence in situ hybridization is an alternative to detect specific abnormalities like RUNX1::RUNX1T1, CBFB::MYH11, KMT2A (MLL), and MECOM (EVI1) gene fusions, or myelodysplasia-related chromosome abnormalities, eg, loss of chromosome 5q, 7q, or 17p material (Table 1).
Molecular genetic testing should screen for all the genetic abnormalities that define disease and risk categories or that are needed for targeted treatment modalities (Table 4). These tests can be performed by commercially available gene panel diagnostics or platforms simultaneously testing for mutations and rearrangements. When AML with germline predisposition is suspected, a dedicated gene panel including known predisposing alleles should be used. However, caution should be used in interpreting data from tumor-based panels, because hematopoietic tissues undergo somatic reversion frequently leading to false-negative results, and panel-based testing is often not able to detect germline copy number variants, which are relatively common predisposition alleles.
For patients with mutant NPM1 and core-binding factor (CBF)-AML, it is recommended to perform baseline molecular assessment by quantitative polymerase chain reaction (qPCR) or droplet digital PCR (dPCR) to facilitate MRD monitoring after treatment.
Biobanking
At least in clinical studies, but preferably also outside this context, bone marrow and blood samples should be obtained at time of diagnosis, at remission, and at relapse and stored under appropriate conditions (DNA and RNA stored at −80°C and viable cells stored at −196°C). Broad informed consent should be obtained to allow for performance of correlative laboratory studies. In addition, a sample from healthy tissue should be stored to enable delineation of germline from somatic mutations.
2022 European LeukemiaNet genetic risk classification at diagnosis
Since 2017, new data have emerged that prompted the need to adjust the risk classification. In addition to baseline genetic characterization, the importance of response to initial therapy and assessment of early MRD in individual risk assignment are highlighted.67 In clinical practice, a patient with favorable-risk AML may be reclassified as intermediate-risk or vice versa, based on the presence or absence of MRD, respectively. For instance, this is particularly relevant for patients with NPM1-mutant AML.68-70
The most important changes made to the previous risk classification are outlined in Table 6. (1) The FLT3-ITD allelic ratio is no longer considered in the risk classification; consequently, AML with FLT3-ITD (without adverse-risk genetic lesions) are now categorized in the intermediate-risk group, irrespective of the allelic ratio or concurrent presence of NPM1 mutation. The reason for this change relates to methodological issues with standardizing the assay to measure the FLT3-ITD allelic ratio, the modifying impact of midostaurin-based therapy on FLT3-ITD without NPM1 mutation,71 and the increasing role of MRD in treatment decisions. (2) AML with myelodysplasia-related gene mutations is now categorized in the adverse-risk group. These mutations, typically associated with AML following an antecedent hematologic disease, are also prevalent in de novo AML and indicate adverse risk even in the absence of myelodysplasia-related cytogenetic abnormalities.6,26,42,44,45 Beyond the previously considered ASXL1 and/or RUNX1 genes, this category of myelodysplasia-related gene mutations now includes pathologic variants in at least one of the ASXL1, BCOR, EZH2, RUNX1, SF3B1, SRSF2, STAG2, U2AF1, or ZRSR2 genes. (3) The presence of adverse-risk cytogenetic abnormalities in NPM1-mutated AML now defines adverse risk. A meta-analysis has shown that NPM1-mutated AML with adverse cytogenetic abnormalities is associated with a poor outcome.72 Whether other genetic abnormalities (eg, myelodysplasia-related gene mutations) also confer unfavorable outcome to NPM1-mutated AML is under investigation. (4) As mentioned previously, recent studies have shown that in-frame mutations affecting the basic leucine zipper region of CEBPA confer the favorable outcome, irrespective of their occurrence as biallelic or monoallelic mutations and therefore are now categorized in the favorable-risk group.32,34,35 (5) Additional disease-defining recurring cytogenetic abnormalities are included in the adverse-risk group, including t(3q26.2;v) involving the MECOM gene,31,73 or t(8;16)(p11.2;p13.3) associated with KAT6A::CREBBP gene fusion.14 (6) Finally, hyperdiploid karyotypes with multiple trisomies (or polysomies) are no longer considered complex karyotypes and as adverse risk.74
Although numerous reports have studied mutations in other genes, for example, IDH1/IDH2 or DNMT3A, current evidence does not yet warrant their assignment to a distinct ELN prognostic group. Also, the emerging therapeutic use of targeted inhibitors might impact prognostic outcome in IDH1/IDH2-mutated AML. Finally, it should be emphasized that the ELN AML risk classification has been developed based on data from intensively treated patients and may warrant modifications for patients receiving less intensive therapies.
Monitoring of measurable residual disease
MRD assessment in AML is used to (1) provide a quantitative methodology to establish a deeper remission status; (2) refine postremission relapse risk assessment; (3) identify impending relapse to enable early intervention; and (4) as a surrogate end point to accelerate drug testing and approval.75
Currently, the 2 most extensively evaluated methodologies are multiparameter flow cytometry-based MRD (MFC-MRD) and molecular MRD (Mol-MRD) assessed by qPCR.76 Emerging exploratory technologies are next-generation sequencing (NGS) and dPCR (Table 7).77 The current update of the ELN recommendation on MRD includes new technical recommendations for standardized MFC-MRD and Mol-MRD analysis, MRD thresholds, definitions of MRD response, and suggestions for clinical implications.67
Multiparameter flow cytometry
Integration of diagnostic LAIP that distinguish AML cells from normal hematopoietic cells in an individual patient and the more generally defined “different from normal” aberrant immunophenotype (DfN) allow both for tracking of diagnostic and emerging clones and should include core MRD markers (Table 5).67 MFC-MRD assessment should be performed with a qualified assay based on guidelines for rare event detection.78 Evaluation of residual leukemic stem cells (LSC) by MFC-MRD is still investigational but is recommended for evaluation in clinical studies. The prognostic value of LSC-MRD has been associated with a higher sensitivity and lower false negativity.79,80 LSC can be immunophenotypically defined as CD34+/CD38low cells combined with an aberrant marker not present on normal HSCs (eg, CD45RA [PTPRC], CLL-1 [CLEC12A], or CD123 [IL3RA]).81
Molecular MRD
The technique used, including qPCR and dPCR, should reach a limit of detection of at least 10−3. Either peripheral blood or bone marrow may be used, although sensitivity in blood is generally lower by an order of magnitude compared with bone marrow. Leukemia-related abnormalities suitable for qPCR monitoring include mutated NPM1; CBFB::MYH11, RUNX1::RUNX1T1, KMT2A::MLLT3, DEK::NUP214, and BCR::ABL1 gene fusions; and WT1 expression.67 Validation is most robust for NPM1-mutated, as well as CBFB::MYH11 and RUNX1::RUNX1T1-positive AML.82
If using NGS, error-corrected targeted panel-based approaches are preferred.83 Care must be taken to recognize and exclude germline mutations. Mutations consistent with premalignant clonal hematopoiesis (eg, DNMT3A, TET2, ASXL1) should not be considered as MRD.84 Further study is required to identify and distinguish mutations truly indicative of residual AML from clonal hematopoiesis related abnormalities.85,86 It is important to note that NGS-based strategies currently lack standardization as a stand-alone technique for MRD assessment.
Implementation of MRD testing/decision making in AML
The prognostic value of MRD detection in complete remission (CR) or CR with incomplete hematologic recovery (CRi) has been demonstrated both in patients treated with intensive and more recently less-intensive treatment modalities.87-89 Various studies and a systematic meta-analysis of 81 publications have shown the prognostic value of MRD for relapse and overall survival (OS).68,87,90-93 Although MRD estimates furnish critical prognostic insights, they are imperfect, because relapse still occurs in a minority of MRD-negative patients. Thus, a negative MRD test result may not indicate complete disease eradication but refers to disease below the MRD test threshold in the tested sample. Conversely, not all patients who are MRD positive will relapse. Of note, Mol-MRD may remain detectable at low levels (CRMRD-LL) without prognostic significance, and therefore, are called negative operationally if the MRD values are below the threshold linked to prognosis.67 For instance, in CBF-AML and NPM1-mutant AML, the transcripts may show persistent low-level expression after treatment, but this is not prognostic of relapse.68,70,94-96 The presence of detectable MRD before transplant is an independent unfavorable predictor of posttransplant outcome.97-100 However, there is currently no evidence showing benefit of additional courses of intensive chemotherapy prior to transplant in CR1 patients who are MRD positive. If fit enough, such patients should be considered candidates for a myeloablative conditioning (MAC) regimen or an early taper of posttransplant immunosuppression.98
Definitions of MRD response categories and molecular relapse are listed in Table 8. In Figure 2, the recommended time points for MRD evaluation and clinical decision making are depicted for NPM1-mutated, CBF-AML, and AML assessed by MFC.
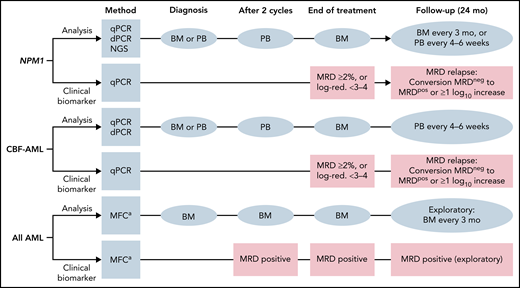
Algorithm of MRD assessment and time points at which MRD is considered a clinically relevant biomarker. Blue squares indicate timepoints of assessment and source of material; pink squares indicate timepoints for treatment modification based on a clinical relevant biomarker: for example, if the level of molecular MRD as assessed by qPCR is ≥2% or if there is failure to reduce mutant transcript levels by 3 to 4 log after completion of consolidation chemotherapy, treatment modifications (eg, allogeneic hematopoietic cell transplantation) may be considered; similarly, if patients are still MRD positive by MFC after 2 cycles of intensive chemotherapy or at end of treatment. For patients receiving less intensive therapy, timepoints for assessment and clinical decision making are not yet established. Modified from 2021 ELN MRD recommendations67 BM, bone marrow; CBF, core-binding factor. aMFC as assessed by LAIP or the DfN method.
Algorithm of MRD assessment and time points at which MRD is considered a clinically relevant biomarker. Blue squares indicate timepoints of assessment and source of material; pink squares indicate timepoints for treatment modification based on a clinical relevant biomarker: for example, if the level of molecular MRD as assessed by qPCR is ≥2% or if there is failure to reduce mutant transcript levels by 3 to 4 log after completion of consolidation chemotherapy, treatment modifications (eg, allogeneic hematopoietic cell transplantation) may be considered; similarly, if patients are still MRD positive by MFC after 2 cycles of intensive chemotherapy or at end of treatment. For patients receiving less intensive therapy, timepoints for assessment and clinical decision making are not yet established. Modified from 2021 ELN MRD recommendations67 BM, bone marrow; CBF, core-binding factor. aMFC as assessed by LAIP or the DfN method.
Response criteria and outcome measures
Response criteria
CR, CRi, partial remission (PR), and morphologic leukemia-free state (MLFS)
The criterion “absence of blasts with Auer rods” was eliminated.
CR with partial hematologic recovery
The term CR with partial hematologic recovery (CRh) has been introduced for patients with morphologic bone marrow blast clearance and partial recovery of both neutrophils (≥0.5 × 109/L [500/µL]) and platelets (≥50 × 109/L [50 000/µL]) because those represent clinical benefit to the patient; other CR criteria need to be met. Thus far, CRh has only been used in the context of trials evaluating less-intensive therapies. It is recommended that future studies validate the role of CRh as a surrogate measure of survival after intensive and less-intensive therapies.
Response criteria with MRD assessment
The 2017 ELN recommendations included the term CR without MRD (CRMRD−) to recognize the increasing role of MRD technologies in stratifying prognosis of patients in CR.1 The current response criteria expand MRD classification to include patients achieving CRh or CRi without MRD (CRhMRD− or CRiMRD−).
Time window for response assessment
To recognize the potential for continuing improvements in blood counts after myelosuppressive therapy, response definitions for patients with marrow blast clearance (<5%) may be adjusted to reflect the best hematologic response achieved prior to commencement of the next treatment cycle. Aspirate reports that include MLFS, CRh, or CRi should note the potential for post-marrow blood counts to alter the final response designation.
No response
Patients evaluable for response but not meeting the criteria for CR, CRh, CRi, MLFS, or PR will be categorized as having “no response.”
Nonevaluable for response
For accurate reporting of response, it is necessary to include all registered/randomized patients on an intention to treat principle. Therefore, patients nonevaluable for response should be included in the denominator of response assessment analyses. This category may include patients yet to have a response assessment, suffering early death, exiting the study early, or those with a technically suboptimal bone marrow sample precluding assessment. Patients previously categorized as having death in aplasia or from indeterminate causes are now designated as nonevaluable for response.
Treatment failure
Relapsed disease is defined as ≥5% leukemic blasts in the bone marrow, reappearance of leukemic blasts in peripheral blood (PB) in at least 2 PB samples at least 1 week apart, or development of new extramedullary disease.
Refractory disease
If a specified response has not been achieved by a defined landmark (ie, failure to achieve response after 2 cycles of intensive chemotherapy or a predetermined landmark, eg, 180 days after commencing less-intensive therapy), the patient will be designated as having refractory disease.
CR, CRh, or CRi with MRD relapse
Outcome measures
Systematic reporting of early death (eg, 30 and 60 days) is recommended to enable assessment of treatment-related mortality with new therapies being relevant for the therapy under consideration.
Although the primary end point for registrational studies in AML has historically been OS, the increased availability of poststudy treatment options with potential to confound OS interpretation may encourage adoption of alternative end points, such as event-free survival (EFS; or relapse-free survival [RFS] for postremission studies) as comparative outcome measures in registrational studies (see also “Clinical trials”). In a retrospective patient-level analysis of 8 randomized trials evaluating intensive chemotherapy conducted by the US Food and Drug Administration (FDA), EFS had the best correlation with OS when response was limited to a strict CR (R2 = 0.87; 95% confidence interval [CI], 0.47-0.98); EFS with the definition of response broadened to include CRi and CR with incomplete platelet recovery was also shown to correlate, albeit less strongly, with OS (R2 = 0.59; 95% CI, 0.13-0.93).101 Limitations of the analysis included relatively small sample sizes, heterogeneity among trials, and lack of multivariate analyses.
For drugs that add myelosuppression (eg, venetoclax, CPX-351, gemtuzumab ozogamycin), the sole use of a strict CR in the definition of EFS is increasingly challenged. We recommend broadening the definition of EFS to include CRh or CRi in response. Patients not achieving response by the predetermined landmark (refractory disease) should have the event recorded on day 1 of registration in nonrandomized trials (or day 1 of random assignment in randomized trials). Patients who die before reaching the response landmark and prior to/without response assessments are considered treatment failures and should have the event recorded at day 1 of registration/randomization. Patients alive but nonevaluable for response are censored at day 1 of registration/randomization. To enable consistency in trial reporting, a response landmark for failure to achieve response should be prespecified. Furthermore, the response landmark should be relevant for the treatment received; for example, after completion of 2 cycles of intensive therapy or 180 days after commencing less-intensive approaches.
The incorporation of MRD outcomes as a measure of treatment failure necessitates the inclusion of new terms incorporating molecular MRD relapse into time to event definitions for EFSMRD, RFSMRD, and cumulative incidence of relapse (CIRMRD; Table 9). For each study, clear definitions regarding how MRD relapse is determined should be specified in the statistical analysis plan.
Therapy for AML
The goal of treatment is control and, whenever possible, eradication of disease. This outcome is accomplished ideally by inducing a CR with initial therapy, followed by consolidation and/or maintenance efforts to deepen the remission and maximize response duration. The role of HCT and post-HCT therapies is discussed in the section on allogeneic HCT. Results of genetic analyses should be available as rapidly as possible, preferably within 3 to 5 days, to identify therapeutically actionable targets (Table 4). A short delay in starting treatment to stabilize patients and identify the best treatment option is recommended to optimize clinical outcome.102 If hyperleukocytosis is present, immediate cytoreduction is advised (see Management of special situations). If a patient cannot tolerate an active intensive or nonintensive treatment option, the purpose of therapy is to optimize quality of life and decrease the incidence of cytopenia-related complications with transfusion and other supportive care measures and early involvement of palliative care services as appropriate.
The survival of patients with AML that are related to previous therapy overall remains poor, which is mainly due to the high frequency of adverse (cyto)genetic features,103,104 but also to the sequelae of prior therapy and sometimes persistent primary disease. In general, patients should be managed according to the same general therapeutic principles depending on whether they are candidates for intensive or nonintensive therapy and allogeneic HCT.104,105 CPX-351 offers a new option for the treatment of these patients (see below).
Patients considered fit for intensive therapy
Induction therapy
Anthracyclines and cytarabine remain the backbone of intensive chemotherapy. Alternative options are fludarabine, cytarabine, granulocyte colony-stimulating factor, and idarubicin (FLAG-IDA) and mitoxantrone-based cytarabine regimens (Table 10). It has become standard to incorporate the kinase inhibitor midostaurin into first-line therapy for patients with FLT3-mutant AML. Midostaurin improved 4-year OS by 7.1%, from 44.3 to 51.4% when used as an adjunct to daunorubicin-cytarabine induction and high-dose cytarabine consolidation in patients 18 to 59 years of age.106 Although study treatment incorporated single-agent maintenance for 12 monthly cycles, the value of adding maintenance therapy remains uncertain.107 In a prospective nonrandomized study, midostaurin also showed a beneficial effect in patients up to 70 years of age in comparison with a historical control group.108
Newer and potentially more potent FLT3 inhibitors are currently under randomized evaluation as therapeutic alternatives to midostaurin.109,110 A placebo-controlled phase 3 trial enrolled 539 patients to either quizartinib or placebo in combination with intensive induction and consolidation chemotherapy followed by single agent quizartinib maintenance for up to 36 cycles in patients 18 to 75 years of age with FLT3-ITD–positive AML. Post-HCT maintenance was permitted. Although peer-reviewed results are not yet available, a preliminary meeting abstract reported prolonged OS for quizartinib compared with placebo. Grade ≥3 treatment-emergent neutropenia was more frequent in the quizartinib arm; early death (≤30 days) was 5.7% and 3.1% in the quizartinib compared with placebo arms, respectively.111
Gemtuzumab-ozogamicin (GO) is a humanized anti-CD33 IgG4 antibody chemically linked to a calicheamicin-based cytotoxic warhead. Following a history of initial FDA approval followed by retraction based on questionable clinical benefit, a subsequent randomized study demonstrated an EFS advantage among patients 50 to 70 years with de novo AML, with benefit limited to favorable or intermediate cytogenetic risk disease.112,113 Although 4 other open-label randomized studies individually failed to demonstrate improved survival for GO added to front line therapy in AML, a meta-analysis of all 5 studies indicated a benefit, particularly in patients with CBF-AML.114 In another randomized study, a reduction of the relapse probability and greater mutant NPM1 molecular clearance was shown in patients with NPM1-mutated AML, but with no EFS difference.70,115 GO dosed at 3 mg/m2 (capped at 5 mg) D1, 4, and 7 of induction and day 1 of consolidation has been approved for patients with previously untreated CD33 antigen positive AML in combination with daunorubicin and cytarabine, but a single dose of GO delivered on day 1 of induction may also be efficacious.114,116,117
CPX-351 is a dual-drug liposomal formulation that encapsulates cytarabine/daunorubicin in a 5:1 fixed molar ratio.118 In an open label phase 3 randomized study in newly diagnosed patients aged 60 to 75 years with disease subtypes including therapy-related AML, a history of MDS or CMML, or de novo AML with myelodysplasia-related cytogenetic abnormalities CPX-351 improved the clinical response rate and OS compared with induction with cytarabine-daunorubicin, followed by “5 + 2” consolidation.119 Five-year OS in the CPX-351 arm was improved from 10% to 18% compared with patients receiving “7 + 3.”120 CPX-351 delayed the median time to neutrophil and platelet recovery by approximately 7 days and increased the risk of bleeding. Early 30-day mortality, however, was not increased by CPX-351 (5.9%) compared with “7 + 3” (10.6%), and less mucositis was noted. Randomized data are lacking for patients under 60 years and for AML following prior MPN.
Consolidation therapy
After attainment of CR (or CRh/CRi), patients are consolidated ideally with regimens that include intermediate-dose cytarabine.121 Consecutive administration on days 1 to 3, rather than on alternate days (days 1, 3, and 5) may hasten blood count recovery.122,123 Although high-dose cytarabine (3000 mg/m2) is still used in some centers, its greater toxicity and failure to improve survival argues against its continued use.124-126
In addition to baseline risk factors, assessment of MRD in CR (or CRh/CRi) is recommended for patients with nonadverse risk in first remission to inform consolidation treatment choice. For patients with an estimated relapse risk exceeding 35% to 40%, consolidation with allogeneic HCT remains the preferred postremission option.127 These include patients with adverse-risk AML or nonadverse-risk disease with MRD persistence. Autologous HCT, although not widely used, offers an alternative postremission option for patients with favorable- or intermediate-risk disease with an adequate MRD response or for whom allogeneic HCT is not available.128 In the subset of patients receiving induction with a FLT3 inhibitor, GO or CPX-351, these agents may be incorporated into consolidation (Table 10).
Maintenance therapy
There is no generally accepted definition of “maintenance therapy.” In most previous trials, maintenance therapy has been administered for a defined period of time in patients who achieved remission after intensive chemotherapy. The FDA defines maintenance therapy for AML as an extended but time-limited course of treatment, that is usually less toxic, given after achievement of CR with the objective of reducing the risk of relapse. Thus, a trial designed to demonstrate the efficacy of maintenance therapy would need to include a specified induction and consolidation treatment followed by randomization to a predefined duration of treatment.129
The main objective of maintenance therapy is to deliver a minimally toxic therapy capable of reducing the risk of leukemic relapse. In a randomized study in newly diagnosed older patients in first remission after 2 cycles of intensive induction, azacitidine maintenance therapy, administered subcutaneously for up to 12 cycles, improved disease-free survival compared with no maintenance.130 An orally administered version of azacitidine, CC-486, given over 14 days in 28-day cycles as continuous postremission therapy, was shown subsequently in a randomized placebo-controlled trial to reduce relapse risk and improve median OS (from 14.8 to 24.7 months) among patients ≥ 55 years not considered candidates for allogeneic HCT.131 Oral azacitidine prolonged OS independently of the MRD status as assessed by MFC (47% of patients were MRD positive and 53% were MRD negative at study entry).132 Oral azacitidine is approved for continued treatment of patients with AML in first CR/CRi following intensive induction chemotherapy who are not able to complete intensive curative therapy, including allogeneic HCT. However, there are limitations to the trial design that prohibit generalizability of the data.133 First, data regarding the role of oral azacitidine in younger populations or patients with CBF-AML are lacking; furthermore, only few patients had AML with adverse-risk cytogenetics (14%). Second, because the trial did not specify prior induction and consolidation therapy, there was considerable variability in therapy prior to selection for maintenance (ie, 45% of patients had received 1 consolidation cycle, 31% had 2 cycles, and 20% had no consolidation).
Patients who received midostaurin during induction and consolidation may continue these agents in maintenance in line with the reported phase 3 experience.106
Patients not considered candidates for intensive therapy
There are no generally accepted or validated criteria to consider a patient ineligible for intensive chemotherapy. In the context of clinical trials, criteria have been used that consider a patient not eligible for intensive chemotherapy (for instance as defined in Table 11) that may also offer guidance in routine practice.
Substantial progress has been made in the management of patients considered unfit for intensive chemotherapy (Table 11). Compared with azacitidine alone, addition of the BCL2 inhibitor venetoclax improved clinical response (CR/CRi, 66.4% vs 28.3%) and median OS (14.7 vs 9.6 months), establishing a new standard of care for older or unfit patients with AML.134 To limit prolonged myelosuppression and the risk of tumor lysis syndrome associated with this regimen, management recommendations are outlined in Table 12.135 Although not evaluated in randomized clinical trials, phase 1b/2 studies suggest that clinical outcomes with decitabine plus venetoclax are similar to the azacitidine plus venetoclax combination.136 For patients failing frontline venetoclax-based therapy, prognosis appears very poor.137 For patients unable to receive a hypomethylating agent (HMA), low-dose cytarabine (LDC) in combination with venetoclax represents an alternative treatment option.138 Although an open-label randomized study showed improved survival for the hedgehog inhibitor glasdegib in combination with LDC, compared with LDC alone, the relatively low response rate (CR/CRi 24%) with this regimen does not favor its use as an alternative nonintensive option.139
For newly diagnosed patients with IDH1 mutation, results from a randomized study indicate that the IDH1 inhibitor ivosidenib plus azacitidine improves EFS (hazard ratio, 0.33; 95% CI, 0.16-0.69), clinical response (CR/CRh, 52.8 vs 17.6%), and median OS (24.0 vs 7.9 months) compared with azacitidine plus placebo.140 To identify patients suitable for ivosidenib at initial diagnosis, rapid IDH1 mutation screening in older patients with AML is recommended. Patients with IDH1/2-mutated AML who are considered too frail to tolerate HMA-based treatment may be offered best supportive care or monotherapy with targeted IDH1/IDH2 inhibitors.141
In patients receiving HMA-based combination therapy (with venetoclax, ivosidenib, other investigational agents), response should be assessed early during the first cycle (eg, on day 14-21) due to high rates of early responses seen with HMA combinations and the need to delay or modify dosing in the setting of persistent cytopenias in a leukemia-free marrow (Table 12). A second assessment is commonly performed after 3 cycles and then repeated every 3 cycles for patients in remission or at the discretion of the physician outside of a clinical trial. In the absence of treatment intolerance, nonintensive treatment approaches have commonly been continued until disease progression, but for the time being, there are no data supporting the advantage of an open-ended duration approach over therapy for a confined period.
Relapsed and refractory disease
Common salvage regimens for patients with refractory or relapsed disease are given in Table 10. At clinical progression, it is important to highlight the potential for clonal evolution and emergence of actionable targets not detected at diagnosis. Currently, these include emergence of IDH1/IDH2 mutations or new or expanded FLT3-ITD or FLT3 tyrosine kinase domain clones.142-146 Therefore, molecular re-evaluation at relapse is important to identify patients who may be suitable for targeted salvage options. In the interest of therapeutic progress, it is recommended to enter these patients into clinical trials whenever possible. Patients failing to achieve remission after 2 cycles of induction (including at least 1 cycle of intermediate-dose cytarabine) are defined as having primary refractory AML. Patients are unlikely to benefit from further cycles of conventional chemotherapy and instead should be referred for consideration of allogeneic HCT or participation in clinical trials.147
Factors associated with reduced survival at AML relapse include shorter RFS (<6-12 months), nonfavorable risk karyotype at diagnosis, older age (>45-55 years), or prior history of HCT.148,149 In general, after cytoreduction has been achieved, allogeneic HCT is recommended. If HCT is not a realistic option (eg, in the older patient), disease control using a nonintensive option, such as HMA with or without venetoclax, may be appropriate. For patients with relapsed/refractory FLT3-mutated disease, the kinase inhibitor gilteritinib has been approved based on a randomized trial showing improved response rates (CR, 21.1% vs 10.5%) and median OS (9.3 vs 5.6 months) in the gilteritinib arm compared with physician’s choice of salvage therapy.109,150 Although more patients receiving gilteritinib were bridged to HCT (25.5% vs 15.3%) and these patients were permitted to restart gilteritinib 30 to 90 days after HCT, the clinical benefit of post-HCT gilteritinib remains uncertain. In addition, only 5.7% of patients received prior midostaurin as first-line therapy in this study, making generalization of treatment outcomes after this and other FLT3 inhibitors difficult. In a randomized trial evaluating the FLT3 inhibitor quizartinib in patients with relapsed/refractory FLT3-ITD–positive AML, quizartinib also showed improved OS compared with conventional care regimens.110 However, after evaluation of the trial data, neither FDA nor the European Medicines Agency granted approval.
For patients with relapsed/refractory IDH1/IDH2-mutant AML, salvage with ivosidenib or enasidenib is a possibility because these IDH inhibitors induce CR rates in the range of 20% and overall response, including hematologic improvement in approximately 40%.151-153 Median time required to attain CR is ∼3 to 4 months, with 80% of cumulative responses attained after completion of 6 cycles of therapy.151,152 Among responders, molecular clearance with ivosidenib was observed in 21% and was associated with longer remission duration and prolonged survival.151 Although responders to enasidenib may also achieve molecular clearance, targeting IDH2 in a nonblinded randomized trial did not show improvement in OS compared with conventional care options among patients ≥60 years failing 2 or 3 prior lines of therapy.154 For management of adverse events associated with novel agents, see the section on supportive care below and Table 12.
Allogeneic hematopoietic cell transplantation
AML is the most frequent indication for allogeneic HCT.155,156 Advances allowing for the use of partially matched unrelated donors, cord blood, and haplo-identical family members mean that an allogeneic donor can be found for most patients in need. Nonmyeloablative and reduced intensity conditioning (RIC) regimens make allogeneic HCT possible in patients up to age 80 at experienced centers.157,158 With newly approved methods to prevent and treat both infections and graft-versus-host disease (GVHD), outcomes following transplant continue to improve, leaving disease recurrence as the major cause of treatment failure.159 Despite its central role in the management of adult AML, only a minority of patients for whom transplantation is indicated undergo the procedure.156 Reasons for underutilization include biologic factors, personal and physician choice, and lack of access.160
Indications for allogeneic HCT
The decision to perform allogeneic HCT during first remission depends on the risk-benefit ratio (ie, nonrelapse mortality [NRM] and disability/reduction in relapse risk) based on cytogenetic and molecular genetic features of disease at presentation and response to initial therapy, as well as patient, donor, and transplant factors. Allogeneic HCT should be considered when the relapse probability without the procedure is predicted to be >35% to 40%.127 For patients with favorable-risk disease, allogeneic HCT in CR1 is generally not recommended except for those with inadequate clearance of MRD.69,161-163 In contrast, allogeneic HCT is recommended for patients with adverse-risk AML and for the majority of those with intermediate-risk disease, although quite a few centers rely on the presence of MRD to guide their decision based on the predicted risk of relapse. For patients who are age 60 or older, mostly based on retrospective comparisons, allogeneic HCT in first remission is recommended for those with intermediate-risk or adverse-risk disease willing and able to undergo remission-inducing therapy.164,165 Judicious patient selection is important in patients over 60 especially regarding the presence of comorbidities and support at home. Allogeneic HCT is the only curative therapy for patients with primary refractory disease and offers the best chance for cure in those who relapse after initial chemotherapy.166 Other factors including comorbidities, donor source, and individual patient goals must be considered.
Comorbidities and risk scores
Several transplant-related models address the impact of comorbidities and disease risk.167 The HCT comorbidity index (which has been modified to include age) sums a patient’s comorbidities into a single score that predicts the likelihood of NRM following transplantation independent of the disease being treated.168,169 A disease-risk index based on disease-stage and cytogenetics predicts the likelihood of disease recurrence following transplantation independent of patient comorbidities.170 The modified European Society of Blood and Marrow Transplantation risk score combines both patient and disease risk factors thus predicting OS rather than NRM or relapse risk.171
Preparative regimen intensity
Transplant preparative regimens run the gamut from nonmyeloablative, which would result in only mild, temporary depression of blood counts without transplant, to RIC regimens of varying intensity, to high-dose true MAC. Prospective randomized trials yield inconsistent results, but in general, NRM is increased, and relapse rates are diminished with higher-dose regimens. The best evidence supporting the use of MAC regimens in patients aged 18 to 65 years comes from the randomized phase 3 BMT CTN 0901 study, which showed improved survival with MAC compared with RIC because of a marked reduction in disease recurrence.172,173 In a retrospective analysis, the benefit of MAC was greatest in patients with genomic evidence of residual disease before transplant, as determined by NGS at the time of transplant.98,100
Donor selection/GVHD prophylaxis
Registry analyses show approximate equivalence in outcomes for patients transplanted using a well-matched unrelated donor compared with those using a matched sibling donor.174,175 However, many patients lack a suitable sibling or volunteer unrelated donor. The recent demonstration that posttransplant cyclophosphamide GVHD prophylaxis is tolerable and results in encouraging outcomes using mismatched unrelated and haplo-identical donors substantially widens the donor pool.176-179 The use of single or double cord blood units with a high nucleated cell dose also results in excellent outcomes, particularly in patients with evidence of pretransplant MRD.179,180 Current data support the utilization of a matched sibling donor or well-matched unrelated donor as the preferred donor option in adults with AML.177 Recognition of germline predisposition in the patient with AML and family members influences donor selection, and the use of relatives with deleterious germline variants should be avoided (see Germline predisposition). Randomized trials comparing outcome after transplantation using a matched unrelated donor vs a haplo-identical donor are underway.
Pre- and posttransplant strategies to prevent posttransplant relapse
Disease relapse is the major cause of treatment failure in adults allografted for AML.181 For patients who are in CR1 following 2 cycles of intensive therapy, there is no evidence that additional chemotherapy prior to transplantation reduces the risk of relapse regardless of pretransplant MRD status. There is increasing interest in the use of pharmacological or cellular therapy posttransplant to prevent disease recurrence. In patients allografted for FLT3-mutated AML, randomized studies show that maintenance with the FLT3 inhibitor sorafenib, although sometimes challenging to deliver, reduces the risk of relapse, suggesting that the use of a FLT3 inhibitor is a reasonable option.182,183 A randomized trial examining the benefit of posttransplant maintenance with the second-generation FLT3 inhibitor gilteritinib in this patient population is in progress. There is less evidence supporting the use of other agents as posttransplant maintenance in AML. A randomized study of maintenance using subcutaneous azacitidine showed no benefit and is not recommended based on available evidence184; oral azacitidine (CC-486) is currently under study.
Relapse after transplant
Ninety percent of those who relapse after an allogeneic HCT for AML do so by 2 years. The outcome of patients with morphologic relapse within the first 12 months is very poor, although a rapid taper of immunosuppression or donor lymphocyte infusion may salvage a proportion of patients with early molecular or cytogenetic relapse.185,186 For patients relapsing after an allogeneic HCT for FLT3-mutated AML, giltertinib is the preferred treatment option with evidence of an emergent FLT3 mutant clone. In the pivotal study, giltertinib improved survival in patients with early relapses and was at least equivalent compared with intensive chemotherapy in relapses occurring beyond 6 months.109,150 Azacitidine, with or without donor lymphocyte infusion, and venetoclax-based salvage regimens may produce remissions in a small proportion of patients with less toxicity than intensive chemotherapy.187 Those who achieve a second CR can sometimes still be cured with either donor lymphocyte infusion or a second allograft.188
Clinical trials
It is recommended to enroll patients with AML onto clinical trials whenever a suitable trial opportunity is available. Real-time availability of rapid biomarker screening has become a basic requirement to enable timely enrollment of patients to clinical trials targeting defined AML subpopulations. Routine biobanking of patient samples should be standard practice to maximize clinical research.
Trial design
The execution of clinical trials for drug development in AML has become progressively challenging. There is an increasing number of novel AML therapeutics that warrant evaluation of safety and efficacy, in single agent and combination format, with many requiring prospective allocation to biologically defined genotypes. As AML is already a relatively rare disease, timely completion of adequately powered phase 3 clinical trials within smaller disease subsets has become more challenging, highlighting the growing need for intercontinental trials.
Early-phase clinical development
Innovation in clinical trial design is needed. Phase 1 exploration of new AML drugs in the relapsed/refractory setting remains a formidable task, with high levels of drug resistance, rapid disease progression, and complications related to severe cytopenias representing key hurdles to success. In such settings, a pharmacodynamic primary end point verifying the drug’s proposed mechanism of action may represent an appropriate objective during the single agent dose-finding stage, followed by rapid transition to combination testing to demonstrate clinical efficacy. In phase 2, multiarm biomarker-stratified studies permitting parallel investigation of several drugs simultaneously will facilitate more efficient screening of new drugs and combinations for clinical activity.189
Phase 3 trials
Randomized trials are the cornerstone of drug approval, especially in newly diagnosed patients. Accelerated recruitment to such trials is of central importance to improvements in outcome for patients with AML and yet paradoxically this remains a notoriously slow process. For example, the regulatory approval of midostaurin in 2017 for FLT3-mutant AML using OS as the primary study end point took almost a decade. The increasing number of new therapies in AML coupled with genomic stratification is creating significant challenges to the timely recruitment of patients to practice informing trials. As more effective salvage therapies are now available, the OS end point is complicated further by subsequent lines of AML-directed therapy; crossover of patients from the control arm to novel agents has confounded the interpretation of OS increasingly in comparative studies (see “Outcome measures”). EFS as a primary study end point will not only eliminate the confounding effect of poststudy therapies, but as an additional advantage, it will shorten study completion timelines. In this regard, the use of the restricted “traditional” CR as one of the key events in EFS has become subject of debate. Because of frequent myelosuppression with novel drug combinations, and in addition, the need to proceed with therapy before full hematologic recovery, from a therapeutic point, it has become increasingly unrealistic to consider failure to attain CRh/CRi as events in EFS estimates even though the level of survival after CRh/CRi may be below that following CR.101 Another way of expediting earlier assessment of drug efficacy is to base outcomes on standardized MRD measurements.67 To facilitate incorporation of MRD as an efficacy end point, CR (or CRh/CRi) with MRD response and EFS with molecular MRD relapse as an event represent promising new study end points. This will allow for direct comparisons between the quantitative depth of response of investigational and reference therapies as indicators of relative therapeutic value.
Acceleration of drug development could also benefit from using a validated control population, thus omitting the concurrent standard control arm so that all patients recruited to the trial receive investigational therapy. To realize such an approach, a well annotated and contemporary external reference cohort is required and efforts to establish real-world databases for this purpose are being explored. Finally, it remains of utmost importance to override geographic and interstudy group barriers and continue efforts to stimulate the formation of “global” alliances and networks to expedite completion of registration-enabling clinical studies within a markedly condensed time window.
New therapies
Clinical investigation of new therapies and new combinations is of critical importance in continuing to improve AML patient outcomes.190 Drug development strategies have focused until now primarily on single-agent dose finding studies in the relapsed setting, which have led to successful approvals of targeted therapies, such as FLT3, IDH1, and IDH2 inhibitors, and is the pattern for the current evaluation of menin inhibitors for patients with KMT2A rearrangements or NPM1 mutations.109,151,152,191
Other agents (ie, epigenetically targeted therapies) and immunotherapy approaches including bi-specific T-cell engaging antibodies, checkpoint inhibitors, and chimeric antigen receptor T cells or natural killer cells are likely to be most effective in the setting of MRD, in frontline, or early salvage combination approaches.190,192,193 Although of limited single agent activity, the CD47 inhibitor magrolimab has demonstrated preliminary activity in combination with azacitidine in patients with newly diagnosed MDS and AML, even in the setting of TP53-mutated disease. Trials of various inhibitors of the CD47-SIRP-α macrophage checkpoint are currently under various stages of early clinical evaluation.194
Due to the changing therapeutic environment, which now includes HMAs in combination with small molecule inhibitors like venetoclax or targeted therapies, future development of frontline combinations is now more complex. The evaluation of so-called “triplet” therapies is an increasingly common clinical trial design for “chemotherapy-ineligible” patients, which involves the evaluation of a third agent (either approved or investigational) to the HMA and venetoclax “backbone.” New combination trials in intensive chemotherapy eligible patients typically involve the incorporation of a new target or agent in combination with standard chemotherapy, such as the ongoing clinical trials of the FLT3 inhibitor gilteritinib with standard “7 + 3” vs “7 + 3” and midostaurin or the spleen tyrosine kinase (SYK) inhibitor with “7 + 3” vs “7 + 3” alone for NPM1-mutant AML.
In addition, oral formulations of the HMAs are now approved for AML maintenance (oral azacitidine)131 and high-risk MDS (oral decitabine/cedazuridine),195 respectively, and given the increased patient convenience of oral formulations, it is likely that these agents will be increasingly used in future HMA-based combination trials.
Management of special situations and supportive care
A white blood cell count (WBC) > 100 × 109/L is generally defined as hyperleukocytosis and associated with increased induction mortality mainly due to hemorrhagic events, tumor lysis syndrome, and the risk for clinical leukostasis syndrome.196 Hydroxyurea (up to 50-60 mg/kg per day) is most commonly used to lower the WBC below 25 × 109/L, particularly before the commencement of HMA- or venetoclax-based treatments. Clinical leukostasis syndrome is a medical emergency requiring the WBC to be promptly lowered without delay by either hydroxyurea or planned induction therapy and a restrictive transfusion policy for red blood cells. Retrospective studies suggest a beneficial effect of dexamethasone, which may counteract effects of leukostasis.197 Although leukapheresis may be performed in parallel with chemotherapy in patients with leukostasis syndrome,198 current evidence does not support the use of leukapheresis in asymptomatic patients with hyperleukocytosis.199,200
Other special situations requiring therapeutic intervention are the presence of disseminated intravascular coagulation (DIC), tumor lysis syndrome, and differentiation syndrome. DIC can be screened for using a scoring system and is present in 8.5% to 25% of patients with non–APL, with another ∼15% also developing DIC soon after the initiation of chemotherapy.201 Special attention to tumor lysis syndrome is required in patients with hyperleukocytosis or with venetoclax-based treatments (Table 12). Close monitoring for signs of differentiation syndrome such as unexplained fever, lung edema, weight gain, pulmonary infiltrates, hypoxia, and dyspnea is necessary, particularly in patients on treatment with IDH inhibitors.202
Supportive care
Anti-infectious prophylaxis
For prophylaxis and treatment of infections, prevailing institutional infectious organisms and their drug resistance pattern should be considered primarily. There is good evidence to recommend antifungal prophylaxis with posaconazole during remission induction therapy,203 whereas there is not enough evidence from randomized trials on antiviral prophylaxis for herpes simplex virus in patients with acute leukemia,204 and no evidence for a beneficial effect of Pneumocystis jirovecii pneumonia prophylaxis. For prophylaxis of infectious disease in the setting of allogeneic HCT, we refer to respective guidelines.205
Vaccination for influenza206 and COVID-19 viral infections is recommended for all patients to reduce the risk of severe infections.
The use of growth factors is not routinely recommended unless in individual patients (eg, in case of severe infections) or particular treatment settings (eg, to reduce the hematologic recovery times in consolidation cycles).1,122,123
Transfusions
The availability of several effective novel agents may lead to a higher proportion of patients treated on an outpatient basis. If blood count checks are not possible at regular intervals in the outpatient setting, platelet and hemoglobin transfusion triggers should be elevated to ensure adequate support until the next outpatient visit. Besides the platelet count, mucosal bleeding, infection, severe mucositis, and fever should be considered in the assessment of bleeding risk and should increase the platelet level transfusion threshold. Otherwise, it is generally accepted to keep the hemoglobin level above 8 g/dL, and a platelet count of <10 × 109/L remains the trigger for prophylactic platelet transfusions.
Acknowledgments
The authors gratefully acknowledge Rüdiger Hehlmann for his continuous generous support of these recommendations on behalf of the European LeukemiaNet and Partrycja Gradowska, Axel Benner, Maral Saadati, and Julia Krzykalla for reviewing the sections “Response criteria” and “Outcome measures.”
H. Döhner is supported by SFB 1074 “Experimental models and clinical translation in leukemia” funded by the Deutsche Forschungsgemeinschaft. A.H.W. is supported by the Australian National Health and Medical Research Council, Victorian Cancer Agency, Metcalf Family Fellowship, and the Medical Research Future Fund. C.D.D. is supported by the LLS Scholar in Clinical Research award.
Clara Bloomfield, Elihu Estey and Francesco Lo-Coco have died since the publication of the 2017 ELN AML Recommendation edition. The authors acknowledge the impressive seminal contributions that Clara Bloomfield, Elihu Estey, and Francesco Lo-Coco made throughout many years to improve the treatment of AML and advance our understanding of the pathobiology of the disease. They were founding coauthors of the ELN AML Recommendation editions that first appeared in 2010 and (pro)actively participated with a high level of commitment in this endeavor. Each one in their own unique way contributed their invaluable clinical expertise, knowledge, and scientific rigor and were instrumental in establishing these recommendations as a widely accepted standard of reference.
Authorship
Contribution: All authors reviewed the literature and wrote first drafts of specific sections. H. Döhner and B.L. assembled the sections and wrote the final version of the manuscript. All authors reviewed and approved the final version of the manuscript.
Conflict-of-interest disclosure: H. Döhner had an advisory role with honoraria for AbbVie, Agios, Amgen, Astellas, AstraZeneca, Berlin-Chemie, BMS, Celgene, GEMoaB, Gilead, Janssen, Jazz, Novartis, Servier, and Syndax and received clinical research funding (to institution) from AbbVie, Agios, Amgen, Astellas, Bristol Myers Squibb, Celgene, Jazz Pharmaceuticals, Kronos Bio, and Novartis. A.H.W. had an advisory role with honoraria for AbbVie, Agios, Amgen, Astellas, AstraZeneca, Roche, BMS, Celgene, Gilead, Pfizer, Janssen, Jazz, Novartis, and Servier; received clinical research funding (to institution) from AbbVie, Servier, Celgene-BMS, Astra Zeneca, Amgen, and Novartis; and receives a fraction of royalty payments from the Walter and Eliza Hall Institute of Medical Research related to venetoclax. F.R.A. has consulted for Jasper Biote. C.C. had an advisory role with honoraria for AbbVie, Amgen, Astellas, AstraZeneca, Berlin-Chemie, BMS, Celgene, Daiichi-Sankyo, Eurocept, Gilead, Janssen, Jazz, and Novartis and received clinical research funding from AbbVie, Bristol Myers Squibb, Celgene, and Jazz Pharmaceuticals. C.D.D. received honoraria/consulting fees from AbbVie, Agios/Servier, Astellas, Celgene/BMS, Cleave, Foghorn, Genentech, GenMab, GSK, Novartis, Notable Labs, Takeda and research grants (to institution) from AbbVie, Agios/Servier, Astex, Calithera, Celgene/BMS, Cleave, Foghorn, ImmuneOnc, and Loxo. H. Dombret received honoraria/consulting fees from AbbVie, Amgen, Astellas, Celgene-BMS, Daiichi Sankyo, Incyte, Jazz Pharmaceuticals, Pfizer, and Servier and research funding from Amgen, Astellas, Celgene-BMS, Incyte, Jazz Pharmaceuticals, and Pfizer. B.L.E. received consulting fees from GRAIL and research funding from Celgene, Deerfield, Novartis, and Calico and is a member of the scientific advisory board and shareholder for Neomorph Therapeutics, TenSixteen Bio, Skyhawk Therapeutics, and Exo Therapeutics. P.F. received honoraria and, as French MDS group chairperson, research support from Celgene/BMS, Novartis, AbbVie, Jazz, Janssen, and Agios. R.A.L. has been a consultant or advisor to Amgen, Ariad/Takeda, Astellas, Celgene/BMS, CVS/Caremark, Epizyme, Immunogen, MedPace, MorphoSys, Novartis, and Servier and has received clinical research support to his institution from Astellas, Celgene, Cellectis, Daiichi Sankyo, Forty Seven/Gilead, Novartis, Rafael Pharmaceuticals and royalties from UpToDate. R.L.L. was on the supervisory board of Qiagen and an scientific advisor to Imago, Mission Bio, Zentalis, Ajax, Auron, Prelude, C4 Therapeutics, and Isoplexis, for which he receives equity; received research support from Calico, Constellation, Zentalis, and Ajax; consulted for Incyte, Janssen, Morphosys, and Novartis; and received honoraria from Roche, Lilly, and Amgen for invited lectures and from Gilead for grant reviews. Y.M. received honoraria from Nippon-Shinyaku, Bristol-Myers Squib, Novartis, Sumitomo Pharma, Kyowa-Kirin, AbbVie, Daiichi-Sankyo, Takeda, Janssen Pharmaceutical, Astellas, Pfizer, Eizai, and Otsuka Pharmaceutical and research funding from Sumitomo-Dainippon. G.O. had an advisory role with honoraria for AbbVie, Astellas, Bristol Myers Squibb, Celgene, Gilead, Servier, JAZZ, and Novartis. C.R. had an advisory role with honoraria for AbbVie, Amgen, Astellas, BMS, Celgene, Jazz, Novartis, Pfizer, and Servier and received clinical research funding from AbbVie, Novartis, and Pfizer. J.S. had an advisory role with honoraria for AbbVie, Astellas, AstraZeneca, Berlin-Chemie, Celgene, AvenCell-GEMoaB, Janssen, Jazz Pharmaceuticals, Novartis, and Bristol Myers Squibb and received clinical research funding from AbbVie, Astellas, Jazz Pharmaceuticals, and Jose Carreras International Leukemia Foundation. E.M.S. was on the advisory board for Novartis, PinotBio, Janssen, Bristol Myers Squibb, Agios, Jazz, Menarini, Genentech, Genesis, AbbVie, Neoleukin, Gilead, Syndax, OnCusp, CTI Biopharma, Foghorn, Servier, Calithera, Daiichi, Aptose, Syros, Astellas, Ono Pharma, and Blueprint; received honoraria from Kura; performed safety monitoring for Epizyme and Cellectis; and received research funding for Eisai and Bristol Myers Squibb and equity from Auron. M.S.T. was on the advisory boards for AbbVie, Daiichi-Sankyo, Orsenix, KAHR, Oncolyze, Jazz Pharma, Roche, Biosight, Novartis, Innate Pharmaceuticals, Kura, Syros Pharmaceuticals, Ipsen Biopharmaceuticals, and Cellularity and received research funding from AbbVie, Orsenix, Biosight, Gylcomimetics, Rafael Pharmaceuticals, and Amgen and royalties from UpToDate. H.-F.T. had an advisory role with honoraria for AbbVie, Alexion, Celgene, Daiichi Sankyo, Novartis, and Roche and received research funding from Celgene. J.W. had an advisory role with honoraria for AbbVie. A.W. had an advisory role with honoraria for AbbVie, Astellas, BMS, Celgene, Gilead, Janssen, Jazz Pharmaceuticals, Novartis, and Servier and received clinical research funding from Jazz Pharmaceuticals. B.L. had an advisory role with honoraria for AbbVie, Astellas, Bristol Myers Squibb/Celgene, Catamaran Bio, AvenCell/GEMoaB, Frame Pharmaceuticals, Gilead, Kronos Bio, Oxford Biomedica, and Servier/Agios and received royalties from UpToDate and equity from Frame Pharmaceuticals. All remaining authors declare no competing financial interests.
Correspondence: Hartmut Döhner, Department of Internal Medicine III, Ulm University Hospital, Albert-Einstein-Allee 23, 89081 Ulm, Germany; e-mail: hartmut doehner@uniklinik-ulm.de.
There is a Blood Commentary on this article in this issue.

