Key Points
Using RNA-seq in pediatric AML patients, 5 gene rearrangements were newly identified, including NPM1 and RUNX1 gene rearrangements.
RNA-seq unmasked the complexity of gene alterations in pediatric AML by identifying disease-causing alterations in nearly all patients.

Abstract
Recent advances in the genetic understanding of acute myeloid leukemia (AML) have improved clinical outcomes in pediatric patients. However, ∼40% of patients with pediatric AML relapse, resulting in a relatively low overall survival rate of ∼70%. The objective of this study was to reveal the comprehensive genetic background of pediatric AML. We performed transcriptome analysis (RNA sequencing [RNA-seq]) in 139 of the 369 patients with de novo pediatric AML who were enrolled in the Japanese Pediatric Leukemia/Lymphoma Study Group AML-05 trial and investigated correlations between genetic aberrations and clinical information. Using RNA-seq, we identified 54 in-frame gene fusions and 1 RUNX1 out-of-frame fusion in 53 of 139 patients. Moreover, we found at least 258 gene fusions in 369 patients (70%) through reverse transcription polymerase chain reaction and RNA-seq. Five gene rearrangements were newly identified, namely, NPM1-CCDC28A, TRIP12-NPM1, MLLT10-DNAJC1, TBL1XR1-RARB, and RUNX1-FNBP1. In addition, we found rare gene rearrangements, namely, MYB-GATA1, NPM1-MLF1, ETV6-NCOA2, ETV6-MECOM, ETV6-CTNNB1, RUNX1-PRDM16, RUNX1-CBFA2T2, and RUNX1-CBFA2T3. Among the remaining 111 patients, KMT2A-PTD, biallelic CEBPA, and NPM1 gene mutations were found in 11, 23, and 17 patients, respectively. These mutations were completely mutually exclusive with any gene fusions. RNA-seq unmasked the complexity of gene rearrangements and mutations in pediatric AML. We identified potentially disease-causing alterations in nearly all patients with AML, including novel gene fusions. Our results indicated that a subset of patients with pediatric AML represent a distinct entity that may be discriminated from their adult counterparts. Based on these results, risk stratification should be reconsidered.
Introduction
Recent advances in the genetic understanding of cancer have drastically improved clinical outcomes in pediatric patients. However, ∼40% of patients with childhood acute myeloid leukemia (AML) relapse, resulting in a relatively low overall survival (OS) rate of ∼70%. AML is caused by various chromosomal aberrations, gene mutations/epigenetic modifications, and deregulated/overregulated gene expression, leading to increased proliferation and decreased differentiation of hematopoietic progenitor cells.1,,-4 Several clinically important molecular markers have been discovered in patients with AML, which assisted in improving risk stratification. However, the evaluation of risk remains challenging even after incorporating these molecular markers in clinical practice.
Patients with biallelic CEBPA mutations, NPM1 mutations, RUNX1-RUNX1T1 fusions, or CBFB-MYH11 fusions have relatively good outcomes in response to treatment with chemotherapy-based consolidation regimens.5,6 However, the prognosis of patients with FUS-ERG, NUP98-NSD1, CBFA2T3-GLIS2, NUP98-KDM5A, or KMT2A-MLLT3 with high MDS1 and EVI1 complex locus protein EVI1 (MECOM, synonyms EVI1 and PRDM3) expression was dismal, because many of these patients could not be during the first complete remission (CR), even with allogeneic transplantation.7,,-10
FMS-like tyrosine kinase 3-internal tandem duplication (FLT3-ITD) is a major alteration in pediatric and adult AML that is significantly associated with poor prognosis. However, the prognosis of patients with this alteration is not necessarily dismal, depending on concomitant genetic alterations.11,-13 In our previous studies, the overexpression of PRD1-BF1-RIZ1 homologous domain containing 16 (PRDM16, also known as MEL1) was highly recurrent in patients with both pediatric AML and adult AML with intermediate- and high-risk cytogenetic profiles while being independently associated with an adverse outcome.14,15
Recently, Bolouri et al reported the seminal comprehensive molecular landscape and characteristics of pediatric AML in Children’s Oncology Group AML trials.16 However, the biologic basis differs among pediatric AML patients in the United States, Europe, and Japan. The objective of this study was to reveal the genetic alterations of all Japanese patients with pediatric AML and the differences in the genetic background between favorable and adverse groups using RNA sequencing (RNA-seq) by identifying new cytogenetic aberrations and comparing gene expression.
Methods
Study design and participants
In this study, we enrolled 485 patients with de novo AML who registered in the Japanese Pediatric Leukemia/Lymphoma Study Group (JPLSG) AML-05 trial between November 2006 and December 2010. The trial was registered with the University Hospital Medical Information Network Clinical Trials Registry (#UMIN000000511; http://www.umin.ac.jp/ctr/index.htm), and details of the schedules and treatment regimens have been described previously (supplemental Figure 2).17 Patients diagnosed with acute promyelocytic leukemia or AML with Down syndrome were excluded from this study. Among the 485 patients with AML who were enrolled in the AML-05 trial, 116 were excluded because of sample insufficiency for diagnosis (n = 7), not meeting the criteria (n = 6), misdiagnosis (n = 25), lack of RNA samples (n = 74), or other reasons (n = 4). Therefore, a total of 369 patients were genetically analyzed. In these 369 analyzed patients, the age and initial white blood cell count were higher, whereas the proportion of M7 patients was lower compared with the 74 nonanalyzed patients. However, there were no significant differences observed between these groups in terms of mortality (20% vs 24%, log-rank P = .84) (supplemental Table 1). The present study was conducted in accordance with the Declaration of Helsinki and approved by the institutional review boards of Gunma Children’s Medical Center and the participating institutes and the ethical review board of the JPLSG AML-05 trial. Written informed consent was provided by all patients or their parents/guardians.
Sample preparation
All leukemic samples were obtained from the bone marrow or peripheral blood at the time of diagnosis. DNA and total RNA samples were prepared using the AllPrep DNA/RNA Mini Kit (Qiagen, Hilden, Germany) according to the manufacturer’s instructions. Complementary DNA was prepared using 0.8 to 1.0 µg of total RNA and Ready-To-Go reverse transcription polymerase chain reaction (RT-PCR) beads (GE Healthcare, Buckinghamshire, United Kingdom).
RNA-seq
We performed RNA-seq for 139 out of the 369 patients with pediatric AML in order to obtain a complete registry of gene rearrangements, other genetic lesions, and gene expressions in pediatric AML. The RNA-seq data were available at the European Genome-Phenome Archive (EGAS00001003701). The cytogenetic characteristics of the analyzed patients are shown in supplemental Table 2. The study population mainly included patients with a normal karyotype (60/70 patients), FLT3-ITD (33/47 patients), KMT2A-PTD (12/13 patients), and high PRDM16 expression (65/84 patients). All 137 patients with core binding factor AML (CBF-AML) and many patients with KMT2A rearrangements were excluded from RNA-seq, because these fusions are already known to affect leukemogenesis. The main reasons behind the inability to perform analyses were assay failure, sample availability, and/or sample quality. The quality of the extracted RNA was assessed using TapeStation system (Agilent Technologies, Santa Clara, CA). Sequencing libraries were prepared using an NEBNext Ultra RNA Library Prep Kit for Illumina (New England Biolabs, Ipswich, MA), and prepared libraries were run on a HiSeq 2500 high-throughput sequencing system. Sequencing reads were aligned using bowtie and blat, and fusion genes were analyzed using Genomon-fusion.18 Candidate gene fusions were validated by RT-PCR. Obtained reads were also analyzed using an in-house pipeline, GenomonExpression (https://github.com/Genomon-Project/GenomonExpression), to obtain fragments per kilobase million (FPKM) values. We used Cluster 3.0 to perform hierarchical clustering.19 Briefly, genes that were not expressed (FPKM = 0) in >20% of samples and genes with a standard deviation <10 were excluded. For the remaining genes, their FPKM values were log transformed. Finally, an average linkage was calculated. Results were visualized using Java TreeView.20
Targeted gene mutation analysis
We performed targeted gene mutation analysis of KIT (exons 8 and 17), CEBPA (all coding regions), NPM1 (exon 12), WT1 (exons 7-10), RUNX1 (exons 2-6), NRAS (exons 1 and 2), KRAS (exons 1 and 2), ASXL1 (exon 11), ASXL2 (exons 11 and 12), and GATA2 (exons 4-6). The allelic ratio (AR) of FLT3-ITD to wild-type alleles was further examined using GeneScan (Applied Biosystems, Foster City, CA), and a defined mutant/wild-type >0.40 was considered as high AR according to previous studies.12,21,,,,-26 KMT2A-PTD was analyzed using previously described multiplex ligation-dependent probe amplification methods.27 Gene fusions, namely, RUNX1-RUNX1T1, CBFB-MYH11, KMT2A rearrangements, NUP98-NSD1, CBFA2T3-GLIS2, NUP98-KDM5A, FUS-ERG, and DEK-NUP214, were also analyzed in all patients. In addition to these conventional gene fusions, we performed RT-PCR using samples from all patients to detect the MYB-GATA1, NPM1-CCDC28A, MLF1-NPM1, NPM1-TRIP12, TBL1XR1-RARB, ETV6-CTNNB1, ETV6-NCOA2, ETV6-MECOM, RUNX1-FNBP1, RUNX1-CBFA2T2, and RUNX1-CBFA2T3 alterations identified through RNA-seq.
Quantification of PRDM16 and MECOM gene expression
Quantitative RT-PCR analysis of the PRDM16 and MECOM genes was performed using the 7900HT Fast Real Time PCR System, TaqMan Gene Expression Master Mix, and TaqMan Gene Expression Assay (Applied Biosystems), which have been previously described.28
Data collection
A standardized form was used to record clinical variables, including patient demographic information. Every 6 months, data forms were forwarded to the JPLSG AML-05 trial data coordination center at the National Center for Child Health and Development (Tokyo, Japan), reviewed for internal consistency and face validity, and transferred into an Excel database (Microsoft Corporation, Redmond, WA). The clinical data of patients in each risk group were followed up until December 2013 (censored for 3 years from the date of final registration). The JPLSG conducted a central review of morphological classification and karyotyping based on the World Health Organization’s classification, French-American-British (FAB) classification and cytogenetic analysis using conventional G-banding. OS was defined as the time from AML diagnosis to death or censorship at the last follow-up. Event-free survival (EFS) was defined as the time from AML diagnosis to treatment failure, relapse, death, or last follow-up.
Statistical analysis
Univariate and multivariate Cox regression analyses were calculated using EZR (version 1.37; Saitama Medical Center, Jichi Medical University, Saitama, Japan), which is a graphical user interface for R (version 3.4.1; The R Foundation, Vienna, Austria)29 to extract the adverse and favorable risk factors related to OS. We defined the risk factors with a hazard ratio (HR) of <0.20 as favorable and those with a ratio of >2.0 as adverse. Continuous variables are presented as means ± standard deviations and/or medians with ranges. Categorical variables are represented by frequencies and percentages. For all analyses, the P values were 2 tailed, and P < .05 was considered statistically significant. A multivariate Cox regression analysis was used to identify independent genetic factors related to OS. Initially, we included all genetic variables in the first model and then sequentially removed the nonsignificant variables (P ≥ .05). To test the probability of co-occurrence and mutual exclusivity of 2 parameters, we performed a Monte Carlo simulation using the total number of patients and the number of patients who were positive for each parameter.30
Results
Overview of RNA-seq
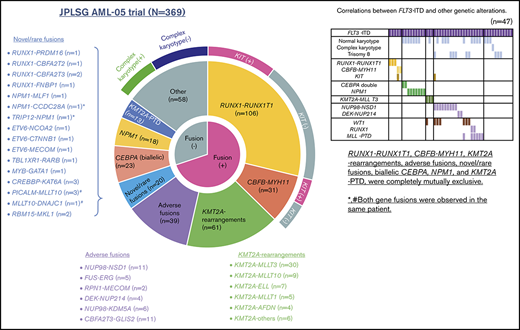
We performed RNA-seq in 139 patients with pediatric AML and identified a total of 54 in-frame fusions and 1 RUNX1 out-of-frame gene fusion in 53 patients. Many of the recurrent gene fusions identified in this study have been previously reported as targets in AML (Figure 1). Among these 54 gene fusions, 5 gene rearrangements were newly identified, namely, NPM1-CCDC28A, TRIP12-NPM1, MLLT10-DNAJC1, TBL1XR1-RARB, and RUNX1-FNBP1. We also identified 17 rare gene rearrangements, including MYB-GATA1, NPM1-MLF1, ETV6-NCOA2, ETV6-MECOM, ETV6-CTNNB1, RUNX1-PRDM16, RUNX1-CBFA2T2, and RUNX1-CBFA2T3 (Figures 1 and 2). The characteristics of the patients with these fusions are shown in Table 1. We identified 5 RUNX1 rearrangements other than RUNX1-RUNX1T1, 4 NPM1 rearrangements, 3 ETV6 fusions, and 1 TBL1XR1-RARB fusion in FAB-M3 patients without the PML-RARA fusion.

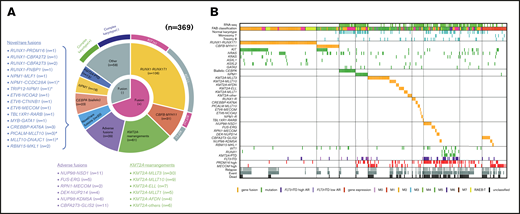
Clinical and genetic profiles of the 369 patients enrolled in the JPLSG AML-05 trial. (A) Pie chart represented the frequency of each gene alterations. (B) Landscape of 369 de novo pediatric AML patients. Each column indicates 1 patient. *,#Both gene fusions were observed in the same patient. RUNX1-R, RUNX1 rearrangement; NPM1-R, NPM1-rearrangement.
Clinical and genetic profiles of the 369 patients enrolled in the JPLSG AML-05 trial. (A) Pie chart represented the frequency of each gene alterations. (B) Landscape of 369 de novo pediatric AML patients. Each column indicates 1 patient. *,#Both gene fusions were observed in the same patient. RUNX1-R, RUNX1 rearrangement; NPM1-R, NPM1-rearrangement.
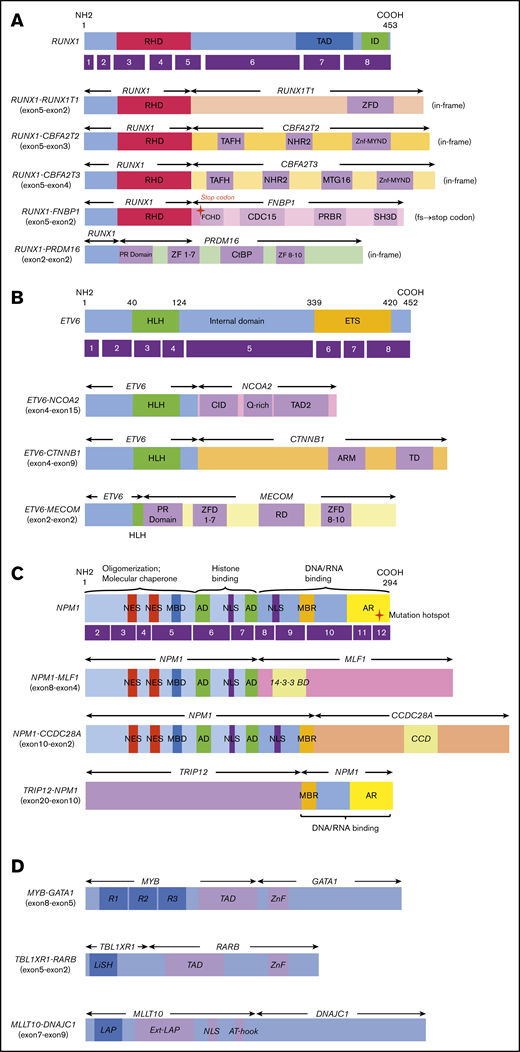
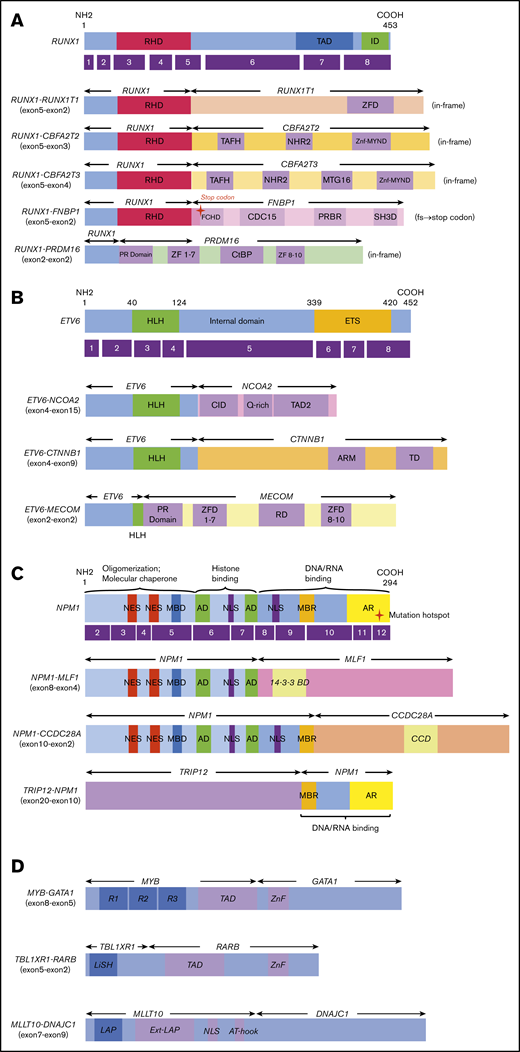
Novel/rare fusion genes identified in patients with de novo pediatric AML. (A-D) The structure of the detected fusion proteins. (A) Five RUNX1 rearrangements. (B) Three ETV6 rearrangements. (C) Three NPM1 rearrangements. (D) Other novel gene fusions. R1, R2, and R3 are 51 ± 52 amino acid tandem repeats that comprise the DNA-binding domain Ext–leukemia-associated protein (LAP). 14-3-3 BD, 14-3-3 binding domain; AD, acidic domains; AT-hook, adenine-thymine hook; CCD, coiled-coil domain; CDC15, CDC15 homology region; CID, CBP interaction domain; CtBP, C-terminal binding protein; ETS, ETS domain; FCHD, FER-CIP1 homology domain; HLH, helix-loop-helix; ID, transcription inhibition domain; LiSH, Lis homology domain; MBD, metal-binding domain; MBR, moderately basic region; MTG16, myeloid translocation gene 16; NES, nuclear export signal; NHR2, nervy homology region 2; NLS, nuclear localization signal; PRBR, putative ρ-binding region; RD, runt domain; RHD, runt homolog domain; SH3D, Src homology 3 domain; TAD, transcription activation domain; TAFH, TAF homology; ZF, zinc finger; ZFD, zinc-finger domain; Znf-MYND, MYND zinc-finger domain.
Novel/rare fusion genes identified in patients with de novo pediatric AML. (A-D) The structure of the detected fusion proteins. (A) Five RUNX1 rearrangements. (B) Three ETV6 rearrangements. (C) Three NPM1 rearrangements. (D) Other novel gene fusions. R1, R2, and R3 are 51 ± 52 amino acid tandem repeats that comprise the DNA-binding domain Ext–leukemia-associated protein (LAP). 14-3-3 BD, 14-3-3 binding domain; AD, acidic domains; AT-hook, adenine-thymine hook; CCD, coiled-coil domain; CDC15, CDC15 homology region; CID, CBP interaction domain; CtBP, C-terminal binding protein; ETS, ETS domain; FCHD, FER-CIP1 homology domain; HLH, helix-loop-helix; ID, transcription inhibition domain; LiSH, Lis homology domain; MBD, metal-binding domain; MBR, moderately basic region; MTG16, myeloid translocation gene 16; NES, nuclear export signal; NHR2, nervy homology region 2; NLS, nuclear localization signal; PRBR, putative ρ-binding region; RD, runt domain; RHD, runt homolog domain; SH3D, Src homology 3 domain; TAD, transcription activation domain; TAFH, TAF homology; ZF, zinc finger; ZFD, zinc-finger domain; Znf-MYND, MYND zinc-finger domain.
Complete genetic landscape of the JPLSG AML-05 trial
We showed the demographic characteristics and genetic landscape of 369 patients with AML enrolled in the JPLSG AML-05 trial (Figure 1). RUNX1-RUNX1T1 and KIT mutations were more frequently observed and CBFB-MYH11, trisomy 8, and NPM1 were less frequently observed as a molecular characteristic in the Japanese cohort compared with the Children’s Oncology Group (supplemental Figure 1). We also showed the result of univariate and multivariate Cox regression analysis in Table 2. In the univariate analysis, factors significantly associated with OS included CBFB-MYH11 (HR, 0.11, 95% confidence interval [CI], 0.02-0.80), biallelic CEBPA (HR, 0.16; 95% CI, 0.02-1.13), RUNX1-RUNX1T1 (HR, 0.19; 95% CI, 0.09-0.39), NPM1 (HR, 0.19; 95% CI, 0.03-1.35), FLT3-ITD (HR, 2.92; 95% CI, 1.83-4.65), CBFA2T3-GLIS2 (HR, 3.00; 95% CI, 2.54-6.48), PRDM16 overexpression (HR, 3.09; 95% CI, 2.06-4.65), and NUP98-NSD1 (HR, 5.07; 95% CI, 2.54-10.1). The multivariate analysis revealed that CBFB-MYH11 (HR, 0.08; 95% CI, 0.01-0.58), biallelic CEBPA (HR, 0.10; 95% CI, 0.01-0.69), RUNX1-RUNX1T1 (HR, 0.17; 95% CI, 0.08-0.35), NPM1 (HR, 0.09; 95% CI, 0.01-0.64), FLT3-ITD (HR, 2.53; 95% CI, 1.57-4.08), FUS-ERG (HR, 4.54; 95% CI, 1.81-11.4), and RPN1-MECOM (HR, 5.44; 95% CI, 1.32-22.4) were independent prognostic factors associated with OS. Concerning CBF-AML, 9 of 137 patients died of primary disease or other causes (4 and 5 patients, respectively) (Table 3). Although CBF-AML patients with the KIT mutation exhibited a higher relapse rate than those without the KIT mutation, most of these patients were rescued by hematopoietic stem cell transplantation.31 No fusion genes were identified in patients with the NPM1 or biallelic CEBPA mutation, and their prognosis was absolutely favorable irrespective of the presence of FLT3-ITD. Among the 41 patients with the biallelic CEBPA or NPM1 mutation, only 2 patients died, and the causes of death were acute respiratory disorder syndrome (ARDS) and primary disease (Table 3). On the other hand, the prognosis of patients with adverse gene fusions (eg, FUS-ERG, NUP98-NSD1, RPN1-MECOM, KMT2A-AFDN, DEK-NUP214, CBFA2T3-GLIS2, and NUP98-KDM5A) and adverse mutations (eg, FLT3-ITD, RUNX1, and KMT2A-PTD) was dismal (5-year OS, 31%; EFS, 9%). The characteristics of patients with unfavorable gene fusions are shown in supplemental Table 3. Although all of patients with FUS-ERG, RPN1-MECOM, KMT2A-AFDN, and DEK-NUP214 were predicted by chromosomal G-banding, NUP98-NSD1, NUP98-KDM5A, and CBFA2T3-GLIS2 were cryptic. No disease-causing genetic alterations were identified in 45 patients out of 369 patients with AML (29 patients with FAB non-M7 and 16 patients with FAB-M7) (supplemental Table 4). Ten patients were cytogenetically normal, whereas the remaining 35 patients had structural alterations in chromosomes, including complex karyotype (n = 23), monosomy 7 (n = 4), and trisomy 8 (n = 3). The prognosis of these patients tended to be dismal (3-year OS, 67%; 3-year EFS, 42%); thus, they should be treated as high-risk patients. Further research is warranted to confirm these observations.
Characteristics of patients with pediatric AML with normal karyotype
Sixty out of 70 patients with a normal karyotype were analyzed using RNA-seq. Although some cryptic gene fusions were identified (eg, NUP98-NSD1 and CBFA2T3-GLIS2), fewer gene fusions were identified in this subset of patients. In particular, gene fusions were not identified in patients with normal karyotype with biallelic CEBPA, NPM1, and KMT2A-PTD aberrations, which are frequently observed in the normal karyotype (supplemental Figure 3).
Characteristics of patients with pediatric AML with KMT2A rearrangements
Sixty-one KMT2A rearrangements were identified in 369 patients with AML (16%). Among them, 7 KMT2A fusions not identified through conventional chromosomal G-banding were detected through RNA-seq. KMT2A-MLLT3 was the most commonly observed alteration (30/61 patients). Intriguingly, the prognosis of these patients was highly dependent on the expression of the MECOM gene. Seven of 11 patients with high MECOM expression died compared with only 1 of 19 patients with low MECOM expression (P < .001). KMT2A-ELL and KMT2A-MLLT1 were the second most commonly observed alterations in patients with pediatric AML (supplemental Table 5).
Characteristics of patients with pediatric AML with FAB-M7
In this study, we performed RNA-seq in only 9 of 34 patients with acute megakaryoblastic leukemia. Among them, we confirmed 3 types of recurrent gene fusions identified through RT-PCR, including 2 RBM15-MKL1, 3 CBFA2T3-GLIS2, and 1 NUP98-KDM5A. In the remaining 3 patients with acute megakaryoblastic leukemia, there were no gene fusions identified. Among 34 patients registered and treated in the JPLSG AML-05 trial, 19 of 34 patients showed high expression of the MECOM gene. Although high MECOM gene expression was associated with poor prognosis in other FAB patients, this association was not observed in patients with FAB-M7 (Figure 1). Interestingly, patients with the CBFA2T3-GLIS2 and NUP98-KDM5A alterations, which were associated with poor prognosis, showed low MECOM gene expression, indicating the presence of a different pathogenesis mechanism.
Gene expression profiling
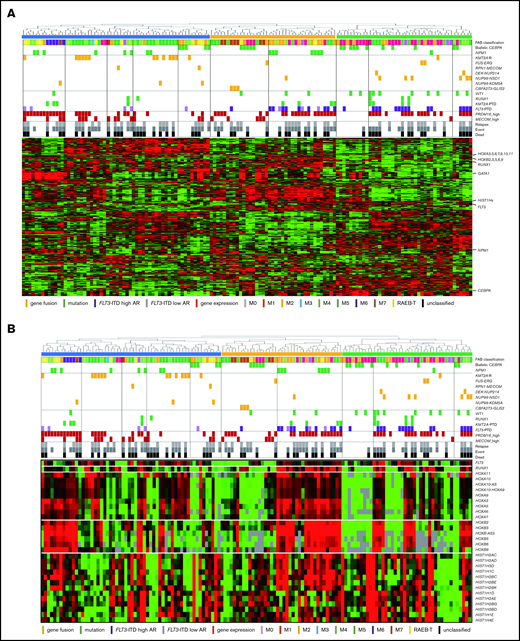
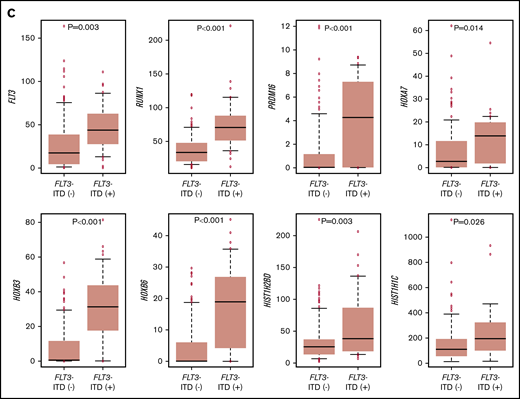
The RNA-seq expression profiles of 139 patients were subjected to all-gene–based unsupervised hierarchical clustering, and 3 clusters were generated (blue, orange, and green) (Figure 3A). Thirteen out of 14 patients with KMT2A rearrangement were classified into the blue cluster, and all patients with FLT3-ITD with high AR were classified into the orange or green cluster. On the contrary, 6 of 7 patients with NUP98-NSD1 and 2 patients with DEK-NUP214 were classified into the green cluster. The gene expression pattern of NUP98-KDM5A was different from the other NUP fusions in this study (Figure 3A). Furthermore, HOXA7, HOXB3, HOXB6, HOXB7, RUNX1, FLT3, PRDM16, HIST1H1C, and HIST1H2BD were overexpressed in patients with FLT3-ITD (Figure 3B-C), and HOX genes were underexpressed in patients with biallelic CEBPA. Intriguingly, FLT3 and HOXAs were overexpressed in patients with KMT2A-rearrangements. Herein, we showed the detail of gene expression pattern focused on HOXAs, HOXBs, PRDM16, FLT3, RUNX1, and HIST1s in Figure 3B.
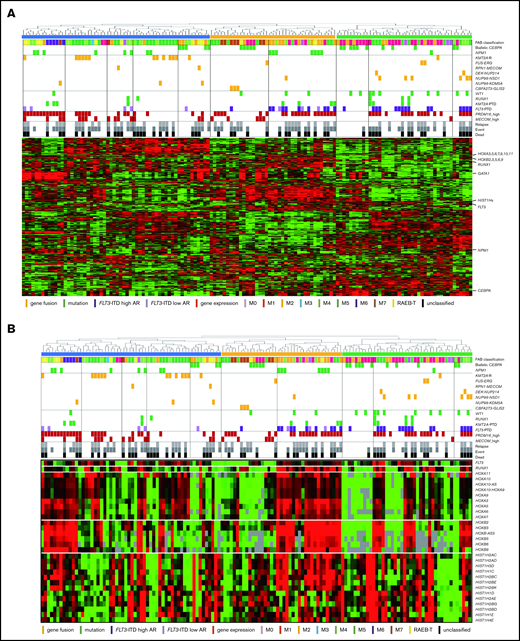
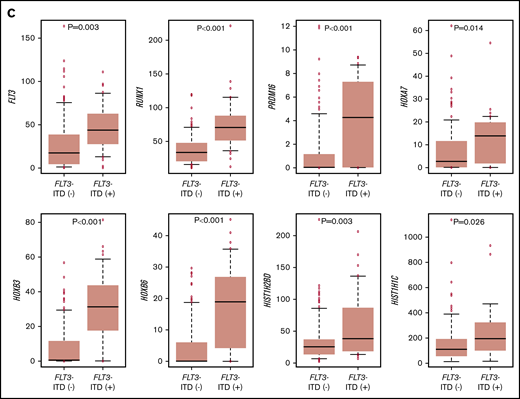
Unsupervised hierarchical clustering analysis of the 139 patients with pediatric AML. (A) Two-dimensional hierarchical clustering analysis of the 139 patients was performed using 3106 probe sets that were differentially expressed in 139 patients. (B) We showed the detail of gene expression pattern focused on the FLT3, RUNX1, HOXAs, HOXBs, and HIST1s in 139 patients. Each column represents a patient and each row represents a probe set. FAB classification, biallelic CEBPA, NPM1, WT1, RUNX1, KMT2A-PTD, KMT2A rearrangements, adverse gene fusions, PRDM16 and MECOM gene expression status, and outcome of each patient are indicated. Relative expression levels normalized to the average for each probe set are indicated by color, where red and green represent high and low expression, respectively. (C) Messenger RNA expression levels of FLT3, RUNX1, PRDM16, HOXA7, HOXB3, HOXB6, HIST1H2BD, and HIST1H1C in 139 AML patients. KMT2A-R, KMT2A rearrangement.
Unsupervised hierarchical clustering analysis of the 139 patients with pediatric AML. (A) Two-dimensional hierarchical clustering analysis of the 139 patients was performed using 3106 probe sets that were differentially expressed in 139 patients. (B) We showed the detail of gene expression pattern focused on the FLT3, RUNX1, HOXAs, HOXBs, and HIST1s in 139 patients. Each column represents a patient and each row represents a probe set. FAB classification, biallelic CEBPA, NPM1, WT1, RUNX1, KMT2A-PTD, KMT2A rearrangements, adverse gene fusions, PRDM16 and MECOM gene expression status, and outcome of each patient are indicated. Relative expression levels normalized to the average for each probe set are indicated by color, where red and green represent high and low expression, respectively. (C) Messenger RNA expression levels of FLT3, RUNX1, PRDM16, HOXA7, HOXB3, HOXB6, HIST1H2BD, and HIST1H1C in 139 AML patients. KMT2A-R, KMT2A rearrangement.
Discussion
Using RNA-seq, we identified 54 in-frame gene fusions and 1 out-of-frame fusion in 53 of 139 patients with pediatric AML. Moreover, we found ≥258 gene fusions in 369 patients (70%) through RT-PCR and RNA-seq. Only a few fusion genes were detected in patients with AML with a normal karyotype. Only 2 patients had 2 in-frame gene fusions, which resulted from 3-way translocations (PICALM-MLLT10 and MLLT10-DNAJC1; NPM1-CCDC28A and TRIP12-NPM1). This finding suggested that most of the in-frame gene fusions are able to facilitate AML. In the remaining 111 patients, KMT2A-PTD, biallelic CEBPA, and NPM1 mutations were found in 11, 23, and 17 patients, respectively. These mutations were completely mutually exclusive with any gene fusions. The FLT3-ITD alteration was more frequently observed in patients with adverse risk factors than in those with favorable risk factors, such as co-occurring mutations, and was considered to amplify the malignant potential.
In this study, we identified 5 novel gene rearrangements, namely, NPM1-CCDC28A, TRIP12-NPM1, NPM1-DNAJC1, TBL1XR1-RARB, and RUNX1-FNBP1, and 17 rare fusions. Concerning patients with NPM1 rearrangements, TRIP12 encodes E3 ligase, which ubiquitinates CDKN2A. NPM1 is a major component of the nucleolus, with important functions in regulating cell growth, proliferation, and transformation. Intriguingly, as an additional important effect, NPM1 stabilizes the protein levels of CDKN2A.32 CDKN2A is known to suppress aberrant cell growth by activating the p53 response.33 The function of CDKN2A may be inhibited by fusing NPM1 and TRIP12. NPM1-MLF1 has been associated with therapy-related AML and myelodysplastic syndrome with good prognosis.34,35 Our patient was diagnosed as RAEB-T and maintained CR for >3 years (Table 1). Regarding the roles of NPM1-MLF1 and NPM1-CCDC28A, the fusion resulted in the loss of DNA/RNA-binding domains. This may deregulate the function of NPM1 and facilitate leukemogenesis because the mutational hotspot is located within the DNA/RNA binding domain at exon 12. Interestingly, CCDC28A has been reported as the partner gene of NUP98 in T-ALL.36 Regarding the RUNX1-CBFA2T2 gene fusion, the expression of the CBFA2T2 gene was high in AML cells following the activation of the RUNX1 promoter compared with that observed in normal bone marrow. Thus, the RUNX1-CBFA2T2 gene fusion may be associated with leukemogenesis.37 In addition, RUNX1-CBFA2T3 showed a gene expression pattern similar to that of RUNX1-RUNX1T1.38 Moreover, we identified novel out-of-fusion of RUNX1-FNBP1 (Figure 2). The affected RUNX1 is truncated at the end of the runt homolog domain, forming a stop codon within the FNBP1 gene, 88 to 90 bp downstream of the break point (Figure 2A). In this study, patients with RUNX1-CBFA2T3, RUNX1-CBFA2T2, and RUNX1-FNBP1 maintained CR without relapse for >3 years. On the other hand, the patient with RUNX1-PRDM16 did not achieve CR and eventually expired owing to primary disease following hematopoietic stem cell transplantation in non-CR. This patient showed the highest PRDM16 gene expression among the 369 patients with AML, whereas other patients with RUNX1 fusions showed low PRDM16 expressions. This finding indicates that the RUNX1-PRDM16 fusion gene has a function distinct from those of other RUNX1 fusions (eg, RUNX1-RUNX1T1) in terms of pathogenesis. RUNX1-PRDM16 has already been reported in myelodysplastic syndrome, de novo AML, therapy-related AML, and chronic myelogenous leukemia and is associated with high PRDM16 gene expression and dismal outcomes.39,,,-43 We showed the genetic landscape of patients with AML with high levels of PRDM16 expression (supplemental Figure 4). Many adverse risk factors were observed in patients with high PRDM16 gene expression, and numerous patients died, indicating that high PRDM16 expression is associated with high-risk factors and dismal outcomes.4
Previous studies suggested MYB-GATA1 as the cause of acute basophilic leukemia.44 This fusion gene commits myeloid cells to the granulocyte lineage, blocking their differentiation.44 This fusion gene has been reported to be a cause of infant AML FAB-M5 as a result of reducing the expression of the GATA1 gene.45 Recently, this fusion was also reported in a pediatric patient with acute erythroid leukemia.46 The break point was quite the same as in our patient, and the characteristics were very similar, including age, FAB-M5, and favorable outcomes. Thus, the phenotype may depend on the co-occurrence of genomic alterations.
It has been reported that ETV6-NCOA2 in acute leukemia is associated with the coexpression of T-lymphoid and myeloid markers. Our patient was diagnosed with peroxidase-negative acute leukemia including minimally differentiated FAB-M0. As blast cells were positive for CD7, CD13, CD33, CD34, CD56, CD117, and HLA-DR by surface marker analysis, myeloid-natural killer cell acute leukemia should be considered. Consistent with the results of previous studies, this finding suggests that this fusion gene may be specific for a common myeloid-lymphoid progenitor cell.47,48 Intriguingly, similar to ETV6-NCOA2, ETV6-CTNNB1 has been previously reported in patients with T-cell acute lymphoblastic leukemia.49 ETV6-MECOM was previously reported in patients with chronic myelogenous leukemia or therapy-related leukemia.50 Our patient with this fusion showed the eighth highest MECOM expression among the 369 patients. ETV6 may promote the expression of the MECOM gene and turn on the critical role in progression to the blast crisis of AML. Interestingly, all 3 patients with MECOM rearrangements and 1 patient with PRDM16 rearrangement harbored monosomy 7, suggesting that PRDM alterations may be associated with monosomy 7, and showed dismal outcomes (supplemental Table 3).
Of the 61 patients (16%) with KMT2A rearrangements, 16 patients did not show 11q23 breakage through conventional chromosomal G-banding. This suggested that fluorescence in situ hybridization analysis of KMT2A may be useful for the detection of rare KMT2A rearrangements, because >100 partner genes are already known (supplemental Table 5). KMT2A-MLLT3 was the most common KMT2A rearrangement. Moreover, MECOM gene expression was useful in stratifying the prognosis of patients with KMT2A-MLLT3 (low expression [3-year OS, 92%; 3-year EFS, 86%] vs high expression [3-year OS, 39%; 3-year EFS, 9%]). However, the expression of the MECOM gene was not correlated with poor prognosis in patients with FAB-M7. One of the reasons for this may be that MECOM expression is also associated with the differentiation of megakaryocytes, despite the association of high MECOM expression with stem cell maintenance through the expression of the GATA2 gene in other FAB subtypes.51 KMT2A-MLLT10, ELL, and MLLT1 were common partner genes found in 9, 7, and 5 patients with de novo pediatric AML, respectively (supplemental Table 5). Of the 7 patients with KMT2A-ELL, 3 patients expired. Intriguingly, these 3 patients were <1 year old, and 2 of them died due to ARDS. On the other hand, the prognosis of patients with KMT2A-MLLT10 tended to be worse than that of patients with other KMT2A gene fusions (ie, 3 of 9 patients died due to primary disease) (supplemental Table 5). Consistent with the findings of previous studies,52 the prognosis of patients with KMT2A-AFDN was extremely dismal, indicating a different pathogenesis from other KMT2A rearrangements.
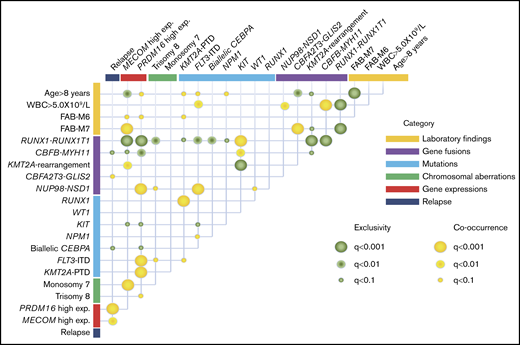
The co-occurrence and mutual exclusivity test identified that various clinical biomarkers may reflect different aspects of the same poor prognostic subgroup of patients with AML (Figure 4). RUNX1-RUNX1T1 and CBFB-MYH11 formed a unique entity that was mutually exclusive of any fusions and mutations other than KIT mutation. High MECOM expression was significantly associated with KMT2A-R and monosomy 7, whereas high PRDM16 gene expression was significantly associated with FLT3-ITD, KMT2A-PTD, and trisomy 8. These could potentially be used as surrogate biomarkers for this distinct subgroup, which shares several molecular signatures and predict the prognosis.
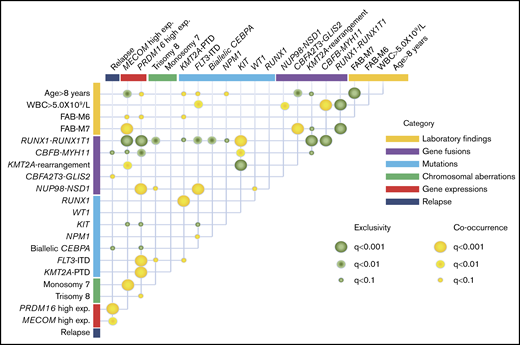
Correlations between established risk factors for pediatric AML. The diagram indicates co-occurrence or mutual exclusivity between pairs of clinical features, genetic alterations, gene expressions, and outcomes. The circles indicate that crossed factors are statistically co-occurrent (yellow) or exclusive (green).
Correlations between established risk factors for pediatric AML. The diagram indicates co-occurrence or mutual exclusivity between pairs of clinical features, genetic alterations, gene expressions, and outcomes. The circles indicate that crossed factors are statistically co-occurrent (yellow) or exclusive (green).
In view of the gene expression results, HOX genes were upregulated in patients with FLT3-ITD or KMT2A-rearrangements. Moreover, overexpression of some HOX is known to enhance the self-renewal of hematopoietic stem and progenitor cells and perturb differentiation.53 Additionally, FLT3, RUNX1, and some HIST1s were upregulated in these patients, suggesting that the aberrant expression of these genes plays a crucial role in leukemogenesis and indicates poor prognosis. Tiberi et al reported that the expression of HIST1 was significantly higher in patients with AML with low levels of H3K27me3 than in those with high levels of H3K27me3.54 Repressing H3K27me3 levels affects the transcriptional repression in patients with AML harboring aberrant expression of HOX, FLT3, RUNX1, and HIST1.
In the present study, it was shown that RNA-seq unmasked the complexity of gene alterations in pediatric AML. We identified potentially disease-causing alterations in nearly all patients with AML, including novel gene fusions. Our results indicated that a subset of patients with pediatric AML represent a distinct entity that may be discriminated from their adult counterparts through an investigation of the spectrum of gene rearrangements and mutations. Furthermore, we found that a complex interplay of genetic events contributes to the pathogenesis of AML in individual patients. These findings suggest that gene rearrangements in conjunction with mutations play essential roles in pediatric AML. Based on the results of the RNA-seq, the prognostic factors currently used for risk stratification in pediatric AML may be reconsidered, particularly for intermediate/high-risk patients.
Acknowledgments
The authors thank Yuki Hoshino for her valuable technical assistance. The authors would like to thank Enago (www.enago.jp) for the English language review. Supercomputing resources were provided by the Human Genome Center, Institute of Medical Science, The University of Tokyo.
This work was supported by a Grant-in-Aid for Scientific Research on Innovative Areas from the Ministry of Health, Labor and Welfare of Japan (15H05909) (S.O.), a grant for project for development of innovative research on cancer therapeutics (P-DIRECT) from the Japan Agency for Medical Research and Development (AMED; JP18ck0106250h0002) (S.O.), the Practical Research for Innovative Cancer Control from AMED (15ck0106066h0002) (M.S.), a grant for project for cancer research and therapeutics evolution (P-CREATE) from AMED (JP18cm0106501h0003) (S.O.), the Japan Society for the Promotion of Science (KAKENHI grants 25893028 [N.S.], 15H05909 [S.O.], 16K20951 [N.S.], and 17K10130 [N.S.]), a research grant from the Japanese Society of Hematology, and the Kawano Memorial Public Interest Incorporated Foundation for Promotion of Pediatrics.
Authorship
Contribution: N.S. and Y. Hayashi designed the study; N.S., K.Y., Y. Hara, G.Y., T.K., H.M., K.O., and A.S. performed the experiments; Y. Hayashi and S.O supervised the work; N.S., K.Y., Y. Hara, G.Y., Y.S., T.K., M.T., Y.O., and K.O. analyzed the results; N.S. constructed the figures; N.S., K.Y., and Y. Hayashi wrote the paper; and all authors critically reviewed the draft and approved the final version for publication.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Yasuhide Hayashi, Institute of Physiology and Medicine, Jobu University, 270-1, Shinmachi, Takasaki 370-1393, Gunma, Japan; e-mail: hayashiy-tky@umin.ac.jp.