

Key points
Antisense oligonucleotide targeting of hepatic THPO gene expression reduces platelet count within the hemostatic range in mice and baboons.
THPO gene silencing suppresses spontaneous metastatic mammary gland carcinoma progression in transgenic MMTV-PyMT mice.

Abstract
In humans, platelet count within the normal range is required for physiological hemostasis, but, adversely, platelets also support pathological thrombosis. Moreover, by releasing growth factors, they may enhance neoplastic proliferation. We hypothesize that platelet count correlates with platelet-dependent pathologies, even within the range of hemostatic competence. Because platelet production is promoted by thrombopoietin signaling through the myeloproliferative leukemia virus oncogene (cMPL), a receptor expressed on megakaryocytes, we evaluated the feasibility of selective targeting of hepatic thrombopoietin production to test this hypothesis. We synthesized murine- and primate-specific antisense oligonucleotides (THPO-ASO) that silence hepatic thrombopoietin gene (THPO) expression without blocking extrahepatic THPO. Repeated doses of THPO-ASO were administered to mice and a baboon, causing a sustained 50% decline in plasma thrombopoietin levels and platelet count within 4 weeks in both species. To investigate whether reducing platelet count within the translationally relevant hemostatic range could alter a neoplastic process, we administered THPO-ASO to 6-week-old transgenic MMTV-PyMT mice that develop early ductal atypia that progresses into cMPL-negative fatal metastatic breast cancer within 2 to 3 months. THPO-ASO treatment increased the average time to euthanasia (primary humane endpoint) at 2 cm3 combined palpable tumor volume. Our results show that THPO-ASO reduced blood platelet count, plasma platelet factor 4, vascular endothelial growth factor, thrombopoietin levels, bone marrow megakaryocyte density, tumor growth rate, proliferation index, vascularization, platelet and macrophage content, and pulmonary metastases vs untreated controls. These findings confirm that sustained and moderate pharmacological platelet count reduction is feasible with THPO-ASO administration and can delay progression of certain platelet-dependent pathological processes within a safe hemostatic platelet count range.
Introduction
Platelets are critical contributors to both hemostasis and thrombosis. Localized accumulation of activated platelets is accompanied by an increased liberation and concentration of soluble and microparticle-bound bioactive molecules. Some of these entities may affect, among others, neoplastic processes, including wound healing, angiogenesis, and cancer progression.1 Paraneoplastic thrombocytosis has long been documented in the medical literature, yet only a limited number of pioneering clinical and experimental studies provide definitive data to convincingly support the hypothesis that platelets directly contribute to cancer progression.2,3 These and other experimental data suggest that select platelet antagonists could inhibit neoplastic processes by reducing angiogenesis.3,4 However, the use of platelet inhibitors as anticancer agents has not yet been established in the clinic, perhaps in part due to the deleterious effects of current antiplatelet therapies on hemostasis. We rationalized that lowering platelet count rather than ubiquitously inhibiting platelet function may achieve separation between the occult role of platelets in cancer and their essential role in hemostasis.
Platelet count is regulated by a class I hematopoietic cytokine, thrombopoietin (TPO), formerly known as the megakaryocyte growth and differentiation factor, which signals through its receptor, the myeloproliferative leukemia virus oncogene (c-MPL, MPL, CD110). Platelets, megakaryocytes, and a subset of bone marrow hematopoietic stem/progenitor cells express c-MPL.5,6 TPO is encoded by the THPO gene and is constitutively expressed and secreted by the liver and some other organs (kidney, etc). Binding of TPO to c-MPL induces tyrosine phosphorylation of the Janus kinase signal transducer and activation of the JAK-STAT transcription signaling pathway that promotes differentiation and subsequent maturation of hematopoietic stem cells into the megakaryocyte lineage.7
Targeting TPO with antibodies has been shown to be antithrombotic, yet may be difficult to control and can, adversely, cause thrombocytopenia in primates.8 Liver transplants from TPO-deficient to wild-type mice cause a 50% reduction in TPO levels, suggesting that about half of TPO is liver-derived in mice.9 Thus, we hypothesized that liver THPO gene silencing could be used for limited platelet count reduction while keeping the platelet count within the hemostatic competence range as a safe antiplatelet approach. Gene silencing with a new generation of antisense oligonucleotide (ASO) has been shown to selectively inhibit hepatic gene expression by promoting ribonuclease H1 (RNase H1)–mediated messenger RNA (mRNA) degradation.10 Because our second-generation therapeutic ASOs are taken up by hepatocytes without significant uptake by other cells and have been used in the clinic, we targeted THPO gene expression in mice and a baboon using THPO-ASOs. We show herein that THPO-ASOs can be used to maintain a reduced platelet count within the hemostatic range in mice and baboons, and inhibit mammary cancer progression in mice.
Methods
THPO-ASO design, synthesis, and screening
THPO-ASOs used in this study were our second-generation ASOs, 20 nucleotides in length, connected sequentially by phosphorothioate internucleotide linkages. The 5 nucleotides at both the 5′ and 3′ ends are composed of 2'-O-(2-methoxyethyl) (2'-MOE)–modified ribonucleotides, which confer an increased affinity to the target mRNA and increased resistance to exo- and endonucleases within the cell.11-13 The central portion is composed of 10 deoxynucleotides, enabling RNase H1 to recognize and cleave the target mRNAs in the ASO:RNA duplex. Synthesis, purification, and testing of second-generation ASOs were done as previously described.14 Both conjugated and unconjugated THPO-ASOs were synthesized. GalNAc3-conjugation facilitates specific delivery and uptake of ASOs by hepatocytes via high-affinity binding to the hepatocyte-specific asialoglycoprotein receptor.15 GalNAc3-conjugation was shown to improve the potency of second-generation “gapmer” ASOs, targeting hepatocyte-specific RNAs by ∼10- to 30-fold more effectively than first-generation ASOs in preclinical models.15,16
Animals
A series of THPO-ASOs, delivered by electroporation, was initially screened for their ability to reduce THPO RNA, ex vivo, in murine Hepa1-6 cells and in primary monkey hepatocytes. The sets of selected murine THPO-ASOs with the highest activity were screened and evaluated in vivo by subcutaneous (SC) administration in 4-week studies in both male and female BALB/C mice for general tolerability and effects on hepatic THPO mRNA expression and platelet count. One unconjugated murine THPO-ASO sequence with favorable general safety and efficacy profile was selected for use in subsequent experiments in mice. One unconjugated primate THPO-ASO sequence and its more potent 5′-GalNAc3-conjugated version were tested sequentially in a single baboon to determine if platelet count responses to TPO level reduction were potentially safe and also feasible in a primate.
Mouse and baboon experiments conducted at Oregon Health & Science University were approved by an Institutional Care and Use Committee. We previously established that arterial-type thrombus formation correlates with platelet count in baboons, and demonstrated the antithrombotic activity of neutralizing TPO antibodies.8,17 Here, we used a juvenile male baboon (15 kg) to determine whether THPO-ASO (unconjugated, 50 mg/kg repeat dose or GalNAc3-conjugated, 8 mg/kg repeat dose) could be used to reduce plasma TPO levels and platelet count within the safe range of hemostatic competence in primates. Treatment with SC injections of unconjugated and, after complete platelet count recovery, GalNAc3-conjugated THPO-ASOs was initiated with 3 loading doses within a week and then maintained with once-a-week dosing until the platelet count settled at 50% of baseline. Blood samples were collected for general safety and TPO level evaluation. The second round of GalNAc3-conjugated THPO-ASO treatment started 6 months after the first experiment, which used the unconjugated THPO-ASO. The animal went off study without adverse events.
We used female MMTV-PyMT transgenic mice to investigate the effect of murine THPO-ASO treatment on blood platelet count, plasma TPO levels, and progression of breast cancer. Hemizygous MMTV-PyMT mice spontaneously developed ultimately fatal metastatic breast cancer in mammary glands, with early microscopic mammary gland epithelial atypia beginning as early as 3 to 4 weeks of age.18 Inbred Friend virus B/Pde6brd1 wild-type (FVB/N WT) and FVB/N-Tg 634Mul/J transgenic (MMTV-PyVT) mice were bred, characterized, and housed under conditions as described elsewhere.19 FVB/N WT mice were used for establishment of methods and to verify that THPO-ASO reduced TPO levels and platelet count in this mouse strain. Hemizygous MMTV-PyMT mice that develop cancer were used as comparators and to also determine if platelet count reduction affects the progression of their cancers. All mice were euthanized with isoflurane, either at the time of a terminal blood draw or when total combined mammary tumor volume reached ≥2 cm3.
Experimental design
THPO-ASO (50 mg/kg SC) or saline treatment was started in 65-day-old FVB/N WT and 40-day-old MMTV-PyMT mice with treatment on the first, third, fifth, and seventh days followed by weekly maintenance injections based on prior safety and dose-finding screening studies in C57BL6 mice. Developing mammary tumors in MMTV-PyMT mice are typically not yet palpable (<2 mm diameter) at 40 days of age. Diagnostic evaluation by palpation was performed every 3 days from day 40, and palpable tumor dimensions were measured with a caliper and recorded from 80 days of age. Following euthanasia, primary tumors, lungs, and plasma were isolated for further evaluation.
In a similar manner, the baboon received loading and maintenance THPO-ASO doses based on weekly monitoring; treatment was discontinued when the platelet count fell to ∼50% of the baseline value.
Blood counts
The tail tip of mice was cut, and 10 µL blood was collected using a micropipette with an EDTA-prewetted tip. Samples were immediately mixed with an equal volume of 10 mM EDTA in phosphate-buffered saline (Sigma-Aldrich, St. Louis, MO). The blood counts were obtained using a scil Vet abc hematology analyzer within 1 hour. After sample preparation, each tail tip was cauterized.
In the baboon, citrate-anticoagulated blood samples were obtained by standard antecubital venipuncture for blood counts, TPO, and clinical chemistry without any other intervention. Blood monitoring was continued for several months after cessation of THPO-ASO administration to monitor recovery.
Plasma tests
In anesthetized mice, terminal blood samples (1 mL) were drawn from the heart into 3.2% sodium citrate (9:1), and plasma was obtained by centrifugation. The samples were apportioned into 100-μL aliquots and stored at −80°C until testing. TPO levels in the plasma were quantified using a mouse Thrombopoietin Quantikine ELISA kit (enzyme-linked immunosorbent assay; R&D Systems, Minneapolis, MN) per the manufacturer's instructions. Plasma platelet factor 4 (PF4) levels were measured using mouse PF4 ELISA kit (Abcam, Cambridge, MA). Vascular endothelial growth factor (VEGF) levels were measured using a mouse VEGF Quantikine ELISA kit (R&D Systems).
Baboon plasma or serum TPO levels were measured using a human Thrombopoietin Quantikine ELISA kit (R&D Systems),8 and standard clinical laboratory tests were periodically performed for general safety and recovery assessment.
Immunostaining
Formalin-fixed mouse tissues from tumors and organs were embedded in O.C.T. compound (ThermoFisher Scientific, Waltham, MA) after soaking with 30% sucrose in phosphate-buffered saline. Femurs were decalcified with 12.5% (vol/wt) EDTA solution for 72 hours at 4°C before soaking. Sections (8 μm) were stained with the following primary antibodies: rat anti-mouse CD41 monoclonal antibody (MWReg30; Biolegend, San Diego, CA), rabbit anti-human von Willebrand factor (VWF) polyclonal antibody (Agilent, Santa Clara, CA), rabbit anti-mouse CD34 monoclonal antibody (EP373Y; Abcam), rabbit anti-MPL polyclonal antibody (Bioss Antibodies Inc, Woburn, MA), rabbit anti–Ki-67 polyclonal antibody (Abcam), and rat anti-mouse F4/80 monoclonal antibody (BM8; Biolegend). For immunofluorescence, Alexa Fluor dye-labeled secondary antibodies (ThermoFisher Scientific) were used and counterstained with Hoechst 33342 (ThermoFisher Scientific). Five random fields per section and 3 sections (100-μm intervals) were imaged on 20× objective lens using an inverted fluorescence microscope IX-71 and Slide Book software (Denver, CO). Megakaryocyte density, platelet accumulation, intratumoral blood vessel density, macrophage infiltration, and Ki-67–positive (S-phase) cells were analyzed with FIJI ImageJ software, and mean values in 15 images were used for quantification. The ratio of Ki-67–positive cells to Ki-67–negative cells was used to determine the proliferation index in primary breast cancer sections.
Pulmonary metastases
Formalin-fixed paraffin-embedded lung sections (5-μm thickness) from the mice were cut at 100-μm intervals and stained with hematoxylin and eosin. Nodules in each section were evaluated via microscopy. The mean number of nodules of 5 sections from each mouse was plotted and used for statistical analysis.
Statistical analyses
All data are presented as the mean ± standard error of the mean. Differences between means were determined at P < .05 using Student t test. Averages of time to mandatory euthanasia were evaluated using the log-rank (Mantel-Cox) test and GraphPad Prism software (San Diego, CA).
Results
THPO-ASO reduces platelet count in FVB/N WT mice and baboons
First, we examined whether THPO-ASO administration would reduce platelet counts in mice. Our data show that THPO-ASO treatment reduced platelet count in FVB/N WT mice by ∼50% after 28 days of treatment (Figure 1A-B; saline = 1.1 ± 0.07 × 106/µL vs THPO-ASO = 0.5 ± 0.03 × 106/µL); the reduction in platelet count remained steady until the end of observation (135 days). The difference in mean platelet volume after 70 days was not statistically significant (Figure 1C). Leukocyte and red blood cell counts were also not affected, similar to observations made in TPO-deficient mice.20 At the 70-day treatment endpoint, mouse plasma and femurs were analyzed for circulating TPO levels and megakaryopoiesis, respectively. There was a significant reduction in plasma TPO levels in THPO-ASO–treated mice (102 ± 6 pg/mL) as compared with vehicle-treated controls (214 ± 54 pg/mL; Figure 1D). Histological analysis revealed a reduced density of CD41+ VWF+ mature megakaryocytes (100 ± 6 cells per mm2) in the femoral bone marrow of THPO-ASO–treated mice as compared with vehicle-treated controls (246 ± 15 cells per mm2; Figure 1E-F).
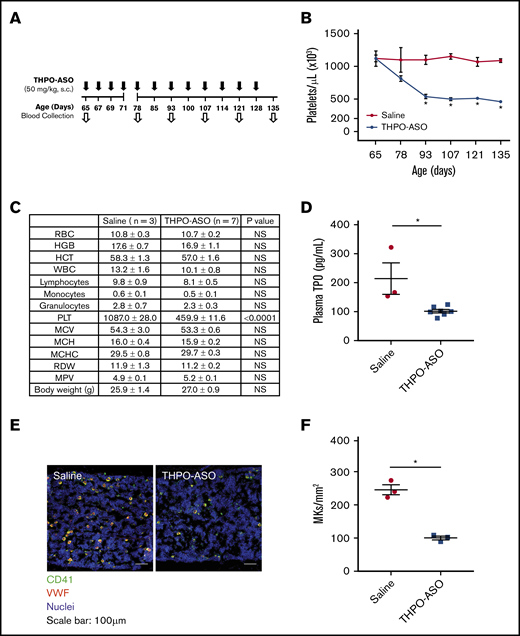
Reduced platelet (PLT) production in THPO-ASO–treated FBV/N mice. (A) THPO-ASO or saline (control) treatment scheme. WT female FBV/N (NIH Jackson–homozygous phosphodiesterase 6Brd1 mutant) mice received repeat injections of saline (controls) or nonconjugated THPO-ASO (50 mg/kg, SC) beginning on day 65 after birth. (B) Changes in mean platelet count over time in saline- (red line, n = 3) or THPO-ASO–treated (blue line, n = 7) mice. (C) Complete blood counts on day 135 after birth. (D) Plasma TPO levels on day 135 after birth. (E) Representative images of megakaryocytes (MKs) in the femoral bone marrow on day 135 after birth in saline- or THPO-ASO–treated mice. Megakaryocytes were visualized via anti-CD41 (GPIIb; green) and anti-VWF (red) antibodies. Nuclei were stained with Hoechst 33342 (blue). Original magnification ×20. Scale bars, 100 µm. (F) Quantitative evaluation of megakaryocyte density in the bone marrow on day 135 after birth. Numeric data are shown as mean ± standard error of the mean. The 2-tailed, unpaired Student t test was used for comparison of means. *P < .05. HCT, hematocrit; HGB, hemoglobin; MCH, mean corpuscular hemoglobin; MCHC, mean corpuscular hemoglobin concentration; MCV, mean corpuscular volume; MPV, mean platelet volume; NS, not significant; RBC, red blood cell; RDW, red blood cell distribution width; S.C., subcutaneous; WBC, white blood cell.
Reduced platelet (PLT) production in THPO-ASO–treated FBV/N mice. (A) THPO-ASO or saline (control) treatment scheme. WT female FBV/N (NIH Jackson–homozygous phosphodiesterase 6Brd1 mutant) mice received repeat injections of saline (controls) or nonconjugated THPO-ASO (50 mg/kg, SC) beginning on day 65 after birth. (B) Changes in mean platelet count over time in saline- (red line, n = 3) or THPO-ASO–treated (blue line, n = 7) mice. (C) Complete blood counts on day 135 after birth. (D) Plasma TPO levels on day 135 after birth. (E) Representative images of megakaryocytes (MKs) in the femoral bone marrow on day 135 after birth in saline- or THPO-ASO–treated mice. Megakaryocytes were visualized via anti-CD41 (GPIIb; green) and anti-VWF (red) antibodies. Nuclei were stained with Hoechst 33342 (blue). Original magnification ×20. Scale bars, 100 µm. (F) Quantitative evaluation of megakaryocyte density in the bone marrow on day 135 after birth. Numeric data are shown as mean ± standard error of the mean. The 2-tailed, unpaired Student t test was used for comparison of means. *P < .05. HCT, hematocrit; HGB, hemoglobin; MCH, mean corpuscular hemoglobin; MCHC, mean corpuscular hemoglobin concentration; MCV, mean corpuscular volume; MPV, mean platelet volume; NS, not significant; RBC, red blood cell; RDW, red blood cell distribution width; S.C., subcutaneous; WBC, white blood cell.
Next, we examined whether THPO administration would reduce platelet counts in nonhuman primates. Indeed, our data confirm that baboon platelet count and TPO levels fell following THPO-ASO administration (loading dose followed by a maintenance dose). Platelet count reduction to ∼50% of the baseline was achieved with both types of ASOs within a month (Figure 2A-B shows 2 consecutive treatments of the same animal). Plasma TPO levels declined by 30% to 35% in both experiments. Dose-response was not established. Recovery after termination of the first treatment on day 28 was followed for 6 months; our data show that the animal naturally normalized its platelet count (Figure 2A), which then allowed for the repeat experiment with the more potent GalNAc3-conjugated THPO-ASO (Figure 2B).
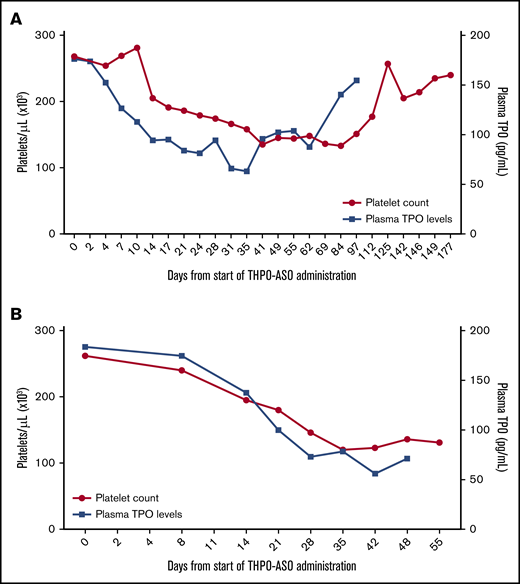
Effect of THPO-ASO–treatment on platelet count and TPO levels in a baboon. (A) Platelet counts and TPO levels during weekly repeat administration of unconjugated THPO-ASO (days 0 to 28) and during the recovery/follow-up period (until day 177) in a juvenile male baboon. (B) Platelet counts and TPO levels during weekly repeat administration of GaINac-conjugated THPO-ASO (days 0 to 28) and during the follow-up period (until day 55) in the same baboon.
Effect of THPO-ASO–treatment on platelet count and TPO levels in a baboon. (A) Platelet counts and TPO levels during weekly repeat administration of unconjugated THPO-ASO (days 0 to 28) and during the recovery/follow-up period (until day 177) in a juvenile male baboon. (B) Platelet counts and TPO levels during weekly repeat administration of GaINac-conjugated THPO-ASO (days 0 to 28) and during the follow-up period (until day 55) in the same baboon.
THPO-ASO reduces platelet count and inhibits mammary cancer progression in MMTV-PyMT mice
The platelet count in MMTV-PyMT mice remained stable and mirrored counts in nontransgenic littermates throughout their lifespan (n = 10 per group) (Figure 3A). THPO-ASO administration at day 40 after birth reduced the platelet count in MMTV-PyMT mice by ∼50% within 2 weeks; maintenance of ∼50% reduction in platelet count was sustained by weekly injections of THPO-ASO until euthanasia. Reduction in platelet count via administration of THPO-ASO at day 40 did not affect the time to appearance of the first palpable tumor (median; vehicle = 58.0 days, THPO-ASO = 58.5 days, P = .65), with all mice developing at least 1 palpable tumor by day 80 (Figure 3B). THPO-ASO–treated mice had fewer palpable tumors on average on day 80 (Figure 3C; vehicle = 6.9 ± 0.7 glands per mouse, THPO-ASO = 4.4 ± 0.5 glands per mouse). The average individual tumor volume was 56% smaller in THPO-ASO–treated mice than in controls (Figure 3D; supplemental Figure 1; vehicle = 68 ± 11 mm3, THPO-ASO = 30 ± 8 mm3). Tumors grew slower in THPO-ASO–treated animals than in controls (Figure 3E) as measured by the time from manual diagnosis to mandatory euthanasia at the maximum allowed combined tumor volume (humane endpoint, 2 cm3) (Figure 3F; median; vehicle = 90 days old, THPO-ASO = 109 days, P < .05). Median time from the average manual diagnosis to euthanasia was 32 days and 50 days in vehicle- and THPO-ASO–treated mice, respectively, indicating slower disease progression at lower platelet count. The average number of metastatic nodules per lung was lower in THPO-ASO–treated (4.4 ± 1.7 nodules per lung) than in vehicle-treated (12.3 ± 2.0 nodules per lung) animals (Figure 3G-H).
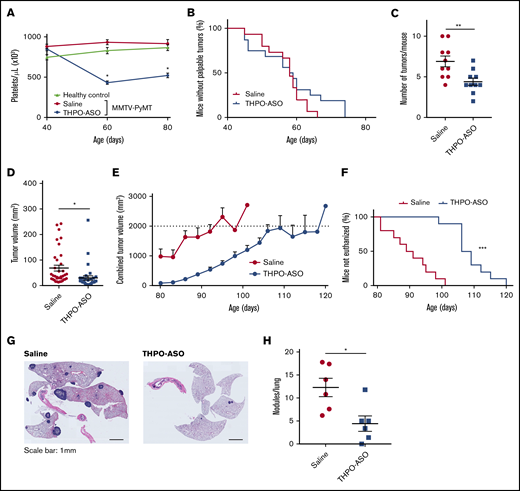
Effect of THPO-ASO treatment on breast cancer propagation in MMTV-PyMT mice. (A) Response to THPO-ASO was determined in a feasibility study by measuring the platelets count in untreated, saline-treated, or THPO-ASO–treated FVB/N-Tg 634Mul/J transgenic (MMTV-PyVT) mice. Weekly treatment with saline (controls, n = 6, red line) or THPO-ASO (n = 6, blue line) was initiated at 40 days of age, and platelet counts were determined at 40, 60, and 80 days after birth. (B) The time of palpable mammary tumor diagnosis during saline (n = 15, red line) and during THPO-ASO treatment (n = 15, blue line). (C) The number of mammary glands with palpable tumor at 80 days of age in saline (n = 10) and THPO-ASO (n = 10) treatment groups. (D) Individual tumor volumes at 80 days of age in saline- (n = 36) and THPO-ASO–treated (n = 33) animals. (E) Growth curves showing the means of total tumor volumes during saline (n = 10, red line) or THPO-ASO (n = 10, blue line) treatment. The combined tumor volume of 2 cm3 was set as the humane endpoint. Any animal reaching this endpoint, shown as a dotted line on the graph, was euthanized in accordance with institutional policies. (F) Age of mice at the time of euthanasia (combined tumor volume of 2 cm3) in saline-treated (n = 10, red line) or THPO-ASO–treated (n = 10, blue line) mice. (G) Representative hematoxylin and eosin–stained lung sections obtained after euthanasia of mice. Scale bars, 1 mm. (H) The number of metastatic nodules in the lung at time of euthanasia in saline-treated (n = 6) or THPO-ASO–treated (n = 6) mice. The 2-tailed, unpaired Student t test was used for comparison of means in panels A and C-D. *P < .05; **P < .01; ***P < .001. Log-rank test was used for comparisons in panels B and F. ***P < .001.
Effect of THPO-ASO treatment on breast cancer propagation in MMTV-PyMT mice. (A) Response to THPO-ASO was determined in a feasibility study by measuring the platelets count in untreated, saline-treated, or THPO-ASO–treated FVB/N-Tg 634Mul/J transgenic (MMTV-PyVT) mice. Weekly treatment with saline (controls, n = 6, red line) or THPO-ASO (n = 6, blue line) was initiated at 40 days of age, and platelet counts were determined at 40, 60, and 80 days after birth. (B) The time of palpable mammary tumor diagnosis during saline (n = 15, red line) and during THPO-ASO treatment (n = 15, blue line). (C) The number of mammary glands with palpable tumor at 80 days of age in saline (n = 10) and THPO-ASO (n = 10) treatment groups. (D) Individual tumor volumes at 80 days of age in saline- (n = 36) and THPO-ASO–treated (n = 33) animals. (E) Growth curves showing the means of total tumor volumes during saline (n = 10, red line) or THPO-ASO (n = 10, blue line) treatment. The combined tumor volume of 2 cm3 was set as the humane endpoint. Any animal reaching this endpoint, shown as a dotted line on the graph, was euthanized in accordance with institutional policies. (F) Age of mice at the time of euthanasia (combined tumor volume of 2 cm3) in saline-treated (n = 10, red line) or THPO-ASO–treated (n = 10, blue line) mice. (G) Representative hematoxylin and eosin–stained lung sections obtained after euthanasia of mice. Scale bars, 1 mm. (H) The number of metastatic nodules in the lung at time of euthanasia in saline-treated (n = 6) or THPO-ASO–treated (n = 6) mice. The 2-tailed, unpaired Student t test was used for comparison of means in panels A and C-D. *P < .05; **P < .01; ***P < .001. Log-rank test was used for comparisons in panels B and F. ***P < .001.
THPO-ASO reduces platelet accumulation in the tumor microenvironment and suppresses tumor angiogenesis
THPO-ASO treatment reduced plasma levels of the systemic platelet activation marker, PF4, as compared with vehicle-treated controls (Figure 4A; vehicle = 220 ± 13 ng/mL, THPO-ASO = 169 ± 16 ng/mL). Circulating plasma VEGF levels were lower in THPO-ASO–treated mice (52.13 ± 8.426 pg/mL) than in untreated mice (104.4 ± 15.14 pg/mL) (supplemental Figure 2). On histology, the percentage of tumor vessels containing bound or trapped platelets was significantly reduced in THPO-ASO–treated mice (Figure 4B-C; vehicle = 54% ± 4% of vessels containing platelets, THPO-ASO = 26% ± 3% of vessels). Intratumoral vessel density was also reduced in THPO-ASO–treated mice (Figure 4D; vehicle = 3.1% ± 0.4% CD34+ area, THPO-ASO = 1.5% ± 0.3% CD34+ area). The deposited platelets stained positive for cMPL, as expected, whereas the extravascular tumor cells were negative for cMPL on immunohistochemistry (supplemental Figure 3).
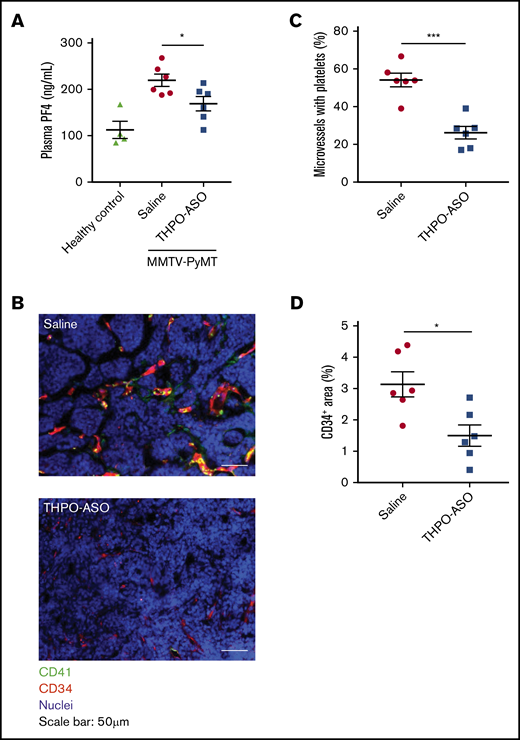
Systemic platelet activation markers, and local platelet deposition and angiogenesis in the tumor microenvironment. (A) Plasma PF4 levels at the time of euthanasia were quantified using an ELISA to assess systemic markers of platelet activation. (B) Representative images of platelet accumulation and vessel density in mammary tumors. Platelets and vascular endothelial cells were visualized with anti-CD41 (green) and anti-CD34 (red) antibodies, respectively. Nuclei were visualized with Hoechst 33342 (blue). Scale bars, 50 µm. (C) Percentage of intratumoral vessels with deposited platelets. Fifteen fields of view per tumor were analyzed for quantification. Each tumor was evaluated from 6 mice per treatment. (D) Percentage of CD34-positive area in the field of view of samples from saline-treated (n = 6) or THPO-ASO–treated (n = 6) mice. Fifteen fields of view per tumor were analyzed for quantification. Every tumor was evaluated in each mouse. A 2-tailed, unpaired Student t test was used for comparison of means. *P < .05; ***P < .001.
Systemic platelet activation markers, and local platelet deposition and angiogenesis in the tumor microenvironment. (A) Plasma PF4 levels at the time of euthanasia were quantified using an ELISA to assess systemic markers of platelet activation. (B) Representative images of platelet accumulation and vessel density in mammary tumors. Platelets and vascular endothelial cells were visualized with anti-CD41 (green) and anti-CD34 (red) antibodies, respectively. Nuclei were visualized with Hoechst 33342 (blue). Scale bars, 50 µm. (C) Percentage of intratumoral vessels with deposited platelets. Fifteen fields of view per tumor were analyzed for quantification. Each tumor was evaluated from 6 mice per treatment. (D) Percentage of CD34-positive area in the field of view of samples from saline-treated (n = 6) or THPO-ASO–treated (n = 6) mice. Fifteen fields of view per tumor were analyzed for quantification. Every tumor was evaluated in each mouse. A 2-tailed, unpaired Student t test was used for comparison of means. *P < .05; ***P < .001.
THPO-ASO reduces monocyte recruitment
When compared at day 80, MMTV-PyMT mice had moderate leukocytosis compared with nontransgenic mice. In the presence of THPO-ASO treatment, the moderate leukocytosis was less pronounced, as measured by a 19.8% decrease in the number of lymphocytes (vehicle = 13.1 ± 0.7 × 103/µL, THPO-ASO = 10.5 ± 0.4 × 103/µL), 27.9% decrease in granulocytes (vehicle = 8.6 ± 0.8 × 103/µL, THPO-ASO = 6.2 ± 0.9 × 103/µL), and 41.7% decrease in monocytes (vehicle = 2.4 ± 0.3 × 103/µL, THPO-ASO = 1.4 ± 0.2 × 103/µL) compared with untreated controls (Figure 5A). On histopathology, the density of F4/80-positive macrophages in the primary tumors of THPO-ASO–treated mice was reduced by 50% compared with untreated controls (Figure 5B-C; vehicle = 2.8% ± 0.5% area, THPO-ASO = 1.4% ± 0.2% area).
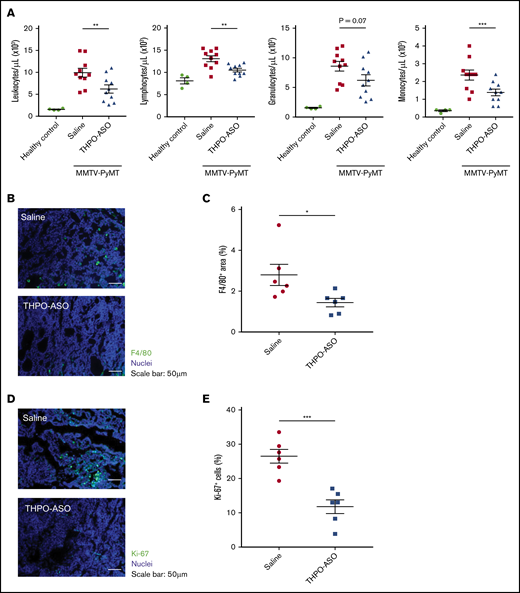
Peripheral leukocyte profile, and intratumoral macrophages and cell proliferation index. (A) Leukocyte profile from complete blood counts at 80 days of age in healthy and MMTV-PyMT mice. (B) Representative images of tumor-infiltrating macrophages on histopathology at the time of euthanasia (complete palpable tumor volume of 2 cm3). Macrophages were visualized with an antibody directed against F4/80, the murine homolog of epidermal growth factor–like module-containing mucin-like hormone receptor-like 1 (green). Nuclei were visualized with Hoechst 33342 (blue). Scale bars, 50 µm. (C) Percentage of F4/80-positive area in the field of view in tumors from untreated (n = 6) and THPO-ASO–treated (n = 6) mice. A 2-tailed, unpaired Student t test was used for comparison of means. *P < .05; **P < .01; ***P < .001. (D) Representative images of proliferating cells in tumors. Proliferating cells were identified by Ki-67, an antibody directed against the MKI67 proliferation-related antigen (green). Nuclei were visualized with Hoechst 33342 (blue). Scale bars, 50 µm. (E) Number of Ki-67–positive (S-phase) cells in magnification field in samples from untreated (n = 6) and THPO-ASO–treated (n = 6) mice. Fifteen fields of view per tumor were analyzed for quantification. Every breast tumor was evaluated for each mouse. A 2-tailed, unpaired Student t test was used for comparison of means. *P < .05.
Peripheral leukocyte profile, and intratumoral macrophages and cell proliferation index. (A) Leukocyte profile from complete blood counts at 80 days of age in healthy and MMTV-PyMT mice. (B) Representative images of tumor-infiltrating macrophages on histopathology at the time of euthanasia (complete palpable tumor volume of 2 cm3). Macrophages were visualized with an antibody directed against F4/80, the murine homolog of epidermal growth factor–like module-containing mucin-like hormone receptor-like 1 (green). Nuclei were visualized with Hoechst 33342 (blue). Scale bars, 50 µm. (C) Percentage of F4/80-positive area in the field of view in tumors from untreated (n = 6) and THPO-ASO–treated (n = 6) mice. A 2-tailed, unpaired Student t test was used for comparison of means. *P < .05; **P < .01; ***P < .001. (D) Representative images of proliferating cells in tumors. Proliferating cells were identified by Ki-67, an antibody directed against the MKI67 proliferation-related antigen (green). Nuclei were visualized with Hoechst 33342 (blue). Scale bars, 50 µm. (E) Number of Ki-67–positive (S-phase) cells in magnification field in samples from untreated (n = 6) and THPO-ASO–treated (n = 6) mice. Fifteen fields of view per tumor were analyzed for quantification. Every breast tumor was evaluated for each mouse. A 2-tailed, unpaired Student t test was used for comparison of means. *P < .05.
THPO-ASO reduces the cell proliferation index the primary tumors
On immunohistopathology, the number of S-phase Ki-67–positive cells was lower in tumors of THPO-ASO–treated mice as compared with untreated controls (Figure 5D-E; vehicle = 26.5% ± 2.0%, THPO-ASO = 11.8% ± 2.0%), suggesting that THPO-ASO treatment suppressed cell proliferation rate.
Discussion
TPO agonists have been successfully used in the clinic to treat thrombocytopenia.21,22 Meanwhile, TPO antagonist antibodies can cause severe thrombocytopenia. We have also demonstrated that TPO antibodies can produce a profound antithrombotic effect in primates.8 TPO deficiency causes severe thrombocytopenia without affecting the activity of the remaining platelets in vitro, suggesting that TPO is unlikely to alter platelet function.8,23,24 Antiplatelet agents uniformly impair platelets, often with detrimental off-target effects. Because both thrombocytopenia and inhibition of hemostatic platelet functions are safety concerns, there remains a need for safer antiplatelet approaches that do not impair hemostasis. We hypothesized that hepatic THPO gene silencing could lower but leave platelet count within the range of hemostatic competence. Second-generation ASOs, particularly GalNAc3-conjugates, selectively target hepatocytes. Thus, to address whether a limited platelet count reduction could affect pathological processes beyond thrombosis, we evaluated the effects of THPO-ASO administration in a model of early spontaneous aggressive breast cancer.
Platelets have been implicated to play a role in angiogenesis, epithelial-mesenchymal transition, oncoprotein expression, and tumor cell growth via a variety of pathways, including via release of platelet-derived growth factors (PDGFs), tumor growth factor-β (TGF-β), insulin-like growth factors, fibroblast growth factors, and other cytokines.1 Moreover, platelets have been suggested to promote metastasis by binding to circulating tumor cells, shield them from immune cells, including natural killer cells, protect them from hemodynamic shear, and mediate their recruitment and binding to the endothelium, thus establishing metastatic sites.25-27 However, direct clinical evidence that platelets promote neoplastic malignancies has not yet been generated in the absence of a significant comorbidity of hemostasis impairment due to platelet function inhibitors or thrombocytopenia. In fact, current cancer treatment guidelines do not caution against the use of platelet transfusions or TPO agonists in patients with cancer.
Because hemostasis impairment, whether due to anticoagulation, platelet inhibition, or other reasons, has also been associated with an effect on cancer propagation and metastasis, separation of the direct effects of platelets from their role in hemostasis while investigating the role of platelets in human cancer has been challenging. Because of a lack of commercial platelet inhibitors that are both safe and platelet-specific, there are only limited clinical data to corroborate causality or significance of the apparent platelet-cancer association. For example, 1 study suggested an association between the lower incidence of colorectal cancer and aspirin use, whereas another found an increased cancer mortality among aspirin users,28,29 both contemplating an antiplatelet effect. However, aspirin is not a platelet-specific inhibitor of cyclooxygenase, and the relationship of its pharmacological effects with cancer is difficult to determine,30 especially in the absence of randomized controlled prospective trials.31-37 Although there are other clinically used platelet inhibitors that are more platelet-specific, they all carry a significant bleeding risk that could outweigh their potential benefits over time, should they be evaluated for safety and efficacy in patients with cancer. There are novel, potentially less antihemostatic, presumably safer platelet inhibitors in clinical development, which may be considered for evaluation in patient populations with specific neoplastic malignancies.38,39 Platelet count correlates with the rate of experimental thrombus propagation, and, thus, platelet count reduction within the range of hemostatic competence may be a mechanistically straightforward antiplatelet treatment approach. The association between platelet count and hemostasis has been well established in humans, documented in baboons, and quantitatively defined using a tail-bleeding assay in mice, which suggests that platelet counts as low as 20% of mean normal are sufficient to maintain hemostasis.24
In this study, we focused on determining whether hepatic THPO gene silencing reduced platelet count within a viable range and interfered with cMPL-negative aggressive metastatic breast cancer progression in MMTV-PyMT mice. Metastatic breast cancer is a prevalent cause of death in women,40 and although platelet count has been associated with metastatic breast cancer prognosis,41 causality in either direction has not been established.42,43 Hemizygous transgenic MMTV-PyMT mice develop microscopic signs of malignancy by ∼1 month of age, and palpable breast cancer by ∼2 months of age. The aggressive disease progresses rapidly to terminal metastatic cancer in mice within 3 to 4 months from birth.19 In our study, steady-state limited platelet count reduction was achieved only after 1 month of starting the THPO-ASO treatment of 40-day-old mice, at which time development of tumors had already started. Nevertheless, we found that tumorigenesis was somewhat delayed at 2 to 3 months of age in THPO-ASO–treated mice, primarily in the caudal mammary glands, which develop later in mice, suggesting that even a marginal platelet count reduction may have beneficial effects.
It is reasonable to suspect that activation of platelets in tumor tissues could have local effects. Leaky vessels may allow for direct and indirect platelet-tumor cell interactions.44 Platelet-derived proangiogenic factors, such as VEGF-A, have been implicated to promote tumor angiogenesis.45 Among others, such as the well-established role of the predominant mitogenic cytokine, PDGF, in promoting cell proliferation through a receptor tyrosine kinase, PDGFR, platelets also release TGF-β.46 The granule contents of activated platelets may even deter the immune response to certain tumors. Platelet-derived TGF-β release has been shown to impair the immune response, as platelet-specific TGF-β-deficient mice show enhanced cytotoxic T-cell activity.47,48 Tumor-associated macrophages are often abundant in mammary tumors and may also promote cancer progression.49 Platelets have been implicated in monocyte recruitment to lipopolysaccharide-inflamed intestinal microvessels in a P-selectin–dependent manner.50 The platelet-derived chemokines PF4 and RANTES have been shown to promote monocyte recruitment at sites of inflammation.51 In our study herein, we observed a significant drop in platelet accumulation in the mammary tumors upon TPO depletion, which correlated with reduced plasma levels of PF4, thus providing a potential mechanism for the observed decrease in monocyte recruitment into mammary tumors as a consequence of THPO-ASO treatment. A direct effect of reduced TPO on the tumors themselves in our case cannot be excluded, but is unlikely because we could not demonstrate the presence of cMPL on the cancer cells themselves (supplemental Figure 2).
Although THPO-ASO appears to have anticancer activity in the MMTV-PyMT breast cancer mouse model, we do not yet know how platelet count within the range of hemostatic competence affects progression of other cancers. We also do not yet know if platelet count affects the immune response to cancer. The role of extrahepatic THPO in driving platelet production in paraneoplastic thrombocytosis is not established either. It is also not known whether selective silencing of hepatic THPO would also reduce platelet count or improve outcomes in cancer-associated thrombocytosis in humans, observed in 15% to 40% of solid tumor patients.52 In a thorough pioneering study, Stone and colleagues have documented reduction of experimental paraneoplastic thrombocytosis and associated antineoplastic effects using a TPO small interfering RNA, with or without also inhibiting interleukin-6.3 This indicated that paraneoplastic thrombocytosis is responsive to THPO gene silencing in some mouse models. Moreover, it was reported that the expansion of megakaryocytes by TPO treatment in BALB/c nude mice resulted in reduced bone metastases in a PC3 prostate cancer intracardiac injection model.53 In addition, TPO-deficient mice, which are hemostatically compromised, exhibited more aggressive bone metastases in the syngeneic 4T1.2 breast cancer model.54 Meanwhile, others have reported a reduced number of lung metastases of IV injected tumor cells in mice with experimental thrombocytopenia-associated hemostasis impairment.38,55 Another study demonstrated that select chemotherapies are more effective in thrombocytopenic mice.56 Together, these studies further suggest that platelets support progression of experimental malignancies. Here, we investigated whether moderately reducing platelet count would slow spontaneous tumor progression, including lung metastases, from the primary tumors in hemostatically competent mice. We found a demonstrable antineoplastic effect of moderate chronic platelet count reduction, in the absence of paraneoplastic thrombocytosis. Future studies are required to validate this approach in other murine tumors or patient-derived tumor xenografts, as well as explore the effect that reducing TPO levels may have on reducing hematopoietic stem cell number or function. Our approach may have translational relevance by virtue of the reasonable safety of the applied antisense technology.
Our findings also raise the theoretical possibility that cytotoxic chemotherapeutic agents that both suppress megakaryopoiesis and induce remission in patients with cancer may be antineoplastic, at least in part, by means of reduction in platelet count. If this is indeed the case, the use of TPO agonists or platelet transfusions to balance chemotherapy-induced thrombocytopenia could, in theory, reduce the efficiency of chemotherapy. TPO agonists or platelet transfusions have been occasionally used to increase platelet count in severely thrombocytopenic patients with cancer who receive nonmyeloablative chemotherapy, but, at present, the availability of data from controlled clinical trials to establish the harm or benefit of TPO agonists or platelet supplementation on long-term outcomes, including survival and quality of life, is limited.57-61
Our present data support the concept that targeted hepatic THPO gene silencing is a feasible approach to therapeutic platelet count reduction within the safe range of hemostatic competence, and also support the hypothesis that platelets may contribute to cancer progression. Reducing platelet count within the hemostatic competence range vs inhibiting platelet functions with agents that impair all circulating platelets has other theoretical advantages, including instant reversibility. We do not yet know if THPO-ASO administration has preventive efficacy in this or other disease models, because THPO-ASO was administered after day 40, and thus, by the time this treatment had achieved a 50% platelet count reduction, the MMTV-PyMT mice had already developed cancer. We contemplate that if therapeutic efficacy of THPO-ASO is verified in additional, less aggressive models of cancer and other platelet-dependent diseases, it could be used alone or in combination with therapies that do not have significant myelotoxicity or antiplatelet effect. Clinically relevant THPO-ASO–induced thrombocytopenia, even if it occurred, could be reversed with platelet transfusion, and TPO deficiency would be readily treatable with TPO agonists.
In conclusion, our data are consistent with the hypothesis that platelets within the normal platelet count range can support tumor progression. We found that partial THPO gene silencing is feasible in both rodents and primates and produced sustained and limited platelet count reduction with a low risk of hemostatic impairment. Therefore, chronic THPO-ASO administration could be a reasonably safe antiplatelet approach to attenuate the progression of pathological processes that are promoted by platelets.
Acknowledgments
This project has been sponsored by Aronora, Inc and Ionis Pharmaceuticals. The investigators were also supported, in part, by National Institutes of Health, National Heart, Lung, and Blood Institute grants HL117589 and HL095315 (A.G. and E.I.T.), and HL101972 (O.J.T.M.), and National Cancer Institute grants CA233280, CA223150, and CA226909 (L.M.C.). Also acknowledged is support from a DOD BCRP Era of Hope Scholar Expansion Award (W81XWH-08-PRMRP-IIRA), the Susan B. Komen Foundation (KG110560), and the Breast Cancer Research Foundation (L.M.C.).
Authorship
Contribution: T.S. contributed to experimental design of the mouse studies, performed the mouse experiments, analyzed and evaluated data, and wrote the manuscript; A.G. conceived, designed, and supervised the project, evaluated the results, and cowrote the manuscript; A.S.R., B.P.M., and A.G. designed, developed, and evaluated rodent and primate THPO-ASOs; J.T., A.T.P.N., A.M., and L.D.H. contributed to mouse breeding and experiments; J.J. performed and M.T.H. oversaw the primate studies; and A.T.P.N., L.M.C., E.I.T., A.M., and O.J.T.M. contributed to experimental design, data interpretation, and manuscript revisions.
Conflict-of-interest disclosure: A.G. and E.I.T. are employees of Oregon Health & Science University, which may have financial interest in the results of this study. A.G., E.I.T. (Aronora, Inc), A.S.R., and B.P.M. (Ionis Pharmaceuticals) are inventors of a pending patent on the chemistry and composition of hepatocyte-specific THPO-ASO. L.M.C. is a paid consultant for Cell Signaling Technologies; receives reagent and/or research support from Plexxikon, Inc, Pharmacyclics, Inc, Decipereha Pharmaceuticals, LLC, Genentech, Inc, Roche Glycart AG, and NanoString Technologies; and is a member of the Scientific Advisory Boards of Syndax Pharmaceuticals, Inc, Carisma Therapeutics, Zymeworks, Inc, and Verseau Therapeutics, Inc. The remaining authors declare no competing financial interests.
Correspondence: András Gruber, Departments of Biomedical Engineering and Medicine, School of Medicine, Oregon Health & Science University, 3303 SW Bond Ave, Portland, OR 97239; e-mail: grubera@ohsu.edu and andras.gruber@aronorabio.com.

