

Key Points
Mouse megakaryocytes can differentially sort and package endocytosed fibrinogen and endostatin into distinct α-granules.
Platelet progenitors contain subpopulations of α-granules.

Abstract
In addition to their primary roles in hemostasis and thrombosis, platelets participate in many other physiological and pathological processes, including, but not limited to inflammation, wound healing, tumor metastasis, and angiogenesis. Among their most interesting properties is the large number of bioactive proteins stored in their α-granules, the major storage granule of platelets. We previously showed that platelets differentially package pro- and antiangiogenic proteins in distinct α-granules that undergo differential release upon platelet activation. Nevertheless, how megakaryocytes achieve differential packaging is not fully understood. In this study, we use a mouse megakaryocyte culture system and endocytosis assay to establish when and where differential packaging occurs during platelet production. Live cell microscopy of primary mouse megakaryocytes incubated with fluorescently conjugated fibrinogen and endostatin showed differential endocytosis and packaging of the labeled proteins into distinct α-granule subpopulations. Super-resolution microscopy of mouse proplatelets and human whole-blood platelet α-granules simultaneously probed for 2 different membrane proteins (VAMP-3 and VAMP-8), and multiple granular content proteins (bFGF, ENDO, TSP, VEGF) confirmed differential packaging of protein contents into α-granules. These data suggest that megakaryocytes differentially sort and package α-granule contents, which are preserved as α-granule subpopulations during proplatelet extension and platelet production.
Introduction
Platelets regulate hemostasis and contribute to inflammation, malignancy, infection, and wound healing.1,2 Although these diverse physiological processes depend upon the targeted release by platelets of chemokines, coagulation factors, and angiogenic factors from α-granules, the packaging and sorting of these key platelet α-granule proteins remain poorly understood.
Megakaryocytes (MKs) produce platelets by extending long, branched structures called proplatelets that sequentially release platelets from their ends. Proplatelet shafts are lined with microtubule tracks that function as assembly lines for platelet production by transporting α-granules from the MK body into the assembling platelet.3 A single platelet contains 40 to 80 α-granules. Proteins are either packaged into platelet α-granules by MKs within the multivesicular body or via selective endocytosis of plasma proteins by platelets while in circulation.4
We have previously shown that the proangiogenic proteins, fibrinogen and vascular endothelial growth factor (VEGF), and antiangiogenic proteins endostatin (ENDO) and von Willebrand factor are organized into separate α-granules in both mouse and human platelets.5-8 Recently, multicolor fluorescence nanoscopy of pro- and antiangiogenic proteins VEGF and platelet factor 4 revealed only minor overlap in staining, further supporting separate α-granule protein storage.9 In addition, Peters and colleagues have identified distinct vesicle-associated membrane proteins (VAMPs) that define separate α-granule subpopulations in platelets.10,11 We and others have also demonstrated that distinct granule contents can be released differentially in response to individual platelet agonists.12,13 The essential role of VAMP-3 in endocytosis was recently demonstrated by Banerjee and colleagues, who established that endocytosis in platelets is dependent on VAMP-3.14
However, the idea of segregated α-granule protein storage is not without controversy. Sehgel and Kamykowski have demonstrated that platelets contain morphologically homogenous α-granules,7,15 wherein proteins are segregated in restricted zones within this homogeneous α-granule population. van Nispen and colleagues have suggested that platelets contain multiple α-granules that are morphologically heterogenous, wherein the diversity of granule content is derived from different structural organization of the α-granules themselves.16 Jonnalagadda and colleagues have proposed that the distribution and release of the protein cargo are random and that granule contents are not differentially packaged or released.17
Although these studies have provided new concepts in platelet granule biology, experimental shortcomings include a predominant focus on granule organization exclusively in platelets, analysis mostly limited to cargo proteins, and the use of a single technique, antibody staining, to interrogate localization. In this article, we build on this body of work with the intent to further refine how MKs organize their granular content for distribution into newly formed platelets. Using super-resolution microscopy and live cell imaging combined with an endocytosis assay, we demonstrate that mouse MKs heterogeneously sort and package α-granule contents into multiple distinct α-granule subpopulations and reveal differential α-granule membrane protein composition, content, and concentration that are preserved during mouse proplatelet extension and platelet production, and in circulating human blood platelets.
Methods
Platelet purification/activation
Blood collection was performed in accordance with the Declaration of Helsinki and ethics regulations with institutional review board/Institutional Animal Care and Use Committee approval. Platelets were isolated from healthy volunteers or mice as described.12 All human platelet experiments were performed in triplicate.
MK cultures
Murine fetal livers were collected from wild-type CD-1 mice (Charles River Laboratories) on embryonic day 13.5 and cultured in the presence of supernatant containing 70 ng/mL recombinant mouse thrombopoietin. Mature MKs were isolated and cultured for 8 hours to allow for proplatelet formation, as described.18,19 All MK experiments were performed in triplicate.
Fixed-cell immunofluorescence microscopy
Mouse proplatelets and resting human platelets were fixed for 15 minutes using 4% formaldehyde and spun onto poly-l-lysine–coated coverslips at 1000g for 5 minutes. Specimens were permeabilized with 0.5% Triton X-100 in Hank’s balanced salt solution and then washed with Hank’s balanced salt solution and phosphate-buffered saline (PBS) for 5 minutes each. Specimens were blocked overnight in immunofluorescence blocking buffer (0.5 g bovine serum albumin [BSA], 0.25 mL 10% sodium azide, and 5 mL fetal calf serum in 50 mL 1× PBS). Specimens were then sequentially incubated in primary antibody for 1 hour, washed, treated with appropriate secondary antibody for 1 hour, and then washed again for each primary-secondary pairing. The following primary antibodies were used at the indicated dilutions: mouse anti-VEGF (MS-1467-PABX, Neomarkers, 1:100), mouse antithrombospondin (TSP; MS-421-P, Neomarkers, 1:100), goat anti–basic fibroblast growth factorAF-233-NA, R&D Systems, 1:100), and rabbit anti-ENDO antibodies (ab3453, Abcam, 1:200). Alexa 488 anti-rabbit, Alexa 568 anti-mouse, and Alexa 405 anti-goat secondary antibodies were purchased from Molecular Probes (Grand Island, NY) and used at a concentration of 1:500. To avoid cross-reactivity between mouse antibodies, the mouse anti-VEGF primary antibody was directly conjugated to Alexa 647 using an Alexa 647 antibody labeling kit (Thermo Fisher Scientific, Waltham, MA), Coverslips were mounted onto glass microscope slides using Aqua-Poly/Mount (Polysciences, Inc, Warrington, PA). Controls were processed identically except for omission of the primary antibody.
For MK studies, mature MKs were mixed with hirudin from leeches (Sigma, St. Louis, MO) at 150 units/mL. MKs were incubated for 2 hours with Alexa 488-fibrinogen (Thermo Fisher Scientific) and Alexa 568-endostatin (Ortho Clinical Diagnostics, Waltham, MA) or Alexa 488-fibrinogen and Alexa 546-fibrinogen (Life Technologies). Fibrinogen was used at 15 μg/mL dilution, whereas ENDO was used at 1:20 000. MKs were washed and fixed 2, 4, and 24 hours after initial addition of fluorescent protein. Samples were spun at 300g (Thermo Fisher Scientific Cytospin 4; Thermo Fisher Scientific) onto poly-l-lysine–coated coverslips, washed with PBS, and mounted onto microscope slides.
Live-cell fluorescence microscopy
Round MKs were incubated overnight with fibrinogen and ENDO as described above. Cells were pipetted onto the center of BSA-coated glass-bottom Petri dishes and overlaid with a sheet of low melting point agarose.20 Pictures were taken on a Zeiss Axiovert 200M microscope (Carl Zeiss, Thornwood, NY) equipped with a 10× (numerical aperture, 0.30) Plan-Fluoro objective using a Hamamatsu charged coupled device (Hamamatsu Photonics, Boston, MA). Differential interference contrast and fluorescent pictures were taken every 5 minutes for 1 hour. Cells were kept at 37°C and 5% CO2 throughout the experiment. Images were analyzed using Metamorph version 7.7.2.0 image analysis software (Molecular Devices, Sunnyvale, CA) and ImageJ software 1.47v (NIH; http://rsb.info.nih.gov.ezp-prod1.hul.harvard.edu/ij/).
Super-resolution microscopy
Mouse proplatelets from primary MK cultures, and resting human blood platelets were isolated, fixed, and probed as previously described.17,18 Images were taken on a Zeiss Elrya microscope (Carl Zeiss) equipped with an α Plan-Apochromat 100×/1.46 Oil differential interference contrast M27 objective. Electronic shutters and image acquisition were under the control of ZEN 2012 Black software (Carl Zeiss). The microscope was calibrated preacquisition using 0.2-µm TetraSpeck microspheres (Molecular Probes). Super-resolution structured illumination images were generated using ZEN 2012 Black and further processed using ImageJ software version 1.47v, using the JACoP (https://imagej.nih.gov/ij/plugins/track/jacop.html) and Coloc2 plugins (https://imagej.net/Coloc_2).
Spinning-disk confocal microscopy
To evaluate fibrinogen localization relative to endosomal markers, mature mouse MKs treated with fibrinogen were fixed at 24 hours as described above. Coverslips were then stained with either anti-rab5 (Cell Signaling, Danvers, MA; C8B1; 1:200) or anti-rab7 (Cell Signaling; D95F2; 1:100) antibodies and imaged at the Nikon Imaging Center at Harvard Medical School using a Nikon-Ti spinning-disk confocal microscope. Images were analyzed using Metamorph version 7.7.2.0 image analysis software (Molecular Devices) and ImageJ software 1.52g.
Immunogold electron microscopy
For preparation of cryosections, isolated human platelets were fixed with 4% paraformaldehyde in 0.1 M PBS, pH 7.4. After 2 hours of fixation at room temperature, the cell pellets were washed with PBS containing 0.2 M glycine to quench free aldehyde groups from the fixative. Before freezing in liquid nitrogen, cell pellets were infiltrated with 2.3 M sucrose in PBS for 15 minutes. Frozen samples were sectioned at −20°C, and the sections were transferred to formvar-carbon–coated copper grids and floated on PBS until the immunogold labeling was carried out. The gold labeling was carried out at room temperature on a piece of parafilm. All antibodies and protein A–gold were diluted with 1% BSA. The diluted antibody solution was centrifuged for 1 minute at 5000g before labeling to avoid possible aggregates. All antibodies were used at a concentration of 1 μg/mL. Grids were floated on drops of 1% BSA for 10 minutes to block for nonspecific labeling, transferred to 5-μL drops of primary antibody, and incubated for 30 minutes. The grids were then washed in 4 drops of PBS for a total of 15 minutes, transferred to 5 L drops of protein A–gold for 20 minutes, and washed in 4 drops of PBS for 15 minutes and 6 drops of double distilled water. For double labeling, after the first protein A–gold incubation, grids were washed in 4 drops of PBS for a total of 15 minutes and then transferred to a drop of 1% glutaraldehyde in PBS for 5 minutes and washed in 4 drops of PBS/0.15 M glycine. The second primary antibody was then applied, followed by PBS washing and treatment with different size protein A–gold as above. Contrasting/embedding of the labeled grids was carried out on ice in 0.3% uranyl acetate in 2% methyl cellulose for 10 minutes. Grids were picked up with metal loops, leaving a thin coat of methyl cellulose. The grids were examined in a Tecnai G2 Spirit BioTWIN transmission electron microscope (Hillsboro, OR) at 30 000 magnification at an accelerating voltage of 80 kV. Images were recorded with an AMT 2k CCD camera.
Results and discussion
Mouse MKs differentially sort and package α-granule content
We have previously shown that the proangiogenic protein fibrinogen and antiangiogenic protein ENDO are organized into separate α-granules in platelets.5-8 To determine whether this organization is initiated within the MK itself, we performed structured illumination super-resolution microscopy of primary mouse MKs that were simultaneously incubated with Alexa 488-fibrinogen and Alexa 568-endostatin (15 µg/mL, protein control) or Alexa 488-fibrinogen and Alexa 546-fibrinogen (15 µg/mL, fluorophore control) overnight. Maximum intensity projections and Imaris 3D spot reconstruction of fluorescent puncta 24 hours after incubation revealed that fibrinogen and ENDO are differentially sorted and packaged by MKs following endocytosis (Figure 1A; supplemental Figures 1 and 2; supplemental videos 1-3). A 2-tailed Student t test was performed, with P = .0498 when comparing variances through an F test (supplemental Figure 1D).
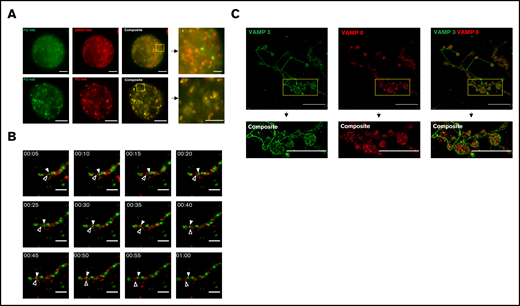
Live cell fluorescence and super-resolution structured illumination microscopy reveal distinct subpopulations of α-granules and VAMPs, in mature mouse MKs. Mouse fetal liver derived MKs were cultured overnight with Alexa 488-fibrinogen (FG488) and Alexa 568-endostatin (ENDO568) or Alexa 488-fibrinogen and Alexa 546-fibrinogen (FG546). (A) Structured illumination microscopy of MKs containing fluorescently tagged ENDO and fibrinogen. The yellow boxes, and their corresponding magnified insets, highlight separation of FG488/ENDO568 in the upper panels and colocalization of FG488/FG546 in the lower panels. Images are maximum intensity projections. For FG488/ENDO568, scale bar is 5 µm for full images, and 2 µm for the inset. For FG488/FG546, scale bar is 10 µm for full images, and 5 µm for inset. (B) Time lapse imaging of a proplatelet containing FG488 and ENDO568. White arrowhead denotes fibrinogen-containing granule; open arrowhead denotes ENDO-containing granule. Images were taken every 5 minutes for 1 hour. Scale bar is 5 µm. (C) Structured illumination microscopy of VAMP-3 (green) and VAMP-8 (red). Note that the proplatelet shown in the insets are circular in structure. Images are maximum intensity projections. Scale bar is 25 µm for full images, and 8 µm for insets. For VAMP-3 and VAMP-8 costaining, the Pearson’s R value is 0.21 ± 0.02. Three proplatelet producing MKs were analyzed.
Live cell fluorescence and super-resolution structured illumination microscopy reveal distinct subpopulations of α-granules and VAMPs, in mature mouse MKs. Mouse fetal liver derived MKs were cultured overnight with Alexa 488-fibrinogen (FG488) and Alexa 568-endostatin (ENDO568) or Alexa 488-fibrinogen and Alexa 546-fibrinogen (FG546). (A) Structured illumination microscopy of MKs containing fluorescently tagged ENDO and fibrinogen. The yellow boxes, and their corresponding magnified insets, highlight separation of FG488/ENDO568 in the upper panels and colocalization of FG488/FG546 in the lower panels. Images are maximum intensity projections. For FG488/ENDO568, scale bar is 5 µm for full images, and 2 µm for the inset. For FG488/FG546, scale bar is 10 µm for full images, and 5 µm for inset. (B) Time lapse imaging of a proplatelet containing FG488 and ENDO568. White arrowhead denotes fibrinogen-containing granule; open arrowhead denotes ENDO-containing granule. Images were taken every 5 minutes for 1 hour. Scale bar is 5 µm. (C) Structured illumination microscopy of VAMP-3 (green) and VAMP-8 (red). Note that the proplatelet shown in the insets are circular in structure. Images are maximum intensity projections. Scale bar is 25 µm for full images, and 8 µm for insets. For VAMP-3 and VAMP-8 costaining, the Pearson’s R value is 0.21 ± 0.02. Three proplatelet producing MKs were analyzed.
To account for endosome labeling during fibrinogen uptake, we stained samples with antibodies against early and late endosomal markers, Rab 5 and 7, respectively, 24 hours postincubation. Spinning-disk confocal microscopy revealed little colocalization, indicating a lack of fibrinogen in endosomes at time of fixation (supplemental Figure 3).
α-Granule subpopulations are preserved during mouse proplatelet extension and platelet production
MKs give rise to platelets through a process of maturation and cytoplasmic changes resulting in long cytoplasmic extensions termed proplatelets. MKs direct α-granules along microtubule tracks within proplatelet shafts into nascent platelets. To determine if α-granule subpopulations are preserved during platelet production, α-granules were visualized by live-cell fluorescence microscopy in proplatelets from primary mouse MKs that had been coincubated with Alexa 488-fibrinogen and Alexa 568-endostatin, as described above. Figure 1B (supplemental video 4) shows distinct α-granule subpopulations moving independent of each other along the proplatelet shaft in a series of time-lapse fluorescence images. The white arrowhead denotes a fibrinogen-containing granule, whereas the black arrowhead denotes an ENDO-containing granule.
VAMP3 and VAMP8 are associated with α-granules.21,22 To establish the presence of multiple distinct α-granule subpopulations during proplatelet extension and platelet production, proplatelet-producing MKs were fixed, permeabilized, and probed for the α-granule membrane proteins VAMP-3 and VAMP-8 (Figure 1C). Pearson’s R values showed high levels of segregation between the membrane proteins, supported by a 1-sample Student t test with P = .0019. In interpreting these data, however, it is important to consider that VAMP-3 also is present on endosomes.14,23 Thus, lack of colocalization between VAMP-3 and VAMP-8 may reflect separation of endosomes from α-granules in addition to or instead of separation of α-granule populations.
Identification of distinct populations of α-granules in mouse proplatelet shafts and human platelets using super-resolution structured illumination microscopy
In the mouse proplatelet shaft, α-granule content proteins were labeled with fibroblast growth factor-basic (bFGF), ENDO, VEGF, and TSP and imaged with structured illumination microscopy (Figure 2A-C; supplemental Figure 4A).11 A 1-way analysis of variance was performed, with P = .005, although multiple comparisons through the Friedman test yielded nonsignificant P values. Although some of these costains showed colocalization, ENDO probed with either TSP or VEGF showed less overlap, indicating the potential for differential sorting of these proteins.
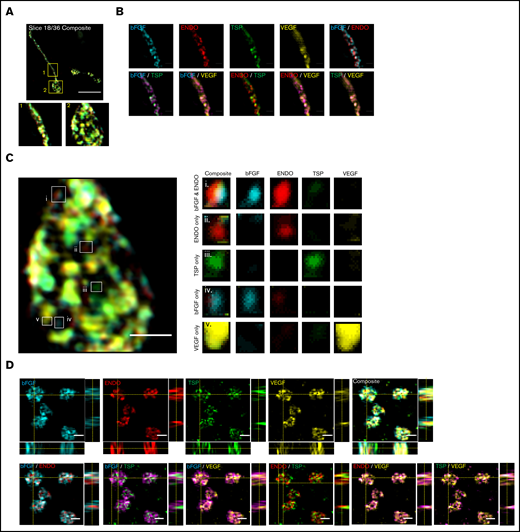
Super-resolution structured illumination and immunogold electron microscopy reveals distinct α-granule subpopulations in mouse proplatelets and human platelets. Mouse fetal liver derived MKs were isolated with a BSA gradient. Released mouse proplatelets were stained with antibodies against bFGF, ENDO, TSP, and VEGF and imaged with structured illumination microscopy. (A) A single slice of a composite stack of a released proplatelet. Scale bar is 10 µm. Magnified images show proplatelet shaft (1) and tip (2). (B) Distribution of α-granule cargo proteins were determined by overlaying different staining combinations. Scale bar is 1 µm. (C) Full image of proplatelet tip (left). White boxes highlight granules of distinct protein cargo, magnified on right. Scale bar is 1 µm. (D) Human platelets were probed with antibodies against bFGF, ENDO, TSP, and VEGF and imaged with structured illumination microscopy. Images are maximum intensity projections. Scale bar is 2 µm. (E) Double immunogold labeling of human platelet sections using anti-ENDO and antifibrinogen primary antibodies followed by staining with protein A–gold conjugated to 10 nm (fibrinogen; red arrows) or 15 nm (ENDO; white arrows). Scale bar is 100 nm.
Super-resolution structured illumination and immunogold electron microscopy reveals distinct α-granule subpopulations in mouse proplatelets and human platelets. Mouse fetal liver derived MKs were isolated with a BSA gradient. Released mouse proplatelets were stained with antibodies against bFGF, ENDO, TSP, and VEGF and imaged with structured illumination microscopy. (A) A single slice of a composite stack of a released proplatelet. Scale bar is 10 µm. Magnified images show proplatelet shaft (1) and tip (2). (B) Distribution of α-granule cargo proteins were determined by overlaying different staining combinations. Scale bar is 1 µm. (C) Full image of proplatelet tip (left). White boxes highlight granules of distinct protein cargo, magnified on right. Scale bar is 1 µm. (D) Human platelets were probed with antibodies against bFGF, ENDO, TSP, and VEGF and imaged with structured illumination microscopy. Images are maximum intensity projections. Scale bar is 2 µm. (E) Double immunogold labeling of human platelet sections using anti-ENDO and antifibrinogen primary antibodies followed by staining with protein A–gold conjugated to 10 nm (fibrinogen; red arrows) or 15 nm (ENDO; white arrows). Scale bar is 100 nm.
To assess the presence of unique α-granule subpopulations in human platelets, we fixed, permeabilized, and simultaneously probed circulating whole-blood platelets for α-granule content proteins bFGF, ENDO, VEGF, and TSP (Figure 2D; supplemental Figures 4B and 5). This revealed some distinct α-granule subpopulations confirming α-granule content organization in circulating blood platelets. A 1-way analysis of variance was performed, with P = .0008. Multiple comparisons through the Friedman test were performed, statistically proving that clear segregation exists between ENDO/TSP and bFGF/TSP, with bFGF/VEGF and TSP/VEGF showing some separation, and ENDO/bFGF as well as ENDO/VEGF also displaying moderate levels of colocalization.
As further evidence of distinct α-granule populations, immunogold electron microscopy indicated that ENDO and fibrinogen are mostly found in separate granules (Figure 2E). Out of 14 granules quantified, 14% were exclusively composed of ENDO, 71% were solely composed of fibrinogen, and 14% contained both.
Conclusion and future work
These findings characterize a new level of heterogeneity within platelet α-granules that is preserved from early stages of platelet production during megakaryopoiesis and proplatelet cytoplasmic extension. This work highlights a need to further understand the physiological relevance of α-granule subpopulations and their organization in MKs and platelets, and how this differential uptake and packaging of protein content is presumably influenced by disease states such as inflammation and malignancy. The mechanism of differential segregation of cargo into α-granules may be explained by the disparate roles of proteins responsible for α-granule biogenesis. There are 3 known proteins the roles of which have been investigated in α-granule biogenesis: VPS33B, VPS16B, and NBEAL2.24-28 Defects in VPS33B and VPS16B cause a complete disruption of α-granule formation; these proteins are thought to sort biosynthesized proteins from the trans-Golgi network to multivesicular bodies. NBEAL2 has been shown to associate with P-selectin to stabilize the α-granule and prevent cargo loss to exocytosis pathways.29 Thus, defects in NBEAL2 result in formation of “ghost granules” or empty α-granules containing P-selectin but lacking other cargo. The different functions of these proteins demonstrate that α-granule biogenesis, and potentially differential packaging, is regulated at multiple stages, including biosynthesis, endocytosis, exocytosis, and transport between various cellular compartments.30 Further characterization of α-granule organization and contents will not only improve our understanding of physiological function of platelets, which is likely more nuanced than originally presumed, but also lead to the development of designer therapies based on directed protein composition of platelet α-granules and their differential release. Our current understanding of α-granule biogenesis is relatively narrow, but a greater understanding of key players of this complex process will contribute to understanding how the MK differentially sorts proteins into these specialized packages.
Acknowledgments
This work was supported by National Institutes of Health (NIH), National Cancer Institute grant 5R01CA200748-02 (E.M.B. and J.E.I.) and NIH award R01H168130 (J.E.I.).
Authorship
Contribution: E.M.B. and J.N.T. designed and carried out experiments, analyzed data, and cowrote the manuscript; R.O. and C.G.P. carried out experiments and analyzed data; P.V., A.R.W., and L.J.N. analyzed data, contributed to design of figures, and contributed to writing the manuscript; and J.E.I. and R.F. contributed to design of experiments, contributed to writing the manuscript, and helped analyze the data.
Conflict-of-interest disclosure: J.N.T. and J.E.I. have financial interest in and are founders of Platelet BioGenesis, a company that aims to produce donor-independent human platelets from human-induced pluripotent stem cells at scale. They are both inventors on this patent. The interests of J.N.T. and J.E.I. were reviewed and are managed by the Brigham and Women’s Hospital and Partners HealthCare in accordance with their conflict-of-interest policies. R.F. has a private equity interest in Platelet Diagnostics, and he is also a founder and consultant for that company. The interests of R.F. are reviewed and managed by the Beth Israel Deaconess Medical Center Office of Compliance and Business Conduct. The remaining authors declare no competing financial interests.
Correspondence: Elisabeth M. Battinelli, Brigham and Women's Hospital, 4 Blackfan Cir, 7th Floor, Harvard Institutes of Medicine, Boston, MA 02115; e-mail: embattinelli@bwh.harvard.edu.

