

Key Points
HLA-A*02:ALNEQIARL derived from NDC80 is a broadly presented cancer immunotherapy target with high abundance in hematologic malignancies.
CAR T cells designed for this target exhibit high sensitivity and specificity with no relevant toxicity toward hematopoietic stem cells.

Abstract
Target identification for chimeric antigen receptor (CAR) T-cell therapies remains challenging due to the limited repertoire of tumor-specific surface proteins. Intracellular proteins presented in the context of cell surface HLA provide a wide pool of potential antigens targetable through T-cell receptor mimic antibodies. Mass spectrometry (MS) of HLA ligands from 8 hematologic and nonhematologic cancer cell lines identified a shared, non-immunogenic, HLA-A*02–restricted ligand (ALNEQIARL) derived from the kinetochore-associated NDC80 gene. CAR T cells directed against the ALNEQIARL:HLA-A*02 complex exhibited high sensitivity and specificity for recognition and killing of multiple cancer types, especially those of hematologic origin, and were efficacious in mouse models against a human leukemia and a solid tumor. In contrast, no toxicities toward resting or activated healthy leukocytes as well as hematopoietic stem cells were observed. This shows how MS can inform the design of broadly reactive therapeutic T-cell receptor mimic CAR T-cell therapies that can target multiple cancer types currently not druggable by small molecules, conventional CAR T cells, T cells, or antibodies.
Introduction
Chimeric antigen receptor (CAR) T cells are immunotherapies approved by the US Food and Drug Administration that have shown remarkable results in patients with hematologic cancers.1,2 However, CAR T-cell products are currently only approved for a small subset of cancer patients because a major limitation in the design of new CAR T-cell candidates is the lack of known cancer-specific cell surface proteins that will provide high sensitivity with limited toxicity to normal tissues.3 Although target identification for CAR T cells against other malignancies seems feasible,4,5 even highly evolved screening strategies cannot always provide single tumor-specific antigens.6 In addition, these target identification strategies are typically designed for a single cancer type, offering few options for a highly desirable, broadly usable, tumor-agnostic approach.
We hypothesized that intracellular proteins that are degraded through the proteasome and presented as peptides on HLA class I molecules can be a valuable source to identify more broadly presented antigens enabling a tumor-agnostic target strategy. Traditionally, such peptide:HLA class I complexes are recognized by T cells through their T-cell receptor (TCR), but as a consequence of thymic selection, self-antigens cannot typically be recognized by TCR T cells.7 In contrast, TCR mimic antibodies offer the unique opportunity to specifically target peptide: HLA complexes in a manner comparable to that of TCRs but independent of the immunogenicity of the presented HLA ligand. Furthermore, TCR mimic antibodies with their respective single-chain variable fragments (scFvs) can be engineered into multiple therapeutic formats, including CAR T cells, which have been proven effective against several targets.8-17
To identify an HLA-A*02–bound peptide present in multiple cancer types, we affinity-purified peptide:HLA complexes from different cancer cell lines with varying HLA-A*02 abundances and performed liquid chromatography–tandem mass spectrometry (LC-MS/MS) to identify the eluted peptide sequences. Network analyses of source proteins from which the HLA ligands were derived, and identification of shared biological functions between the cell lines,18 facilitated the selection of a suitable HLA ligand candidate. This candidate was ALNEQIARL from the “kinetochore NDC80 protein homolog” (referred to as NDC80), an essential and highly expressed protein in tumors, especially within poor prognosis subgroups.19-21
We discovered a TCR mimic scFv (NDC80-clC) that specifically recognized the ALNEQIARL:HLA-A*02 complex and engineered it into a second-generation CAR T-cell format. We describe here the in vitro killing of NDC80-clC CAR T cells against multiple cancer cell lines in a peptide- and HLA-specific manner. Moreover, these CAR T cells suppressed tumor growth and prolonged survival in a hematologic and nonhematologic mouse model while sparing stimulated and unstimulated healthy A*02–positive blood leukocytes, fibroblasts, and cardiomyocytes, as well as hematopoietic stem cells (HSCs) in vitro. Overall, our study shows how non-immunogenic peptide:HLA complexes can be an invaluable source for tumor-specific antigens that are undruggable by conventional therapeutic approaches. The NDC80-clC CAR T cell is a candidate for development into a tumor-agnostic drug capable of targeting highly proliferative HLA-A*02–positive cancer cells such as leukemias or aggressive lymphomas.
Materials and methods
Immunopurification and LC-MS/MS analysis of HLA class I ligands
HLA class I ligands were isolated as described previously.22,23 W6/32 (Bio X Cell, BE0079; RRID: AB_1107730), BB7.2 (MSKCC Antibody Core Facility), or clone C or murine immunoglobulin G1 (mIgG1; Eureka Therapeutics) antibody was used for respective experiments.
scFv clones specific for peptide/HLA-A0201 complexes
A human scFv antibody E-ALPHA phage display library was used for the selection of monoclonal antibody clones specific to ALNEQIARL:HLA-A*02 as described previously.14 In brief, T2 cells pulsed with 50 µg/mL irrelevant control peptides were used to remove any clones that potentially bind to HLA-A*02:01 in the library. Remaining clones were screened for T2 cells pulsed with 50 µg/mL ALNEQIARL peptides. Positive clones were determined by their binding to T2 cells pulsed with ALNEQIARL peptides but not to T2 cells pulsed with control peptides by flow cytometry.
Generation of NDC80-CAR T cells
scFvs were grafted onto a second-generation CAR with CD28 and CD3ζ signaling domains engineered in cis to provide intracellular T-cell stimulation signals. The CAR sequence was cloned into a pCDH lentiviral vector (Systems Biosciences) for delivery into T cells. Human T cells were activated with CD3/CD28 Dynabeads (Thermo Fisher Scientific; 11-161-D). One day after activation, human T cells were transduced with concentrated lentivirus in plates coated with RetroNectin (Takara). Transduced T cells were then expanded in the presence of 100 U/mL interleukin-2 (Sigma) for 8 to 12 days. Transduction efficiency was assessed by direct staining using anti-myc clone 71D10-Al647 (Cell Signaling; #63730).
Animals and in vivo models
Eight- to 10-week-old NOD.Cg-Prkdcscid IL2rgtm1Wjl/SzJ mice (NSG) were purchased from The Jackson Laboratory. Female mice were used for all experiments. For the BV173 model, mice were injected first with 1 million BV173 cells and after 5 days with phosphate-buffered saline, 2 million 4H11 CAR T cells, or 2 million NDC80-clC CAR T cells (8 days after transduction) via tail vein. Starting with day 14, we cheek bled mice and stained the samples for HLA-A*02 as well as murine CD45 to determine the fraction of blast cells in the peripheral blood. For the JMN model, mice were injected intraperitoneally with 300 000 GFP-Luc–transduced JMN cells. Tumor burden was assessed by bioluminescence imaging twice per week before treatment and then after injection of 150 000 NDC80-clC CAR T cells, 4H11 CAR T cells, or phosphate-buffered saline.
Additional and more detailed methods are presented in the supplemental Materials and Methods (available on the Blood Web site).
Results
LC-MS/MS analysis defines the NDC80-derived peptide ALNEQIARL as a highly tumor-associated HLA-A*02 ligand
To discover a tumor-associated HLA-A*02–restricted HLA ligand, we immunopurified HLA complexes from 4 hematologic cancer cell lines (BV173, OCI-AML02, SUDHL6, and MAC2A) and 4 nonhematologic cancer cell lines (JMN, TPC-1, MDA-MB231, and PANC-1) and analyzed the immunopeptidome of the complexes via LC-MS/MS (Figure 1A). We identified >11 000 unique HLA class I–assigned peptides, of which 3289 were considered HLA-A*02:01 binders (supplemental Table 1). Using source proteins from which the HLA ligands were derived (supplemental Table 2), we performed network analyses via the GeneMANIA algorithm through a Cytoscape plugin.24 Of 923 Gene Ontology terms resulting from the analyses of the 8 different cell lines, only 3 processes were shared among all lines: kinetochore, chromosome centromeric region, and protein DNA complex. Finally, from all the proteins involved in these 3 Gene Ontology terms, only peptides from the NDC80 protein were presented in all 8 cell lines. Similarly, three HLA-A*02–restricted HLA ligands from NDC80 were detected at the cell surface (ALNEQIARL, GLNEEIARV, and HLEEQIAKV), but only the ALNEQIARL peptide ligand was shared between all the tested cancer lines. Of note, although the HLA ligand GLNEEIARV also exhibited high presentation frequency (7 of 8 cell lines), we considered it a less suitable candidate because public immunopeptidome data from healthy human tissues showed that the GLNEEIARV ligand was presented in multiple essential healthy tissues, including lung, esophagus, ovary, uterus, cerebellum, and colon.25 In the same study, the ALNEQIARL peptide was only detected in ovary, thymus, and bone marrow, which we considered potentially acceptable off-tumor targets; these reactivities were investigated later in this study.
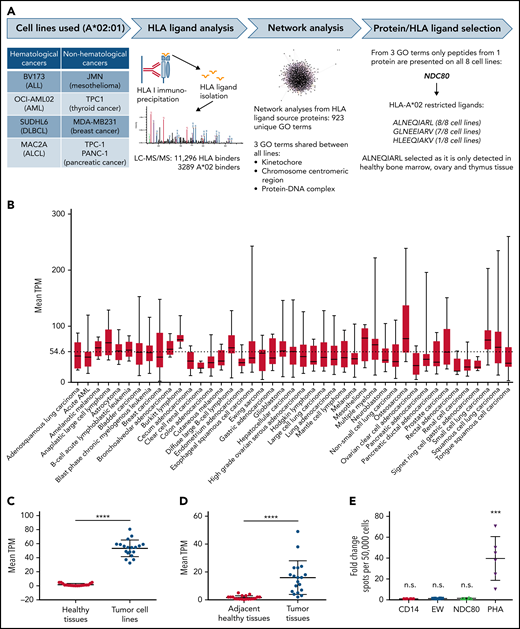
Identification of the NDC80-derived ALNEQIARL peptide as a tumor-associated HLA ligand. (A) Experimental strategy. (B) Mean NDC80 expression levels in different cancer cell lines. Only cancer types with at least 5 data points were considered. Whiskers indicate minimum to maximum. (C) Mean NDC80 expression of healthy tissues and corresponding cancer cell lines. (D) Mean NDC80 expression of adjacent healthy tissues and corresponding primary cancer tissues. (E) ELISpot results from 3 healthy donors and 2 biological replicates per donor. Data were normalized to results from CD14+ cells alone. EW served as control peptide. Error bars in panels C, D, and E denote standard deviation. ***P < .001 (Mann-Whitney test), ****P < .0001 (Wilcoxon matched-pairs signed-rank test). ALCL, anaplastic large cell lymphoma; ALL, acute lymphoblastic leukemia; DLBCL, diffuse large B-cell lymphoma; GO, Gene Ontology. EW, Ewing sarcoma-derived HLA-A0201-binding peptide; EW, QLQNPSYDK; PHA, phytohemagglutinin.
Identification of the NDC80-derived ALNEQIARL peptide as a tumor-associated HLA ligand. (A) Experimental strategy. (B) Mean NDC80 expression levels in different cancer cell lines. Only cancer types with at least 5 data points were considered. Whiskers indicate minimum to maximum. (C) Mean NDC80 expression of healthy tissues and corresponding cancer cell lines. (D) Mean NDC80 expression of adjacent healthy tissues and corresponding primary cancer tissues. (E) ELISpot results from 3 healthy donors and 2 biological replicates per donor. Data were normalized to results from CD14+ cells alone. EW served as control peptide. Error bars in panels C, D, and E denote standard deviation. ***P < .001 (Mann-Whitney test), ****P < .0001 (Wilcoxon matched-pairs signed-rank test). ALCL, anaplastic large cell lymphoma; ALL, acute lymphoblastic leukemia; DLBCL, diffuse large B-cell lymphoma; GO, Gene Ontology. EW, Ewing sarcoma-derived HLA-A0201-binding peptide; EW, QLQNPSYDK; PHA, phytohemagglutinin.
Encouragingly, NDC80 is overexpressed in a plethora of different cancer types, especially within subgroups with a poor prognosis.19-21 To underline its strong tumor association, we retrieved expression data from 934 cancer cell lines from the Cancer Cell Line Encyclopedia26 and calculated mean transcripts per million (TPM) for all cancer types in which ≥5 cell lines were available (Figure 1B). These mean TPM were significantly higher compared with the NDC80 expression levels of the corresponding healthy tissues published by the GTEX consortium27 (Figure 1C; supplemental Figure 1A). However, because cancer cell lines are preselected for high proliferation capacity, we additionally analyzed data from cancer tissues and matched adjacent healthy tissues from the PCAWG (Pan-Cancer Analysis of Whole Genomes) project28 and found a similar, highly significant overexpression of NDC80 (Figure 1D; supplemental Figure 1B). Of note, NDC80 overexpression reached up to 1300-fold in glioblastoma and astrocytoma cell lines vs healthy brain tissue and >100-fold for pancreatic adenocarcinoma vs adjacent healthy tissue (supplemental Figure 1C-D). Nevertheless, the mean TPM of NDC80 in cancer cell lines was significantly higher compared with that of cancer tissues, suggesting an artificial enhancement in the cancer cell lines (supplemental Figure 1E). In line with this broad and high expression of NDC80 in cancer cells, a literature search for MS identification of NDC80 peptides and testing of additional cell lines and primary tissues via MS in our laboratory (>90% positivity rate for A*02–positive cell lines) confirmed the frequent presentation of the ALNEQIARL peptide on the cell surface (Table 1).
MS evidence of HLA-A*02:ALNEQIARL in cell lines and primary tissue samples
| Tumor type | Cell line | Primary tissue | References |
|---|---|---|---|
| Hematologic malignancies | |||
| AML | AML14, OCI-AML02, THP-1 | Yes | 39; authors’ own data for cell lines and primary samples |
| B-ALL | JY, BV173, Nalm6, ALL-3 | Yes | 40-42 |
| Multiple myeloma | U266 | NT | Authors’ own data |
| DLBCL | DB, SUDHL4 | NT | Authors’ own data |
| T-cell lymphoma | MAC2A | NT | Authors’ own data |
| MCL | — | Yes | 43 |
| Nonhematologic malignancies | |||
| Melanoma | A375, SKMEL5 MEWO, MEL624 | Yes | 44-47 |
| Breast cancer | MDA-MB231 | Yes | 48; authors’ own data for cells and primary tissue |
| Colon cancer | — | Yes | 29 |
| Prostate cancer | LnCAP | NT | Authors’ own data |
| Pancreatic cancer | PANC-1 | NT | Authors’ own data |
| Thyroid cancer | TPC-1 | NT | Authors’ own data |
| Mesothelioma | JMN, MESO37 | NT | Authors’ own data |
| Glioblastoma | — | Yes | 49 |
| Tumor type | Cell line | Primary tissue | References |
|---|---|---|---|
| Hematologic malignancies | |||
| AML | AML14, OCI-AML02, THP-1 | Yes | 39; authors’ own data for cell lines and primary samples |
| B-ALL | JY, BV173, Nalm6, ALL-3 | Yes | 40-42 |
| Multiple myeloma | U266 | NT | Authors’ own data |
| DLBCL | DB, SUDHL4 | NT | Authors’ own data |
| T-cell lymphoma | MAC2A | NT | Authors’ own data |
| MCL | — | Yes | 43 |
| Nonhematologic malignancies | |||
| Melanoma | A375, SKMEL5 MEWO, MEL624 | Yes | 44-47 |
| Breast cancer | MDA-MB231 | Yes | 48; authors’ own data for cells and primary tissue |
| Colon cancer | — | Yes | 29 |
| Prostate cancer | LnCAP | NT | Authors’ own data |
| Pancreatic cancer | PANC-1 | NT | Authors’ own data |
| Thyroid cancer | TPC-1 | NT | Authors’ own data |
| Mesothelioma | JMN, MESO37 | NT | Authors’ own data |
| Glioblastoma | — | Yes | 49 |
B-ALL, B-cell acute lymphoblastic leukemia; DLBCL, diffuse large B-cell lymphoma; MCL, myeloid cell leukemia; NT, not tested.
Furthermore, we investigated 12 publicly available pairs of colorectal carcinoma (CRC) and nonmalignant colon (NMC) samples for a quantitative comparison of HLA ligand presentation.29 Using Skyline software (MacCoss Lab Software; version 21.1) and the known retention times from successful MS/MS identifications, we calculated absolute and relative precursor intensities in these paired data sets that were normalized for equal peptide yields before acquisition. However, we note that even for unsuccessful MS/MS identifications, these intensities have been calculated to reflect baseline intensities; we assume in these instances, however, that the peptide is not presented at the surface. Using this approach, we observed significantly higher average intensities in the CRC compared with the NMC samples (supplemental Figure 2A). In 7 of 12 matched samples, the relative intensities between CRC and NMC for our target peptide were 3- to 90-fold higher (supplemental Figure 2B).
Finally, to investigate whether the ALNEQIARL ligand could also be targeted by a human TCR-directed T cell, we tested T-cell reactivity in healthy A*02–positive blood donors. No reactivity was detected in several donors after multiple stimulations in line with previous published results (Figure 1E). Although the cited publication29 described one successful attempt to prime T cells with this peptide, preexisting T-cell responses in 17 cancer patients or 15 healthy donors were not observed. Overall, these data suggest that the human immune system is largely tolerant to ALNEQIARL and that this peptide is a highly tumor-associated HLA ligand, which ideally could be targeted by a TCR mimic–based strategy.
Phage library screen identifies an ALNEQIARL:HLA-A*02–reactive clone
We used an established phage library screening platform (E-ALPHA Phage Display, Eureka Therapeutics; diversity, 1 × 1011clones) comprising both naive and semisynthetic human scFv B-cell antibodies to select clones reactive with the ALNEQIARL:HLA-A*02 complex (Figure 2A). First, the library was screened for clones that selectively bound T2 cells pulsed with the ALNEQIARL peptide but not T2 cells pulsed with a mixture of 20 irrelevant endogenous HLA-A2 peptides derived from various disease-related and housekeeping proteins. After 3 rounds of panning to ALNEQIARL:HLA-A*02 complex, 347 positive clones were identified by using flow cytometry, which correspond to 60 unique clones by sequence. These 60 unique clones were further screened for selectively binding to T2 cells pulsed with the ALNEQIARL peptide but not T2 cells pulsed with 4 homologous peptides (ALNEKLVNL, ALNELLQHV, MLANDIARL, and TLADIIARL) from widely expressed proteins (EIF3F, TLN1, EHD1, and NYNR1), which have been identified in our MS experiments as well as multiple healthy tissues.25 Using this approach, we aimed to avoid cross-reactivity with these peptides and to ensure binding of the scFvs to central amino acid positions #3 to #8. Ultimately, 6 clones (ie, clones A-F) (supplemental Figure 3A) were selected based on these binding data and engineered into lentiviral second-generation CAR T-cell constructs.
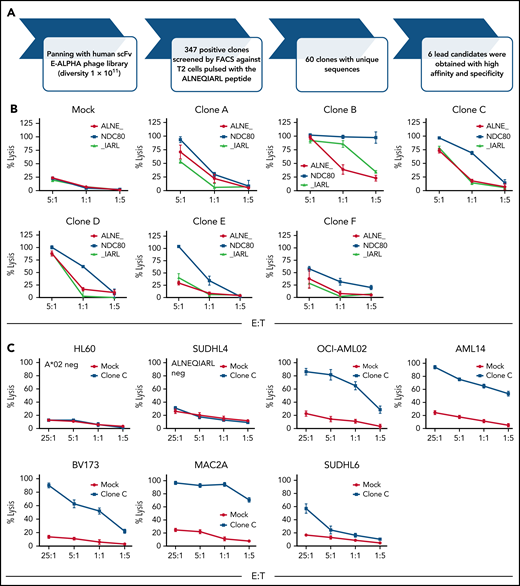
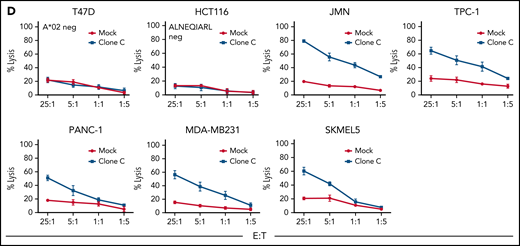
Phage library screen discovers an ALNEQIARL-specific scFv with efficacy against various cancer cell lines in CAR T-cell format. (A) Phage library screen procedure. (B) Characterization of six NDC80 CAR T-cell clones (ie, A-F) against peptide pulsed T2 cells. (C) Efficacy and specificity of NDC80-clC CAR T cells against hematologic cancer cell lines. (D) Efficacy and specificity of NDC80-clC CAR T cells against nonhematologic cancer cell lines. Error bars in panels B through D denote standard deviation. FACS, fluorescence-activated cell sorter; E:T, effector-to-target ratio.
Phage library screen discovers an ALNEQIARL-specific scFv with efficacy against various cancer cell lines in CAR T-cell format. (A) Phage library screen procedure. (B) Characterization of six NDC80 CAR T-cell clones (ie, A-F) against peptide pulsed T2 cells. (C) Efficacy and specificity of NDC80-clC CAR T cells against hematologic cancer cell lines. (D) Efficacy and specificity of NDC80-clC CAR T cells against nonhematologic cancer cell lines. Error bars in panels B through D denote standard deviation. FACS, fluorescence-activated cell sorter; E:T, effector-to-target ratio.
CAR T cells were generated with high efficiency for clones A to F (supplemental Figure 3B). Then, killing against T2 cells pulsed with either the target peptide ALNEQIARL or control peptides ALNEKLVNL and MLANDIARL was assessed (Figure 2B). Clones A, B, and D were sorted out due to cross-reactivity toward either the ALNEKLVNL peptide (clones A and D) or the MLANDIARL peptide (clone B). Although also showing some reactivity to the ALNEKLVNL peptide, clone F was not considered further due to lower reactivity toward the target peptide. These decisions were mostly made based on the data points with an effector-to-target ratio of 1:1 and the areas under the curve, as the 5:1 setting generally led to substantial killing in most of the clone/peptide combinations. Therefore, clones C and E proceeded to additional evaluation.
We selected 7 cell lines each from hematologic and non-hematologic origin and HLA type based on MS data and HLA type (Table 2). Of note, 5 of the cell lines in each group were ALNEQIARL:HLA-A*02 positive, one in each group was HLA-A*02 positive but negative by MS for ALNEQIARL (SUDHL4 and HCT116), and one cell line each was negative for both HLA-A*02 and the target peptide (HL60 and T47D). Strikingly, clone C (NDC80-clC) CAR T cells were able to kill all cell lines in a dose-dependent and target-specific manner (Figure 2C-D). In contrast, clone E (NDC80-clE), although also showing the same high specificity, exhibited much lower efficacy compared with NDC80-clC (supplemental Figure 4A-B). Both clones exhibited more effective killing against cell lines of hematologic origin, probably due to higher antigen density of HLA-A*02 molecules on the cell surface (supplemental Figure 4C). This hypothesis was also supported by the observation that after engineering scFv clone C into a murine IgG1 format, binding against cancer cell lines by flow cytometry was only observed for cell lines of very high antigen density (eg, BV173, AML14, or MAC2A) (supplemental Figure 4D) but not for nonhematologic cell lines. IgG binding to some cell lines allowed an estimate of target complexes on a per-cell level using quantitation beads: 503 sites per cell on MAC2A cells, 876 sites per cell on AML14 cells, and 1356 sites on BV173 cells. Taken together, these data illustrated high sensitivity of the NDC80-clC CAR T cells toward multiple cancer cell lines independent of their cancer type. However, as we showed earlier, primary tissues have, on average, lower levels of NDC80; we thus now wanted to test the ability of NDC80-clC CAR T cells to eliminate patient-derived HLA-A*02–positive or –negative but CD33-positive (PDX) acute myeloid leukemia (AML) cells in vitro (supplemental Figure 5A). Target cells were labeled with carboxyfluorescein diacetate succinimidyl ester (CFSE) and after 24 hours of coincubation with either MOCK-transduced T cells or NDC80-clC CAR T cells at an effector-to-target ratio of 5:1, killing was assessed by using flow cytometry. Again, HLA-A*02–positive PDX tumor cells were efficiently eliminated in contrast to HLA-A*02–negative tumor cells (supplemental Figure 5B).
Cell line killing based on MS evidence of HLA-A*02:ALNEQIARL
| Cell line | HLA-A*02/MS | NDC80-clC killing | NDC80-clE killing |
|---|---|---|---|
| Hematologic malignancies | |||
| AML14 | +/+ | +++ | + |
| OCI-AML02 | +/+ | +++ | + |
| BV173 | +/+ | +++ | + |
| SUDHL6 | +/+ | + | − |
| MAC2A | +/+ | +++ | + |
| SUDHL4 | +/− | − | − |
| HL60 | −/− | − | − |
| Nonhematologic malignancies | |||
| JMN | +/+ | +++ | + |
| TPC-1 | +/+ | ++ | − |
| MDA-MB231 | +/+ | ++ | − |
| PANC-1 | +/+ | + | − |
| SKMEL-5 | +/+ | + | − |
| HCT116 | +/− | − | − |
| T47D | −/− | − | − |
| Cell line | HLA-A*02/MS | NDC80-clC killing | NDC80-clE killing |
|---|---|---|---|
| Hematologic malignancies | |||
| AML14 | +/+ | +++ | + |
| OCI-AML02 | +/+ | +++ | + |
| BV173 | +/+ | +++ | + |
| SUDHL6 | +/+ | + | − |
| MAC2A | +/+ | +++ | + |
| SUDHL4 | +/− | − | − |
| HL60 | −/− | − | − |
| Nonhematologic malignancies | |||
| JMN | +/+ | +++ | + |
| TPC-1 | +/+ | ++ | − |
| MDA-MB231 | +/+ | ++ | − |
| PANC-1 | +/+ | + | − |
| SKMEL-5 | +/+ | + | − |
| HCT116 | +/− | − | − |
| T47D | −/− | − | − |
NDC80-clC CAR T cells exhibit high target specificity
Although we performed a strict screening process, there was a risk that NDC80-clC CAR T cells could still mediate additional off-target binding. To further investigate the specificity of our NDC80-clC CAR T cell, we first characterized its binding characteristics through an alanine screening assay on peptide-pulsed T2 cells (Figure 3A). These peptides were also tested for T2 stabilization, which was determined by HLA-A*02 staining. As expected, alanine substitutions at the anchor positions 2 and 9 of the peptide only allowed moderate stabilization of the HLA-A*02 complexes on T2 cells, although all other peptides stabilized the A*02 protein to similar levels as the unmodified peptide (supplemental Figure 6A). The murine IgG1 (mIgG1) antibody derived from clone C exhibited binding to the center of the peptide, as peptides substituted at positions 3, 4, and 5 decreased binding ∼50%. In addition, modifications at positions 6, 7, and 8 abrogated binding completely. For clone E, antibody binding was similarly impaired by the substitutions, although position 7 seemed to be irrelevant for antibody binding (supplemental Figure 6B).
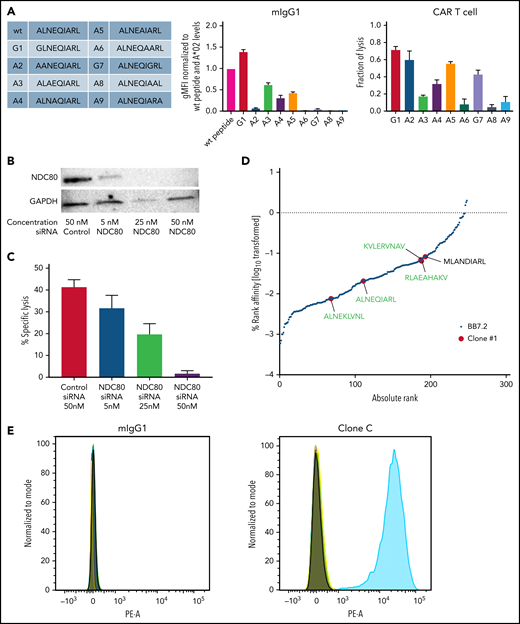
Clone C CAR T cells recognize the ALNEQIARL peptide with high specificity. (A) Alanine screen: peptides used for T2 pulsing (left), mIgG1 binding (middle), and CAR T-cell killing (right). (B) Western blot of NDC80 knockdown using small interfering RNA (siRNA). (C) CAR T-cell killing against JMN NDC80 knockdown cells corresponding to panel B. (D) HLA ligands identified via MS after immunoprecipitation with either HLA-A*02–specific BB7.2 antibody (black) or mIgG1 clone C (red). Peptides are ranked by predicted binding to HLA-A*02 using netMHCpan 4.0 in EL mode. Peptides in green were identified in 2/2 MS runs; peptide in black was identified in 1/2 runs. (E) Flow cytometry of clone C or mIgG1 isotype binding of T2 cells pulsed with either the ALNEQIARL peptide or the potential off-targets from panel D. Two NDC80-derived peptides (GLNEEIARV and HLEEQIAKV) were used as internal negative controls. Error bars denote standard deviation in panels A and C. GAPDH, glyceraldehyde-3-phosphate dehydrogenase; gMFI, geometric mean fluorescence intensity. PE-A, phycoerythrin.
Clone C CAR T cells recognize the ALNEQIARL peptide with high specificity. (A) Alanine screen: peptides used for T2 pulsing (left), mIgG1 binding (middle), and CAR T-cell killing (right). (B) Western blot of NDC80 knockdown using small interfering RNA (siRNA). (C) CAR T-cell killing against JMN NDC80 knockdown cells corresponding to panel B. (D) HLA ligands identified via MS after immunoprecipitation with either HLA-A*02–specific BB7.2 antibody (black) or mIgG1 clone C (red). Peptides are ranked by predicted binding to HLA-A*02 using netMHCpan 4.0 in EL mode. Peptides in green were identified in 2/2 MS runs; peptide in black was identified in 1/2 runs. (E) Flow cytometry of clone C or mIgG1 isotype binding of T2 cells pulsed with either the ALNEQIARL peptide or the potential off-targets from panel D. Two NDC80-derived peptides (GLNEEIARV and HLEEQIAKV) were used as internal negative controls. Error bars denote standard deviation in panels A and C. GAPDH, glyceraldehyde-3-phosphate dehydrogenase; gMFI, geometric mean fluorescence intensity. PE-A, phycoerythrin.
To highlight the functional effects, we repeated alanine screens using CAR T cells of the respective clones and assessed killing by using a lactate dehydrogenase assay. NDC80-clE CAR T cells exhibited an almost identical pattern to the antibody binding, with no relevant effect from modifications at position 7 but clearly centralized engagement of the CAR T cells with the ALNEQIARL:HLA-A*02 complex (supplemental Figure 6B). For NDC80-clC CAR T cells, the killing assay also confirmed the importance of positions 3 to 8 for CAR T-cell binding and killing, as all peptide modifications at these sites led to a decrease in killing compared with the unmodified peptide sequence (Figure 3A). Furthermore, we also observed no relevant cross-reactivity of the mIgG1 antibodies with T2 cells pulsed with the target ALNEQIARL or the potential off-target peptides ALNEKLVNL or MLANDIARL (supplemental Figure 6C).
Target sequence specificity was further shown by NDC80 knockdown experiments. Because NDC80 plays an essential role in chromosome segregation and cell division, stable knockout cell lines cannot be maintained. Therefore, we turned to small interfering RNA–based knockdown experiments as they have been described successfully for NDC80.30 After validating successful knockdown of NDC80 in JMN cells in a dose-dependent manner (Figure 3B), we showed complete abrogation of JMN cell line killing at the highest small interfering RNA dose (Figure 3C). Importantly, we know from JMN cell MS experiments that peptides with potential for off-target binding by NDC80-clC CAR T cells (ALNEKLVNL, ALNELLQHV, ALNEEAGRLLL, YLDEYIARM, and LLFEGIARI) were also presented at the cell surface of these target cells but did not mediate killing after knockdown of NDC80. These specificity data were corroborated by immunoprecipitation combined with MS. We prepared a lysate of 3 × 107 BV173 cells, divided it into 3 equal parts, and performed pulldowns with either nonspecific mIgG1 control antibody, NDC80 clone C antibody, or the HLA-A*02–specific BB7.2 antibody. LC-MS/MS analysis of these samples yielded ∼300 HLA-A*02–specific HLA ligands, of which 1 peptide (ILMEHIHKL) was found in the IgG control sample and 5 peptides in the NDC80-specific pulldown. Strikingly, these 5 peptides contained the NDC80-derived ALNEQIARL as well as 4 other HLA ligands with homologous amino acid sequences (ALNEKLVNL, KVLERVNAV, RLAEAHAKV, and MLANDIARL) (Figures 3D; supplemental Figure 6D). However, the risk of these 4 potential off-targets being functionally relevant seems limited as the signal intensity for the off-targets compared with ALNEQIARL was at least 3-fold lower in the MS experiment. After pulsing T2 cells with these 5 peptides (ALNEQIARL plus 4 off-targets) and the other NDC80-derived epitopes (GLNEEIARV and HLEEQIAKV) as controls, binding was only observed for T2 cells pulsed with the ALNEQIARL peptide (Figure 3E).
Overall, these data suggest that NDC80-clC CAR T cells and the clone C mIgG1 bind the ALNEQIARL peptide in the context of HLA-A*02 with very high specificity. No evidence for a functionally or physiologically relevant off-target was detected.
NDC80-clC CAR T cells and clone C antibody reactivity with healthy human cells
Because our new cellular therapy showed encouraging sensitivity and specificity, we next evaluated risks for on-target off-tumor toxicity to different healthy human cells. First, because leukocytes express the highest levels of HLA class I within the different cell types in the body,31 we tested if these cells would stain positive with the mIgG1 clone C antibody, as we have seen positivity for several cancer cell lines. For all tested subsets (T cells [CD3+], B cells [CD19+], and myelomonocytic cells [CD33+ and CD15+]), no positive staining over the IgG background was observed (Figure 4A). However, because CAR T cells were the preferred therapeutic option for our model, and because we have seen discrepancies between antibody binding and killing, we investigated the potential of the NDC80-clC CAR T cell to kill healthy A*02–positive peripheral blood mononuclear cells. Mitogen-stimulated T and B cells were also included in this assay as their increased proliferation could lead to greater expression of NDC80 as well as processing and presentation of the ALNEQIARL HLA ligand. Little to no killing of sorted hematopoietic cells was observed (Figure 4B). Alternatively, we investigated killing of peripheral blood mononuclear cells via flow cytometry with CFSE-labeled cells without prior sorting and also observed minimal killing; in this assay, however, proliferation effects of different cell populations could have biased the results (supplemental Figure 7).
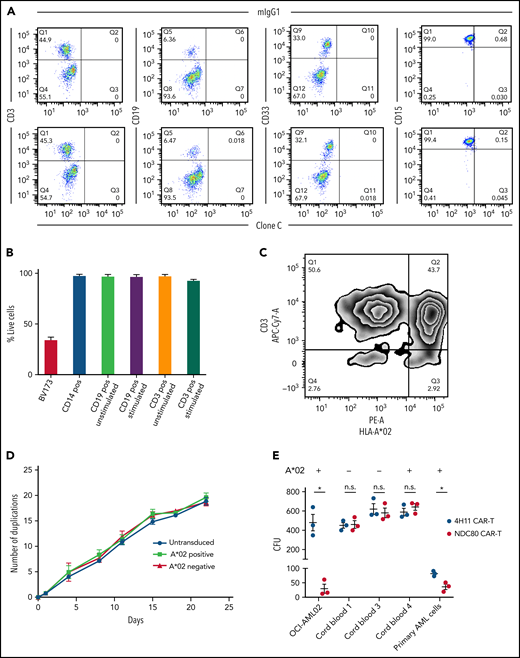
Clone C CAR T cells do not mediate toxicity toward healthy leukocytes, activated lymphocytes, or HSCs. (A) Flow cytometry of clone C or mIgG1 isotype binding of healthy A*02–positive CD3, CD19, CD33, and CD15 positive cells. Data are representative of 3 donors. (B) Lactate dehydrogenase–killing assay with clone C CAR T cells and magnetic-activated cell-sorted CD3, CD14, and CD19 positive cells. CD13 and CD19 positive cells were also tested after 48 hours of activation. BV173 cells served as positive control. (C) Zebra plot after coculture of clone C CAR T cells produced from cells of A*02–positive and –negative blood donors. Flow cytometry was performed after 18 hours’ coculture at 1:1 ratio. Plot is representative of 3 biological replicates. (D) Cell proliferation capacity of simulated but untransduced T cells and HLA-A*02–positive and –negative clone C CAR T cells. (E) Colony-forming unit (CFU) assays of cord blood–isolated CD34+ HSCs, OCI-AML02 cell line as positive control, and N3 primary AML cells. Cells were plated after 18-hour 1:1 coculture of CAR T cells and target cells. MUC16-specific 4H11 CAR T cells served as control. Error bars in panels B, D, and E denote standard deviation. *P < .05 (unpaired t test). APC-Cy7, allophycocyanin-cyanin 7; n.s., not significant; PE-A, phycoerythrin; Q, quadrant.
Clone C CAR T cells do not mediate toxicity toward healthy leukocytes, activated lymphocytes, or HSCs. (A) Flow cytometry of clone C or mIgG1 isotype binding of healthy A*02–positive CD3, CD19, CD33, and CD15 positive cells. Data are representative of 3 donors. (B) Lactate dehydrogenase–killing assay with clone C CAR T cells and magnetic-activated cell-sorted CD3, CD14, and CD19 positive cells. CD13 and CD19 positive cells were also tested after 48 hours of activation. BV173 cells served as positive control. (C) Zebra plot after coculture of clone C CAR T cells produced from cells of A*02–positive and –negative blood donors. Flow cytometry was performed after 18 hours’ coculture at 1:1 ratio. Plot is representative of 3 biological replicates. (D) Cell proliferation capacity of simulated but untransduced T cells and HLA-A*02–positive and –negative clone C CAR T cells. (E) Colony-forming unit (CFU) assays of cord blood–isolated CD34+ HSCs, OCI-AML02 cell line as positive control, and N3 primary AML cells. Cells were plated after 18-hour 1:1 coculture of CAR T cells and target cells. MUC16-specific 4H11 CAR T cells served as control. Error bars in panels B, D, and E denote standard deviation. *P < .05 (unpaired t test). APC-Cy7, allophycocyanin-cyanin 7; n.s., not significant; PE-A, phycoerythrin; Q, quadrant.
Because recognition and killing of activated T cells could strongly affect the production and efficacy of A*02–positive NDC80-specific CAR T cells, we next tested these NDC80-clC CAR T cells for their potential to mitigate fratricide. In an overnight 1:1 mixed lymphocyte culture of A*02–positive and A*02–negative NDC80-clC CAR T cells, only a slight reduction of A*02–positive CAR T cells was observed relative to the A02-negative cells (Figure 4C). To address the potential long-term fratricide effects of HLA-A*02–positive NDC80-clC CAR T cells, HLA-A*02–positive and HLA-A*02–negative CAR T cells were cultivated over 3 weeks, and viability as well as cell numbers were monitored. Again, no signs of significant differences in cell proliferation or viability were observed between these 2 groups, indicating no relevant fratricidal effects (Figure 4D).
Although no toxicities were seen in our experiments with mature hematopoietic cells, the question remained if other important proliferative cells such as HSCs would be affected by the NDC80-clC CAR T cells. This is especially important as MS analysis of the HLA ligandome of healthy individuals showed presentation of the ALNEQIARL peptide in one bone marrow sample.25 We therefore used colony-forming unit assays from cord blood–isolated HLA-A*02–positive and –negative CD34+ HSCs with OCI-AML02 cancer cells and one primary HLA-A*02 AML sample as positive controls. OCI-AML02 cancer cells as well as the primary AML cells formed significantly fewer colonies when treated with the NDC80-clC CAR T cells compared with a control CAR T cell (Figure 4E). In contrast, no differences were observed in cord blood–isolated HSCs independent of their HLA-A*02 status. Furthermore, cell numbers as well as lineage development were likewise not affected by the NDC80-clC CAR T cells (supplemental Figure 8A-C). We corroborated the results from colony-forming unit assays using lactate dehydrogenase assays showing no toxicity for either HLA-A*02–positive or –negative cord blood–derived HSCs (supplemental Figure 8D). Together, these data indicate that NDC80-clC CAR T cells do not cause relevant toxicity toward the CAR T cells themselves, healthy leukocytes, or HSCs.
To expand the list of tissues tested for on-target off-tumor toxicity, we analyzed human thymic fibroblasts, cardiac fibroblasts, and cardiomyocytes. None of these cells stained positively for our NDC80 TCR mimic antibody independent of their HLA-A*02 status, which further highlights its specificity (supplemental Figure 9A). Identical results were obtained after culturing these cells for ∼10 days, although cardiomyocytes did not survive this culture period. In addition, only baseline killing of CFSE-labeled cells was observed after 24 hours of coculture with either MOCK T cells or NDC80-clC CAR T cells (supplemental Figure 9B). We compared our results vs the ratio of remaining target cells observed when this assay was performed with unpulsed T2 cells, which served as a true negative control (supplemental Figure 9C-D).
NDC80-clC CAR T cells control human leukemia and mesothelioma tumor growth in mouse models, leading to prolonged survival
Because our new agent has been proven to selectively identify its target and to mediate killing in a cancer-specific manner, we tested its potency in 2 mouse models: an intravenous BV173 leukemia model and an intraperitoneal JMN mesothelioma solid tumor model. We injected 1 million BV173 cells per mouse intravenously followed by 2 million CAR T cells on day 5; disease burden was monitored through weekly cheek bleeds and subsequently by flow cytometry to determine the fraction of leukemia cells vs CD45+ mouse leukocytes as described previously32 (Figure 5A; gating strategy provided in supplemental Figure 10). MUC16-specific CAR T cells as well as tumor cell injection alone served as controls. We observed significant control of peripheral blast count of the NDC80-clC CAR T cells over controls (Figure 5B; supplemental Figure 11A), which was monitored until day 56 and then followed up by overall survival, which also showed superiority of the NDC80-clC CAR T cells over the control groups (Figure 5C). Diminished killing of GFP-Luc transduced cells compared with wild-type BV173 cells was noted, which positively correlated to the degree of GFP expression (supplemental Figure 11B). Therefore, in these experiments, we chose cheek bleeds over a bioluminescence model, to reduce the risk of biasing the in vivo experiments by this reduction in killing capacity. Wild-type cells were used instead.
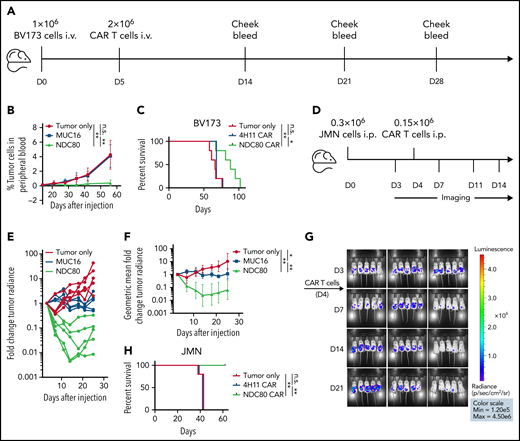
Clone C CAR T cells control tumor growth and prolong survival in leukemia and mesothelioma mouse models. (A) Experimental design for BV173 intravenous (i.v.) leukemia model. (B) Mean tumor burden as defined by percentage of A*02–positive blast cells in mouse blood. Five mice per group. **P < .01 (Mann-Whitney text). Error bars denote standard deviation. (C) Overall survival in BV173 leukemia model. *P < .05, **P < .01 (log-rank test). Experiment was conducted twice, with comparable results. (D) Experimental design for JMN intraperitoneal (i.p.) mesothelioma model. (E) Spaghetti plot depicting individual tumor burden relative to day 3. (F) Geometric mean of average tumor burden spaghetti plot depicting individual tumor burden relative to day 3. Five mice per group. *P < .05, **P < .01 (Mann-Whitney test). Error bars denote 95% confidence interval. (G) Bioluminescence imaging using luciferase pre– and post–CAR T-cell treatment. (H) Overall survival in JMN mesothelioma model. **P < .01 (log-rank test). Experiment was conducted twice, with comparable results. D, day; n.s., not significant.
Clone C CAR T cells control tumor growth and prolong survival in leukemia and mesothelioma mouse models. (A) Experimental design for BV173 intravenous (i.v.) leukemia model. (B) Mean tumor burden as defined by percentage of A*02–positive blast cells in mouse blood. Five mice per group. **P < .01 (Mann-Whitney text). Error bars denote standard deviation. (C) Overall survival in BV173 leukemia model. *P < .05, **P < .01 (log-rank test). Experiment was conducted twice, with comparable results. (D) Experimental design for JMN intraperitoneal (i.p.) mesothelioma model. (E) Spaghetti plot depicting individual tumor burden relative to day 3. (F) Geometric mean of average tumor burden spaghetti plot depicting individual tumor burden relative to day 3. Five mice per group. *P < .05, **P < .01 (Mann-Whitney test). Error bars denote 95% confidence interval. (G) Bioluminescence imaging using luciferase pre– and post–CAR T-cell treatment. (H) Overall survival in JMN mesothelioma model. **P < .01 (log-rank test). Experiment was conducted twice, with comparable results. D, day; n.s., not significant.
JMN cells luciferase positive cells were used in the intraperitoneal model experiment. Notably, luciferase transduction of JMN cells did not affect killing by NDC80-clC CAR T cells compared with untransduced JMN cells in vitro (supplemental Figure 11C). Then, 0.3 million JMN cells were injected intraperitoneally and imaged on day 3; this was then followed by the injection of 0.15 million NDC80-clC CAR T cells or MUC16-specific CAR T cells on day 4 (Figure 5D). Bioluminescence imaging revealed profound tumor control, with a 10- to 200-fold decrease in tumor signal in individual NDC80-clC CAR T cell–treated mice and an average 10-fold decrease in tumor signal from day 14 onward in the entire group (Figure 5E-G). This significant tumor control also translated into a survival benefit, with 100% survival of mice to day 60; in contrast, no survivors remained in the control CAR T cell–treated group after 42 days (Figure 5H).
Discussion
Most protein targets within cancer cells are not druggable by conventional small molecule drugs, and cell surface proteins that may be addressable by therapeutic antibodies are mostly not tumor specific. Therefore, the discovery of new targets that may be approached by immunologic agents is urgently needed. In this study, we showed that MS can be used to rationally discover broadly presented tumor-associated HLA ligands that can serve as targets for therapeutic TCR mimic CAR T cells. In addition, the use of HLA ligands found abundantly presented on cancer cells, but not with, or with limited amounts on, normal cells, broadens the spectrum of potential cancer-specific targets tremendously. As a target, this class of antigen does not require that the individual peptide is immunogenic, because engineering of TCR mimic antibodies bypasses the need to have antigens that are immunogenic to the host. This gives them another advantage over conventional TCR T-cell therapies, as naturally occurring TCRs are usually limited in their targeting capacity by negative thymic selection, which rules out a wide range of self-proteins as potential targets. Of note, alternative strategies to target non-immunogenic self-proteins have also been developed in the TCR T-cell field (eg, through allo-restricted T cells33,34 or mouse models with humanized TCR repertoires35). Nevertheless, these strategies are restricted to some degree and do not allow targeting of any stable peptide:HLA combinations as is theoretically possible with a TCR mimic drug.
This approach also opens the door to epitopes that may be found widely among different cancer types compared with the tumor- and patient-specific mutational neoantigens. TCR mimics against various targets have usually focused on well-known immunogenic CD8 T-cell epitopes.8-17 Some of these neoantigen epitopes are presented in cell lines at an abundance of only a dozen molecules per cell or less.11,13 In contrast, we detected ∼500 to 1300 copies of the NDC80 ALNEQIARL peptide on cancer cell lines by flow cytometry, thus reducing the requirement for development of ultrapotent agents. Notably, as the NDC80 epitope was first identified by MS in the immunopeptidome of several unmatched cancer cell lines, adequate cell surface presentation for the target was validated from the start of the discovery process.
Furthermore, most of the described targets of TCR-based agents are cancer-germline antigens, oncofetal antigens, viral antigens, or neoantigens, which are generally limited to selected cancer types. Importantly, we detected the NDC80 target antigen in >90% of the A*02–positive cell lines tested by MS. Hence, this target is much more prevalent than these other public and private neoantigens. Interestingly, the ALNEQIARL peptide has also been identified by MS on different A*02 suballeles (A*02:02,: 03,: 04,: 07,: 11),36,37 thus broadening target patient populations considerably. Finally, because the NDC80 protein is essential for chromosome segregation and cell division, tumor escape by downregulation of the protein or by functionally relevant mutations would be a less likely event.
Using the E-ALPHA phage library platform, we successfully identified an scFv, which in a CAR T-cell format was able to effectively and selectively bind to the ALNEQIARL:A*02 complex, kill cancer target cells in vitro, and mediate tumor control in mice, resulting in prolonged survival in leukemia and solid tumor models. Importantly, although the ALNEQIARL:A*02 complex also might be presented on healthy HLA-A*02–positive dividing cells, we did not observe relevant killing of healthy leukocytes, HSCs, mitogen-stimulated T and B cells, or thymic and cardiac fibroblasts. In the case of HSCs, the reduced expression of HLA on these cells is a possible explanation for this finding.31 In peripheral leukocytes, which are generally not proliferating, there is much lower expression of NDC80 compared with tumor cell lines or primary tissues. Interestingly, although the parent protein NDC80 is an essential protein in dividing cells, and its expression can be measured in many different tissue types, it seems that NDC80 peptides can be cancer-selective targets due to the additional restriction for HLA presentation. This process appears to be associated with the oncogenic state. The lack of normal cell presentation might also in part be explained by the constraints of having sufficient HLA complexes, rather than their ligands, in HLA:peptide formation.38 Therefore, many peptides, although properly processed and transported to the endoplasmic reticulum, might not be presented if they are of lower abundance (eg, in terminally differentiated cells) or if the available rate of HLA complexes is highly limited (eg, observed in HSCs). Still, due to limitations in current methods, we cannot definitively rule out presentation of the ALNEQIARL peptide on all healthy tissues, similar to the GLNEEIARV peptide, which is also derived from NDC80. Although a reasonable therapeutic index seems to be achievable, additional extensive toxicity studies should be performed to ensure safety of this drug before it could be introduced into clinical development. Testing for cross-reactivity in transgenic mice might be useful; however, the production of murine CAR T cells with human constructs was not feasible, and HLA-A*02 transgenic mouse toxicity studies are confounded by graft-versus-host disease effects when using human cells. Future studies of cross-reactivity might require use of a TCR mimic antibody or a bispecific format.
Overall, our study highlights the importance of considering non-immunogenic, broadly expressed, tumor-associated HLA ligands identified by MS as targets for cancer immunotherapy druggable through TCR mimic CAR T cells. This expansion of the potential antigen repertoire thus enabled us to develop the NDC80-clC CAR T-cell construct with broad applicability and strong reactivity against highly proliferative cancers often found among hematologic malignancies.
Acknowledgments
The authors thank Alex Kentsis for access to the Byonic Software and providing the AML PDX cells for killing assays. They also thank the Proteomics Resource Center at The Rockefeller University for the performance of all LC/MS-MS experiments.
This study was supported by the Leukemia and Lymphoma Society, the National Institutes of Health, National Cancer Institute (P30CA 008748, R01 CA55349, P01 CA23766, and R35 CA241894); The Experimental Therapeutics Center of MSK; and Tudor Funds. M.G.K. is supported by the German Research Foundation (DFG) with an individual Research Grant (KL3118/11) and is a fellow of the Berlin Institute of Health Clinician Scientist Program.
Authorship
Contribution: M.G. Klatt, T.D., Z.Y., J.L., S.S.M., M.M.D., H.L., T.J.G., C.B., L.P., Z.E.H.A.,T.K., and M.L. performed and analyzed experiments; M.G. Klatt, T.D., M.G. Kharas, C.L., and D.A.S. designed experiments; M.G. Klatt and T.D. wrote the original draft of the manuscript; T.D. and D.A.S. supervised the project; and D.A.S. provided funding and edited the manuscript. All authors reviewed and contributed to the manuscript.
Conflict-of-interest disclosure: D.A.S. has ownership in, income from, or research funds from: Pfizer, Sellas Life Sciences, Iovance, Eureka Therapeutics, Sapience, OncoPep, Actinium, CoImmune, and Repertoire. T.D. worked as a consultant to Eureka Therapeutics. C.L., Z.Y., and J.L. are employees of Eureka Therapeutics. M.G. Klatt is a consultant to Ardigen. M.G. Kharas received speaker honorarium from AstraZeneca und Kumquat; and serves on the scientific advisory board of 858 Therapeutics. MSKCC has filed for patent protection for some components developed in this study. The remaining authors declare no competing financial interests.
Correspondence: Martin G. Klatt, Department of Hematology, Oncology and Tumorimmunology, Charité - University Medicine Berlin, Hindenburgdamm 30, 12203 Berlin, Germany; e-mail: martin.klatt@charite.de; and David A. Scheinberg, Molecular Pharmacology and Center for Experimental Therapeutics, Memorial Sloan Kettering Cancer Center, 1275 York Avenue, Box 531, New York, NY 10065; e-mail: scheinbd@mskcc.org.
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
