

Key points
Defective autophagy in mesenchymal progenitors associates with BM hypocellularity and mortality after severe stress.
A short 4-day pharmacological in vivo treatment of mice to attenuate CDC42 activation protects against cytopenia and improves survival.

Abstract
The cellular mechanisms required to ensure homeostasis of the hematopoietic niche and the ability of this niche to support hematopoiesis upon stress remain elusive. We here identify Wnt5a in Osterix+ mesenchymal progenitor and stem cells (MSPCs) as a critical factor for niche-dependent hematopoiesis. Mice lacking Wnt5a in MSPCs suffer from stress-related bone marrow (BM) failure and increased mortality. Niche cells devoid of Wnt5a show defective actin stress fiber orientation due to an elevated activity of the small GTPase CDC42. This results in incorrect positioning of autophagosomes and lysosomes, thus reducing autophagy and increasing oxidative stress. In MSPCs from patients from BM failure states which share features of peripheral cytopenia and hypocellular BM, we find similar defects in actin stress fiber orientation, reduced and incorrect colocalization of autophagosomes and lysosomes, and CDC42 activation. Strikingly, a short pharmacological intervention to attenuate elevated CDC42 activation in vivo in mice prevents defective actin-anchored autophagy in MSPCs, salvages hematopoiesis and protects against lethal cytopenia upon stress. In summary, our study identifies Wnt5a as a restriction factor for niche homeostasis by affecting CDC42-regulated actin stress-fiber orientation and autophagy upon stress. Our data further imply a critical role for autophagy in MSPCs for adequate support of hematopoiesis by the niche upon stress and in human diseases characterized by peripheral cytopenias and hypocellular BM.
Introduction
Declining niche homeostasis is an underlying defect contributing to aging and the development of aging-related malignant hematopoietic diseases.1 Functional changes in bone marrow (BM)-derived mesenchymal stem and progenitor cells (MSPCs) include deregulated osteogenic and adipogenic differentiation, bone loss, and osteoporosis.1-3 Intriguingly, compromised niche health in aging and malignant disease is associated with ineffective hematopoiesis and dysfunctional hematopoietic stem cells (HSCs), resulting in cytopenias and progressive BM hypocellularity. Thus, it is important to understand the mechanisms governing cellular health of niche cells.
Efforts studying HSC dysfunction have shown that interconnected cellular maintenance mechanisms, such as mitochondrial quality control,4 macroautophagy (autophagy),5 and lysosomal lysis6,7 are critical for safeguarding HSC division and function. Interestingly, dysfunctional HSCs further show altered noncanonical WNT5A-CDC42 signaling with a loss of polarized cytoskeletal components,8-10 associated with activation of the small GTPase CDC42 in both stem cells8,9 and mature cells.10 Since the cytoskeleton guides autophagosome formation and lysosomal fusion,11 as well as mitochondrial fission,12 correct assembly and orientation of the cytoskeleton is pivotal for cellular maintenance.
In the present study, we wondered whether and how the hematopoiesis-supportive function of the niche depends on CDC42 activation and autophagy under intermittent stress conditions. For this purpose, we studied hematopoiesis and niche cells in mice lacking Wnt5a expression, specifically in MSPCs. In addition, we studied whether similar molecular events operate in MSPCs from human conditions characterized by hypocellular BM and ineffective hematopoiesis. Such features are shared by different hematologic diseases, such as Shwachman-Diamond syndrome (SDS), hypocellular myelodysplastic syndrome (hypocellular MDS), and severe aplastic anemia as well as therapeutically conditioned leukemia/lymphoma patients undergoing allogeneic hematopoietic stem cell transplantation (HSCT).
We show that BM MSPCs from murine Wnt5a-deficient animals poorly support hematopoiesis associated with CDC42 activation and defective autophagy. Furthermore, MSPCs from the human diseases studied show similar CDC42 activation with reduced WNT5A expression and ineffective autophagy. More importantly, we show that pharmacological attenuation of RhoGDI/CDC42 activation not only restores stromal F-actin organization orientation and autophagy in mice but also protects from accelerated cytopenia and mortality after repeated cytotoxic stress.
Methods
Mice
Wnt5afl/fl mice13 were crossed with Osx-GFP::Cre mice14 (Osx-Cre; The Jackson Laboratory, Bar Harbor, ME). Osx-Cre mice express the CRE recombinase under the control of the Osterix (Sp7) promotor. Although Sp7 is expressed in only a small subpopulation of osteoprogenitors, the numerous progenies of these cells include all major mesenchymal populations.15 Litters of Osx-Cre;Wnt5afl/+ mice were crossed with Wnt5afl/fl (5Afl/fl) mice so that litters yielded controls (5Afl/fl and Osx-Cre [O5A+/+]) as well as Wnt5a deleted mutants (O5AΔ/Δ). In all experiments, the results from 5Afl/fl and O5A+/+ mice were combined as controls since both express a functional Wnt5a gene. Details about mouse models are summarized in supplemental Table 1, available on the Blood Web site.
All animal experiments were approved by the Government of Upper Bavaria and performed in accordance with ethical guidelines and approved protocols (Vet_02-14-112, and -17-124). All animals were housed under specific pathogen-free conditions, according to the Federation of Laboratory Animal Science Associations and institutional recommendations. Mice used were 8 to 10 weeks old.
Human BM samples
Human BM samples were collected from 15 healthy individuals and 6 hip replacement patients after written informed consent. In the healthy samples aged over 60 years, the presence of clonal hematopoiesis was excluded by targeted sequencing of 68 genes recurrently mutated in hematologic malignancies.16 Furthermore, BM samples were obtained from patients with different BM failure states, such as SDS, an inherited BM failure syndrome, hypocellular MDS, or severe aplastic anemia. We also included 3 samples from leukemia/lymphoma patients undergoing allogeneic HSCT, 2 of which had received total body irradiation as part of their conditioning regimen, which may contribute to niche damage, and the third patient showed incomplete BM reconstitution after transplant (likely due to extensive pretreatment) (supplemental Table 2). The studies TUM 538/16 (Klinikum rechts der Isar, Technical University Munich, Munich, Germany) and P00020466 (Boston Children’s Hospital, Boston, MA) were approved by the respective institutional review boards in accordance with the Declaration of Helsinki. Characteristics of healthy individuals and patients used in this study are detailed in supplemental Table 2.
Flow cytometry analysis and cell sorting
Hematopoietic lineage- SCA1+ KIT+ (LSK) cells and their HSC-enriched CD34- CD48- CD150+ subpopulations (LT-HSCs) were isolated and labeled as reported.10,17 Stromal subpopulations were sorted or analyzed as nonhematopoietic (CD45- TER119-) cells as described.10,18 All antibodies used in this study are listed in supplemental Table 3, machines in supplemental Table 4, and sorting schemes are shown in supplemental Figure 1.
Peripheral blood (PB) was analyzed by Animal Blood Cell Counter (Scil Vet Abc).
In vivo transplantation assay
Competitive repopulation was performed using transplantation (Tx) of LT-HSCs into lethally irradiated 129Ly5.1 Wild-type (WT)-recipient mice, as described previously.10 Peripheral engraftment of donor cells was analyzed at regular intervals (4, 8, 12, and 16 weeks posttransplant). Sixteen weeks after transplantation, recipient mice were sacrificed, and the hematopoietic organs were analyzed by flow cytometry.
In vivo treatment with pharmacological compounds
For induction of cytotoxic stress, 5-fluorouracil (5-FU) was administered intraperitoneal (IP, 150 mg/kg, Ribosepharm) on day 0 in a single dose or in 2 doses at days 0 and 8, after which mice were analyzed and MSPCs cultured. Animals were monitored twice daily. Surviving O5AΔ/Δ animals were sacrificed at or prior to day 14 due to severe stress.
In some in vivo experiments, the CDC42/RhoGDI inhibitor CASIN (2.4 mg/kg, TOCRIS)19 was administered by IP every 24 hours for 4 consecutive days (days 5, 6, 7, and 8) post first 5-FU treatment on day 0.
Whole-mount immunofluorescence staining
This procedure was performed as described previously.20 Antibodies are listed in supplemental Table 3. For evaluation, fluorescently labeled bone tissues were placed onto a µ-slide 4 well and covered in antifade or phosphate-buffered saline (PBS) to prevent tissue desiccation. The preparations were examined using a Leica TCS SP8 confocal microscope and analyzed with the image analysis software Volocity (v6.2; Perkin Elmer) and ImageJ. In addition, bone matrix and adipocytes were detected using the TLD mode of the microscope. In analyses, the term arteriole includes both arterial and arteriolar cells.
Stromal cell (MSPC) isolation and culture
Flushed long bones from mice were crushed and digested as described.21 Sorted MSPCs or MSPCs from digested compact bone were cultured on 0.1% gelatin-coated plates in MEM α (with ribosides and Glutamax, Invitrogen), supplemented with fetal calf serum (FCS, 10%; PAA), antibiotics and 0.1% β-mercapto-ethanol (Invitrogen).
Human MSPCs were isolated from BM samples of healthy and diseased individuals and cultured using pooled platelet lysate as described previously.3
Cocultures of HSC and stromal cells
MSPCs were grown to confluence and irradiated (15 Gy). Freshly sorted WT LT-HSCs were seeded to the stroma in long-term culture (LTC) medium (M5300; Stemcell Technologies, Vancouver, Canada) with 1 μM hydrocortisone and incubated (37°C, 5% CO2, >95% humidity). After 6 days, cultures were either reseeded at 60 input LT-HSCs equivalents in MethoCult M3434 (Stemcell Technologies) or analyzed by flow cytometry. Hematopoietic colonies were counted using standard criteria.
Immunofluorescence staining (IF, confocal IF)
Hematopoietic cells were prepared and stained as described.10 For staining of stromal cells, either sorted cells were spotted, or cultured MSPCs were reseeded on 0.1% gelatin-coated Superfrost Plus™ slides (Thermo Fisher Scientific) and cultured overnight in MSPC medium. Cells were fixed with 4% PFA for 5 minutes and staining was performed10 using the antibodies listed in supplemental Table 3.
Pictures were taken using a Leica DM RBE microscope with AxioVision software (Carl Zeiss) using standardized exposure and diaphragm settings for all samples. Thirty randomly captured cells per sample were imaged at a 100-fold magnification.
Confocal fluorescence microscopy and deconvolution of the fluorescent images were performed on a Leica SP8 confocal microscope as described earlier.22 Assessment of different parameters is outlined in the supplemental Methods.
Assessment of mitochondrial function
To interrogate mitochondrial function, mitochondrial number and diameter, reactive oxygen species (ROS) production, glycolysis, and oxygen consumption were assessed. ROS was detected by culturing fresh BM cells for 20 minutes at 37°C with either CM-H2DCFDA or MitoTracker. Staining for the mitochondrial outer membrane receptor TOMM20 indicated the number of mitochondria. Analysis of fluorescence intensity was performed using ImageJ. The extracellular acidification rate (ECAR), a measure of glycolysis, and oxygen consumption rate (OCR) were determined using an XF96 Extracellular Flux Analyzer (Seahorse Bioscience) as described.23
Autophagic flux assessment using CytoID
To determine the autophagic flux, MSPCs were cultured at 80% semiconfluence with rapamycin (1 µM), chloroquine (10 µM), CASIN (5 µM), or the vehiculum 0.01% DMSO for 16 hours (37°C, 5% CO2, >95% humidity). The next day, cells were treated with Cyto-ID Autophagy Detection Kit as described by the manufacturer (Enzo Life Science). Autophagic flux was determined as ΔMFI (= MFIchloroquine − MFItest) and measured using mean fluorescence intensity (MFI) on the CyAn ADP LxP8.
Statistics
The variance was similar between groups that were statistically compared. To test for differences between 2 groups, a paired Student's t test was used unless the data point distribution was not normally distributed. In that case, the nonparametric Mann-Whitney U test was used. All statistical analyses were performed with the Prism software package.
Data
For original data, please contact the corresponding authors.
Results
Lack of Wnt5a in MSPCs impairs HSC function upon cytotoxic myeloablation
Maintenance of HSC function depends on the expression of Wnt5a in the environment.10 In mice, WNT5A protein expression is regulated by different stressors, particularly after 5-FU treatment of WT MSPCs (supplemental Figure 2A). To test whether Wnt5a expression in MSPCs is responsible for reduced support of HSC function, we took advantage of a mouse model in which the floxed Wnt5a gene13 was selectively deleted in osteoprogenitors (O5AΔ/Δ).
In O5AΔ/Δ mice, Wnt5a is deleted in MSPCs and osteoblastic cells (OBCs), but is still expressed by CD31+ endothelial cells (ECs) (supplemental Figure 2B-C), and is even elevated in LT-HSCs (supplemental Figure 2D). Moreover, the number of ECs and MSPCs was unchanged, but a relative increase in OBCs was noted (supplemental Figure 2E). O5AΔ/Δ mice showed normal hematopoiesis in comparison with control littermates, except for a significant decrease in LT-HSCs numbers. These LT-HSCs demonstrated a normal repopulation activity (supplemental Figure 2F).
To determine how stress alters niche and HSC function in the absence of stromal Wnt5a, mice were treated with 5-FU (Figure 1A), which selectively recruits quiescent HSCs through the niche.20 Prior to 5-FU treatment, CDC42 activation, F-actin expression, or CDC42-GTP polarization is unchanged in LT-HSCs (supplemental Figure 2G-H). Eight days after 5-FU treatment, the LT-HSC numbers remain similar in O5AΔ/Δ and control animals (supplemental Figure 2I-K). However, treatment with 5-FU in O5AΔ/Δ animals altered LT-HSCs to not only show an elevation of WNT5A but also decreased F-actin with increased levels of active and nonpolarized CDC42-GTP (Figure 1B; supplemental Figure 3A-C), suggestive of HSC dysfunction.8
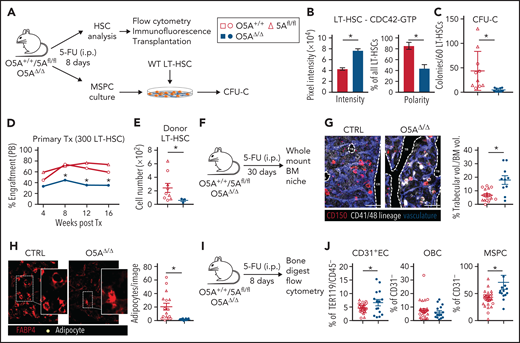
Stress-induced niche remodeling in O5AΔ/Δ mice. (A) Experimental design for IP injection of 5-Fluorouracil (5-FU) in coculture assay of MSPCs and LT-HSC. The following genotypes were analyzed by flow cytometry and IF 8 days after treatment: Control (CTRL): O5A+/+, n = 5, 5Afl/fl, n = 7 and O5AΔ/Δ, n = 10. (B) Protein content and polarity of LT-HSCs stained for CDC42-GTP (n = 30), measured by ImageJ software. (C) Number of colonies from 60 LT-HSCs after coculture for 6 days on MSPCs isolated from 5-FU injected mice with the following genotype: (CTRL): O5A+/+, n = 5, 5Afl/fl, n = 5 and O5AΔ/Δ, n = 10. (D) Primary transplantation (Tx) of 300 sorted LT-HSCs 8 days after 5-FU injection into lethally irradiated 129*Ly5.1 WT recipients. Experimental groups: CTRL: O5A+/+, n = 5 and 5Afl/fl, n = 4 and O5AΔ/Δ, n = 7. Graph showing the donor engraftment in PB. (E) Absolute numbers of donor-type LT-HSCs in the BM, 16 weeks after Tx. For flow cytometry gating strategy, see supplemental Figure 1. (F) Experimental design for IP injection of 5-FU in 8- to 10-week-old mice of following genotypes: CTRL: O5A+/+ n = 2, 5Afl/fl n = 2 and O5AΔ/Δ, n = 2. BM analysis 30 days after treatment. (G) Stacked whole-mount images from epiphyseal/metaphyseal BM. FABP4+ vasculature is shown in blue. CD150 is shown in red, other hematopoietic markers (CD41, CD48, and lineage) are shown in gray. The dashed lines denote the endosteum. Scale bar, 100 µm; n = 10 images (n = 6 images 5Afl/fl PBS) from 2 mice each group. The results represent 2 independent experiments. The graph showing the % of trabecular volume per BM volume. (H) Stacked whole-mount images from epiphyseal/metaphyseal BM. FABP4+ adipocytes are shown in red. Adipocytes are additionally marked by a yellow circle. Extended focus projection images. Scale bar, 100 µm; n = 10 images from 2 mice each group, femora. The graph on the right shows the number of adipocytes per image. (I) Experimental design for analysis of bone-digested stromal cells, isolated from compact bones, 8 days post 5-FU treatment. (J) Relative numbers of CD31+ ECs, OBCs, and immature MSPCs; flow cytometry gating strategy in supplemental Figure 1. *P < .05 (nonparametric Mann-Whitney test: B-G,J). The results represent 2 to 3 independent experiments. Data are represented as dots per mouse or cell and the mean ± SEM. Symbol legends as shown in Figure 1A.
Stress-induced niche remodeling in O5AΔ/Δ mice. (A) Experimental design for IP injection of 5-Fluorouracil (5-FU) in coculture assay of MSPCs and LT-HSC. The following genotypes were analyzed by flow cytometry and IF 8 days after treatment: Control (CTRL): O5A+/+, n = 5, 5Afl/fl, n = 7 and O5AΔ/Δ, n = 10. (B) Protein content and polarity of LT-HSCs stained for CDC42-GTP (n = 30), measured by ImageJ software. (C) Number of colonies from 60 LT-HSCs after coculture for 6 days on MSPCs isolated from 5-FU injected mice with the following genotype: (CTRL): O5A+/+, n = 5, 5Afl/fl, n = 5 and O5AΔ/Δ, n = 10. (D) Primary transplantation (Tx) of 300 sorted LT-HSCs 8 days after 5-FU injection into lethally irradiated 129*Ly5.1 WT recipients. Experimental groups: CTRL: O5A+/+, n = 5 and 5Afl/fl, n = 4 and O5AΔ/Δ, n = 7. Graph showing the donor engraftment in PB. (E) Absolute numbers of donor-type LT-HSCs in the BM, 16 weeks after Tx. For flow cytometry gating strategy, see supplemental Figure 1. (F) Experimental design for IP injection of 5-FU in 8- to 10-week-old mice of following genotypes: CTRL: O5A+/+ n = 2, 5Afl/fl n = 2 and O5AΔ/Δ, n = 2. BM analysis 30 days after treatment. (G) Stacked whole-mount images from epiphyseal/metaphyseal BM. FABP4+ vasculature is shown in blue. CD150 is shown in red, other hematopoietic markers (CD41, CD48, and lineage) are shown in gray. The dashed lines denote the endosteum. Scale bar, 100 µm; n = 10 images (n = 6 images 5Afl/fl PBS) from 2 mice each group. The results represent 2 independent experiments. The graph showing the % of trabecular volume per BM volume. (H) Stacked whole-mount images from epiphyseal/metaphyseal BM. FABP4+ adipocytes are shown in red. Adipocytes are additionally marked by a yellow circle. Extended focus projection images. Scale bar, 100 µm; n = 10 images from 2 mice each group, femora. The graph on the right shows the number of adipocytes per image. (I) Experimental design for analysis of bone-digested stromal cells, isolated from compact bones, 8 days post 5-FU treatment. (J) Relative numbers of CD31+ ECs, OBCs, and immature MSPCs; flow cytometry gating strategy in supplemental Figure 1. *P < .05 (nonparametric Mann-Whitney test: B-G,J). The results represent 2 to 3 independent experiments. Data are represented as dots per mouse or cell and the mean ± SEM. Symbol legends as shown in Figure 1A.
In support of this observation, we found that LT-HSCs cocultured with Wnt5a deleted MSPCs show strongly reduced maintenance of colony-forming cells (Figure 1C). Furthermore, transplantation experiments demonstrated that LT-HSCs from 5-FU-treated O5AΔ/Δ animals show reduced engraftment of both mature and immature cell lineages compared with HSCs from control mice (Figure 1D; supplemental Figure S3D-G), confirming dysfunction of LT-HSCs from 5-FU-treated O5AΔ/Δ mice (Figure 1E; supplemental Figure 3F).
Stress-induced niche remodeling in O5AΔ/Δ mice
To determine whether poor LT-HSC function associates with remodeling of the BM in 5-FU-treated O5AΔ/Δ or control mice, we performed whole-mount BM staining 30 days after 5-FU treatment, an endpoint for BM regeneration in WT mice20 (Figure 1F; supplemental Figure 3H). Our analyses show similar localization of HSCs in O5AΔ/Δ and control BM to endosteal areas, megakaryocytes, or different types of vessels (supplemental Figure 3I-K). However, we found a striking BM remodeling associated with increased trabecular volume of the epiphysis/metaphysis of O5AΔ/Δ mice compared with controls (Figure 1G), a near loss of FABP4high adipocytes with big fat droplets (Figure 1H), and a relative increase in both MSPCs and ECs in flow cytometry, but not in OBCs in the BM of O5AΔ/Δ mice (Figure 1I-J). Despite these changes in BM anatomy and niche cell numbers in vivo, the frequency of CFU-F and the osteo- and adipogenic potential in vitro was unchanged in both freshly isolated and cultured MSPCs from 5-FU-treated O5AΔ/Δ or control mice (supplemental Figure 4A-D).
Intermittent cytotoxic stress causes BM failure and mortality in O5AΔ/Δ mice
Repetitive 5-FU administration leads to HSC exhaustion and death by BM failure.24 Considering impaired HSC function in 5-FU-treated O5AΔ/Δ mice (Figure 1D-E), we wondered whether an additional 5-FU treatment of O5AΔ/Δ mice would deplete HSCs (Figure 2A-B). Strikingly, whereas most control mice (13/18, 72%) survived 2 consecutive applications of 5-FU, few of the O5AΔ/Δ mice (1/11, 9%) survived this treatment until day 14 (Figure 2C). Diminished numbers of white and red blood cells (WBC and RBC) in PB and BM cells indicated a PB and BM cytopenia in surviving O5AΔ/Δ mice (Figure 2D-G; supplemental Figure 4E-F), with a strong decline in mature CD41+ CD42b+ megakaryocytes and LT-HSCs in the BM (Figure 2H; supplemental Figure 4G). The loss of LT-HSC was, however, not caused by a relocation of LT-HSCs to other tissues, such as the spleens (Figure 2I-J).
![Peripheral and BM cytopenia in O5AΔ/Δ mice treated twice with 5-FU. (A) Graphical illustration of our hypothesis of how cytopenia is driven by declining niche homeostasis during consecutive periods of stress. (B) Experimental design for analysis of mice after 2 5-FU treatments (DAYS 0 AND 8). Control (CTRL): O5A+/+, n = 7, 5Afl/fl, n = 11 and O5AΔ/Δ, n = 11. (C) Survival of WT (open symbols, combined results of 5Afl/fl and O5A+/+ mice) and O5AΔ/Δ mice. (D-F) Graphs showing WBC, RBC, and hemoglobin (HGB) measured in PB, respectively, at DAYS 8 AND 14 from WT mice (open symbol, combined results of 5Afl/fl and O5A+/+ mice) and O5AΔ/Δ mice (closed symbols). (G) Total BM cell number 14 days after the first 5-FU treatment in CTRL (n = 9) and O5AΔ/Δ (n = 5) mice. Analysis of BM from 4 flushed long bones. (H) Representative contour plots of the BM after 2 consecutive 5-FU treatments at day 14. Graphs showing the percentage of LSKs (left) and LT-HSCs (right) in CTRL and O5AΔ/Δ mice. (I) Representative contour plots of spleen cells from mice treated twice with 5-FU. (J) Graphs showing the absolute cell number of spleen cells (left) and relative number of LSK cells in spleen (right). *P < .05 (2-sided parametric Student’s t test: [C-F]; Mann-Whitney test: [G-H]). The results represent 2 to 3 independent experiments. Data are represented as dots per mouse or cell and the mean ± SEM. Symbol legend shown in Figure 2B.](https://ash.silverchair-cdn.com/ash/content_public/journal/blood/139/5/10.1182_blood.2021011775/7/m_bloodbld2021011775f2.png?Expires=1769116449&Signature=CshudoBQqFklbasg8TOfaE23olVqREZJnI5l2r-9rH74XsJUc2uknyywSiCjYWIUZtAwa8zguFyRAwDmPUZdXKB0k6~4RsYibUUOLmmffOHvNo6t4GcU6IeKT9RK6FR0JrSzw3vVVeqOCbEeh3-3EGSX9FrEHqTSrMk5uS3jQbpfMZC6J9esi7JFFQAdlCAZmFyDPhlM5gUZMf~1MRtyJB14plYbSgQ4uEx3AffS9FasM9AmohcAF-FdWNqZa-uWRZRTUoMmMADkiglwk0As2znRnXTg96vu6JZl7zHxJ86nioZ-eAmKaQLArnTlVsTqjUwK06WY4hYLrwmPxWuzaA__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
Peripheral and BM cytopenia in O5AΔ/Δ mice treated twice with 5-FU. (A) Graphical illustration of our hypothesis of how cytopenia is driven by declining niche homeostasis during consecutive periods of stress. (B) Experimental design for analysis of mice after 2 5-FU treatments (DAYS 0 AND 8). Control (CTRL): O5A+/+, n = 7, 5Afl/fl, n = 11 and O5AΔ/Δ, n = 11. (C) Survival of WT (open symbols, combined results of 5Afl/fl and O5A+/+ mice) and O5AΔ/Δ mice. (D-F) Graphs showing WBC, RBC, and hemoglobin (HGB) measured in PB, respectively, at DAYS 8 AND 14 from WT mice (open symbol, combined results of 5Afl/fl and O5A+/+ mice) and O5AΔ/Δ mice (closed symbols). (G) Total BM cell number 14 days after the first 5-FU treatment in CTRL (n = 9) and O5AΔ/Δ (n = 5) mice. Analysis of BM from 4 flushed long bones. (H) Representative contour plots of the BM after 2 consecutive 5-FU treatments at day 14. Graphs showing the percentage of LSKs (left) and LT-HSCs (right) in CTRL and O5AΔ/Δ mice. (I) Representative contour plots of spleen cells from mice treated twice with 5-FU. (J) Graphs showing the absolute cell number of spleen cells (left) and relative number of LSK cells in spleen (right). *P < .05 (2-sided parametric Student’s t test: [C-F]; Mann-Whitney test: [G-H]). The results represent 2 to 3 independent experiments. Data are represented as dots per mouse or cell and the mean ± SEM. Symbol legend shown in Figure 2B.
Peripheral and BM cytopenia in O5AΔ/Δ mice treated twice with 5-FU. (A) Graphical illustration of our hypothesis of how cytopenia is driven by declining niche homeostasis during consecutive periods of stress. (B) Experimental design for analysis of mice after 2 5-FU treatments (DAYS 0 AND 8). Control (CTRL): O5A+/+, n = 7, 5Afl/fl, n = 11 and O5AΔ/Δ, n = 11. (C) Survival of WT (open symbols, combined results of 5Afl/fl and O5A+/+ mice) and O5AΔ/Δ mice. (D-F) Graphs showing WBC, RBC, and hemoglobin (HGB) measured in PB, respectively, at DAYS 8 AND 14 from WT mice (open symbol, combined results of 5Afl/fl and O5A+/+ mice) and O5AΔ/Δ mice (closed symbols). (G) Total BM cell number 14 days after the first 5-FU treatment in CTRL (n = 9) and O5AΔ/Δ (n = 5) mice. Analysis of BM from 4 flushed long bones. (H) Representative contour plots of the BM after 2 consecutive 5-FU treatments at day 14. Graphs showing the percentage of LSKs (left) and LT-HSCs (right) in CTRL and O5AΔ/Δ mice. (I) Representative contour plots of spleen cells from mice treated twice with 5-FU. (J) Graphs showing the absolute cell number of spleen cells (left) and relative number of LSK cells in spleen (right). *P < .05 (2-sided parametric Student’s t test: [C-F]; Mann-Whitney test: [G-H]). The results represent 2 to 3 independent experiments. Data are represented as dots per mouse or cell and the mean ± SEM. Symbol legend shown in Figure 2B.
Defects in autophagolysosome formation in MSPCs from O5AΔ/Δ mice
As it has been described that high WNT5A expression is associated with increased autophagy in melanoma25 and pneumonia26 and that, conversely, Wnt5a-deficient cells show decreased autophagy,27Wnt5a expression and autophagy might be linked in MSPCs. While defects in autophagy were shown to be harmful for HSCs,5,28-30 the role of autophagy in maintaining MSPC niche function remains to be established.
Thus, we stained MSPCs and observed strongly increased levels of ATG7, LAMP1, LC3, and SQSTM1 in characteristic punctae, indicating autophagosome accumulation in both freshly sorted and cultured MSPCs from 5-FU-treated O5AΔ/Δ mice (Figure 3A-C; supplemental Figure 5A-C). In addition, accumulation of LAMP1+ lysosomes with larger diameters and reduced colocalization with LC3 in MSPCs from 5-FU-treated O5AΔ/Δ mice indicates associated reduced lysosomal degradation of autophagosomes (Figure 3D-E).
![Defective autophagic vesicle delivery in MSPCs from O5AΔ/Δ mice. (A) Experimental design for IP injection of 5-FU in the following genotypes: CTRL: O5A+/+, n = 5, 5Afl/fl, n = 7 and O5AΔ/Δ, n = 10. Analysis of autophagy relevant mechanisms in compact bone-derived MSPCs 8 days after in vivo treatment. All experiments were performed either with sorted ([B], o/n culture) or compact bone-derived MSPCs ([C-F], passage 4). (B) Fluorescent staining of LC3 (red) and LAMP1 (green) in sorted primary MSPCs from the BM of 5-FU-treated mice, cultured overnight on 0.1%-gelatin-coated coverslips. (C) Fluorescent microscopy images of LC3 (red/left), SQSTM1 (=p62; red/right) and DAPI (blue) staining in compact bone-derived MSPCs (p4). The left graph shows the perinuclear distribution of LC3 measured with ImageJ software. SQSTM1 foci were counted with ImageJ software (graph, right). (D) Confocal images of perinuclear LAMP1 (green) and DAPI (blue) staining. The graphs show the perinuclear distribution of LAMP1 and the diameter of the lysosomes designated as feret's diameter measured with ImageJ software. (E) Confocal images of perinuclear colocalization of LAMP1 (green) and LC3 (red) measured by ImageJ software. Colocalization was measured by ImageJ software (PLUGIN: colocalization) and visualized in white. (F) Representative flow cytometry histogram of basal autophagy and quantification of Cyto-ID dye levels (DMFI: Cyto-ID dye level chloroquine treated-Cyto-ID dye level WITHOUT treatment). Data were measured by ImageJ software. Scale bars, 10 µm. *P < .05 (nonparametric Mann-Whitney test: B-F). The results of each panel represent 2 to 3 independent experiments. Each dot represents the measurement of 1 cell. Data are represented as mean ± SEM. Symbol legends are shown in Figure 3A,F.](https://ash.silverchair-cdn.com/ash/content_public/journal/blood/139/5/10.1182_blood.2021011775/7/m_bloodbld2021011775f3.png?Expires=1769116449&Signature=xgox908ZFxapRQ9Cy~I7ZGA9DoQJhJbAqkfKgE89bh26zcHZNdbpoG8dpf9DYsY3xWCveaxeOuMr4CdRTeOiBBb1IYlQ9rICeJr~kWvfXzh8FO3E-1DESc8bz2XEH4BFRqAwMBoS0JjZ1S97ZJD23eVoSEj9OmCF6IaszzBmbQ0BzR7BsmgTxFXjOjahaS6ZqBA5PdOwLK9H7O7Xa2ny3zebD66l-TWU~qxXRoCed0NfUNpqWrXI5Jt3PtCO6EIYdDYb29cFDTKLM8O3c9-x29Ekf2qHBjq9i8enZM2SQZy9bZCbRZ5yWRCIKhzSWIja87sazsE3y9Lx7Uk4oy8~1A__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
Defective autophagic vesicle delivery in MSPCs from O5AΔ/Δ mice. (A) Experimental design for IP injection of 5-FU in the following genotypes: CTRL: O5A+/+, n = 5, 5Afl/fl, n = 7 and O5AΔ/Δ, n = 10. Analysis of autophagy relevant mechanisms in compact bone-derived MSPCs 8 days after in vivo treatment. All experiments were performed either with sorted ([B], o/n culture) or compact bone-derived MSPCs ([C-F], passage 4). (B) Fluorescent staining of LC3 (red) and LAMP1 (green) in sorted primary MSPCs from the BM of 5-FU-treated mice, cultured overnight on 0.1%-gelatin-coated coverslips. (C) Fluorescent microscopy images of LC3 (red/left), SQSTM1 (=p62; red/right) and DAPI (blue) staining in compact bone-derived MSPCs (p4). The left graph shows the perinuclear distribution of LC3 measured with ImageJ software. SQSTM1 foci were counted with ImageJ software (graph, right). (D) Confocal images of perinuclear LAMP1 (green) and DAPI (blue) staining. The graphs show the perinuclear distribution of LAMP1 and the diameter of the lysosomes designated as feret's diameter measured with ImageJ software. (E) Confocal images of perinuclear colocalization of LAMP1 (green) and LC3 (red) measured by ImageJ software. Colocalization was measured by ImageJ software (PLUGIN: colocalization) and visualized in white. (F) Representative flow cytometry histogram of basal autophagy and quantification of Cyto-ID dye levels (DMFI: Cyto-ID dye level chloroquine treated-Cyto-ID dye level WITHOUT treatment). Data were measured by ImageJ software. Scale bars, 10 µm. *P < .05 (nonparametric Mann-Whitney test: B-F). The results of each panel represent 2 to 3 independent experiments. Each dot represents the measurement of 1 cell. Data are represented as mean ± SEM. Symbol legends are shown in Figure 3A,F.
Defective autophagic vesicle delivery in MSPCs from O5AΔ/Δ mice. (A) Experimental design for IP injection of 5-FU in the following genotypes: CTRL: O5A+/+, n = 5, 5Afl/fl, n = 7 and O5AΔ/Δ, n = 10. Analysis of autophagy relevant mechanisms in compact bone-derived MSPCs 8 days after in vivo treatment. All experiments were performed either with sorted ([B], o/n culture) or compact bone-derived MSPCs ([C-F], passage 4). (B) Fluorescent staining of LC3 (red) and LAMP1 (green) in sorted primary MSPCs from the BM of 5-FU-treated mice, cultured overnight on 0.1%-gelatin-coated coverslips. (C) Fluorescent microscopy images of LC3 (red/left), SQSTM1 (=p62; red/right) and DAPI (blue) staining in compact bone-derived MSPCs (p4). The left graph shows the perinuclear distribution of LC3 measured with ImageJ software. SQSTM1 foci were counted with ImageJ software (graph, right). (D) Confocal images of perinuclear LAMP1 (green) and DAPI (blue) staining. The graphs show the perinuclear distribution of LAMP1 and the diameter of the lysosomes designated as feret's diameter measured with ImageJ software. (E) Confocal images of perinuclear colocalization of LAMP1 (green) and LC3 (red) measured by ImageJ software. Colocalization was measured by ImageJ software (PLUGIN: colocalization) and visualized in white. (F) Representative flow cytometry histogram of basal autophagy and quantification of Cyto-ID dye levels (DMFI: Cyto-ID dye level chloroquine treated-Cyto-ID dye level WITHOUT treatment). Data were measured by ImageJ software. Scale bars, 10 µm. *P < .05 (nonparametric Mann-Whitney test: B-F). The results of each panel represent 2 to 3 independent experiments. Each dot represents the measurement of 1 cell. Data are represented as mean ± SEM. Symbol legends are shown in Figure 3A,F.
To assess autophagosome degradation, we performed autophagosome labeling with Cyto-ID and the degradation inhibitor chloroquine.31 Here, we found a strongly reduced autophagic flux (ΔMFI) in MSPCs from 5-FU-treated O5AΔ/Δ mice (Figure 3F). Combined, these findings indicate that autophagosomes and lysosomes form, but autophagy fails in MSPCs from 5-FU-treated O5AΔ/Δ mice.
Enhanced oxidative mitochondrial activity in dysfunctional niche cells
Since damaged mitochondria are eliminated through autophagy,32,33 we then studied whether mitochondria were altered in O5AΔ/Δ MSPCs. Our experiments show that glycolysis, as measured by the extracellular acidification rate (ECAR), is not affected. But the OCR is 1.7-fold higher in MSPCs from 5-FU-treated O5AΔ/Δ mice compared with controls (Figure 4A-B). Elevated OCR corresponded to high levels of ROS, not only in O5AΔ/Δ MSPCs (Figure 4C), but also in ECs and OBCs (supplemental Figure 5D-F). Furthermore, cultured O5AΔ/Δ MSPCs show alterations associated with oxidative stress, such as increased DNA damage (supplemental Figure 5G). But, these changes did not affect proliferation, apoptosis, or surface phenotype of MSPCs from 5-FU-treated O5AΔ/Δ mice (supplemental Figure 5H-J).
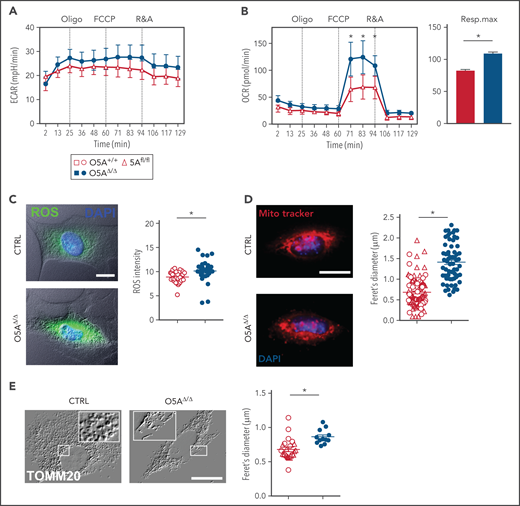
Mitochondria and mitochondrial function in MSPCs (p4) from 5-FU-treated O5AΔ/Δ and control mice. (A-B) Graphs showing 1 experiment out of 2 as an example for extracellular acidification rate (ECAR) (A) and OXPHOS levels measured by oxygen consumption rates (OCR) (B), in cultured MSPCs, respiration maximum (right panel) is shown for MSPCs from both experiments; oligo, oligomycin; FCCP, p-trifluoromethoxyphenylhydrazone; R&A, rotenone and antimycin. (C) Fluorescent microscopy images and intensity of reactive oxygen species (ROS = DCFDA, green) levels and DAPI (blue) in MSPCs. Graph showing pixel intensity measured by ImageJ software. (D) Fluorescent microscopy images and diameter of mitochondria designated as feret's diameter measured by ImageJ software (MitoTrackerRed, red) and DAPI (blue) in MSPCs. (E) Fluorescent microscopy images of mitochondria (TOMM20) in MSPCs illustrated as relief image (Adobe Photoshop: v21.1.1/filter relief) and diameter of mitochondria designated as feret's diameter measured by ImageJ software. Scale bars, 10 µm. *P < .05 (nonparametric Mann-Whitney test; A-E). The results of each panel represent 2 or 3 independent experiments. Data are represented as mean ± SEM. Open symbols and bars represent measurements of control (5Afl/fl and O5A+/+). Closed symbols and bars represent measurements of O5AΔ/Δ MSPCs. Symbol legend shown in Figure 4A.
Mitochondria and mitochondrial function in MSPCs (p4) from 5-FU-treated O5AΔ/Δ and control mice. (A-B) Graphs showing 1 experiment out of 2 as an example for extracellular acidification rate (ECAR) (A) and OXPHOS levels measured by oxygen consumption rates (OCR) (B), in cultured MSPCs, respiration maximum (right panel) is shown for MSPCs from both experiments; oligo, oligomycin; FCCP, p-trifluoromethoxyphenylhydrazone; R&A, rotenone and antimycin. (C) Fluorescent microscopy images and intensity of reactive oxygen species (ROS = DCFDA, green) levels and DAPI (blue) in MSPCs. Graph showing pixel intensity measured by ImageJ software. (D) Fluorescent microscopy images and diameter of mitochondria designated as feret's diameter measured by ImageJ software (MitoTrackerRed, red) and DAPI (blue) in MSPCs. (E) Fluorescent microscopy images of mitochondria (TOMM20) in MSPCs illustrated as relief image (Adobe Photoshop: v21.1.1/filter relief) and diameter of mitochondria designated as feret's diameter measured by ImageJ software. Scale bars, 10 µm. *P < .05 (nonparametric Mann-Whitney test; A-E). The results of each panel represent 2 or 3 independent experiments. Data are represented as mean ± SEM. Open symbols and bars represent measurements of control (5Afl/fl and O5A+/+). Closed symbols and bars represent measurements of O5AΔ/Δ MSPCs. Symbol legend shown in Figure 4A.
We then assessed whether elevated OCR and ROS are associated with a defective mitochondrial clearance in MSPCs from 5-FU-treated O5AΔ/Δ mice. Our experiments showed that mitochondrial mass, number, as well as diameter, in MSPCs from 5-FU-treated O5AΔ/Δ mice were increased (Figure 4D-E). In addition, staining for TOMM20 or the regulator of mitochondrial fission DRP134 showed significant elongation of the mitochondrial network in O5AΔ/Δ MSPCs (Figure 4E; supplemental Figure 5K). This indicates accumulation of mitochondria, reduced mitochondrial fission, and clearance.
Incorrect positioning of critical components of autophagy due to impaired F-actin stress-fiber orientation in MSPCs
The cytoskeleton is critical for correct autophagy.11 Since deletion of Wnt5a in the microenvironment deregulates the cytoskeleton in HSCs,10 we hypothesized that decreased autophagy is associated with deregulated F-actin orientation (Figure 5A). In addition, the GTPase CDC42 is an important regulator of cytoskeletal polymerization and polarization8,10,19 and CDC42 is required for autophagy.35 In line with our hypothesis, we found much less F-actin orientation and perinuclear localization in MSPCs from 5-FU-treated O5AΔ/Δ mice compared with controls (Figure 5B). Moreover, the expression of active GTP-binding CDC42 was strongly increased. Furthermore, while CDC42-GTP localized primarily to cell edges in control MSPCs, it localized mainly to perinuclear regions in MSPCs from O5AΔ/Δ mice (Figure 5C). These results indicate deregulation of F-actin orientation as well as CDC42-GTP localization in MSPCs of 5-FU-treated O5AΔ/Δ mice.
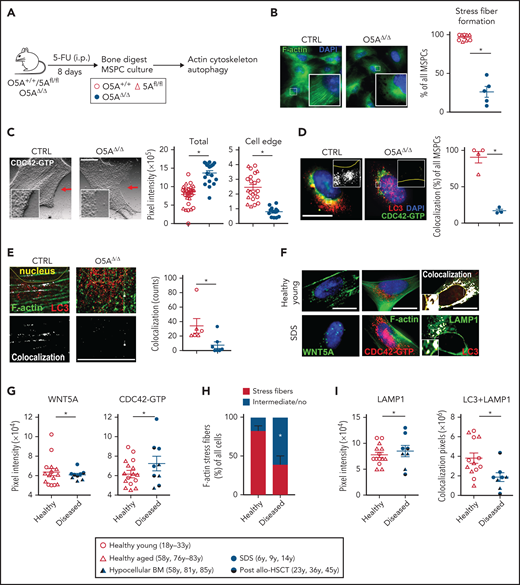
Actin- and CDC42-dependent autophagy in O5AΔ/Δ and human MSPCs. Mouse samples: (A) IP injection of 5-FU in the following genotypes: CTRL: O5A+/+ n = 5, 5Afl/fl n = 7 and O5AΔ/Δ n = 10. Analysis of cytoskeleton-associated proteins and autophagy in compact bone-derived MSPCs, isolated 8 days after 5-FU-treatment and cultured until passage 4. (B) Fluorescent microscopy images of F-actin (Phalloidin/green) and DAPI (blue) staining. The graph shows the results of stress fiber formation in pooled MSPCs of 3 independent experiments. (C) Fluorescent microscopy images of CDC42-GTP in MSPCs illustrated as relief image (Adobe Photoshop: v21.1.1/filter relief). Graphs showing the total pixel intensity of CDC42-GTP (left panel) and the cell edge (right panel) as measured by ImageJ software. (D) Confocal images of CDC42-GTP (green), LC3 (red), and DAPI (blue) in MSPCs. Colocalization was measured by ImageJ software (plugin colocalization) and visualized in white. The graph shows the measurement of colocalization in pooled MSPCs. (E) Fluorescent microscopy images of F-actin (Phalloidin/green) and LC3 (red) in MSPCs. Colocalization was measured by ImageJ software (plug-in: colocalization) and visualized in white. Human samples: (F) Fluorescent microscopy images of cultured human MSPCs (P2) from healthy young donors and SDS patients. Fluorescent staining of WNT5A (green, left), F-actin (green, middle), CDC42-GTP (red, middle), DAPI staining (blue, left and middle), and colocalization of LAMP1 (green, right) and LC3 (red, right) measured by ImageJ software. (G) Graphs showing the protein content of WNT5A (left) and CDC42-GTP (right). (H) Evaluation of the orientation of F-actin stress fibers stained with phalloidin. Cells showing intermediate orientation (for instance, at the cell edge only) or no orientation were taken together (see supplemental Methods). (I) LAMP1 staining (left) and colocalization pixels of LAMP1 and LC3 (right) in MSPCs (P2) from healthy young individuals, n = 7, healthy aged individuals, n = 7, samples from hypocellular BM patients (P_1-3; see supplemental Table 2 for details), n = 3, SDS patients (P_7-9; see supplemental Table 2 for details), n = 3 and post-allo-HSCT patients (P_4-6; see supplemental Table 2 for details), n = 3. Scale bars 10 µm. *P < .05 (nonparametric Mann-Whitney test: B-E,G-I). The results represent 2 to 3 independent experiments. Data are represented as mean ± SEM. Symbol legends shown in Figure 5A and underneath Figure 5H-I.
Actin- and CDC42-dependent autophagy in O5AΔ/Δ and human MSPCs. Mouse samples: (A) IP injection of 5-FU in the following genotypes: CTRL: O5A+/+ n = 5, 5Afl/fl n = 7 and O5AΔ/Δ n = 10. Analysis of cytoskeleton-associated proteins and autophagy in compact bone-derived MSPCs, isolated 8 days after 5-FU-treatment and cultured until passage 4. (B) Fluorescent microscopy images of F-actin (Phalloidin/green) and DAPI (blue) staining. The graph shows the results of stress fiber formation in pooled MSPCs of 3 independent experiments. (C) Fluorescent microscopy images of CDC42-GTP in MSPCs illustrated as relief image (Adobe Photoshop: v21.1.1/filter relief). Graphs showing the total pixel intensity of CDC42-GTP (left panel) and the cell edge (right panel) as measured by ImageJ software. (D) Confocal images of CDC42-GTP (green), LC3 (red), and DAPI (blue) in MSPCs. Colocalization was measured by ImageJ software (plugin colocalization) and visualized in white. The graph shows the measurement of colocalization in pooled MSPCs. (E) Fluorescent microscopy images of F-actin (Phalloidin/green) and LC3 (red) in MSPCs. Colocalization was measured by ImageJ software (plug-in: colocalization) and visualized in white. Human samples: (F) Fluorescent microscopy images of cultured human MSPCs (P2) from healthy young donors and SDS patients. Fluorescent staining of WNT5A (green, left), F-actin (green, middle), CDC42-GTP (red, middle), DAPI staining (blue, left and middle), and colocalization of LAMP1 (green, right) and LC3 (red, right) measured by ImageJ software. (G) Graphs showing the protein content of WNT5A (left) and CDC42-GTP (right). (H) Evaluation of the orientation of F-actin stress fibers stained with phalloidin. Cells showing intermediate orientation (for instance, at the cell edge only) or no orientation were taken together (see supplemental Methods). (I) LAMP1 staining (left) and colocalization pixels of LAMP1 and LC3 (right) in MSPCs (P2) from healthy young individuals, n = 7, healthy aged individuals, n = 7, samples from hypocellular BM patients (P_1-3; see supplemental Table 2 for details), n = 3, SDS patients (P_7-9; see supplemental Table 2 for details), n = 3 and post-allo-HSCT patients (P_4-6; see supplemental Table 2 for details), n = 3. Scale bars 10 µm. *P < .05 (nonparametric Mann-Whitney test: B-E,G-I). The results represent 2 to 3 independent experiments. Data are represented as mean ± SEM. Symbol legends shown in Figure 5A and underneath Figure 5H-I.
LC3 interacts with and regulates CDC42.36 Thus, we explored whether LC3 colocalized with active CDC42-GTP and F-actin. Confocal images revealed punctate LC3 in the perinuclear regions of the O5AΔ/Δ MSPCs (Figure 3C). In O5AΔ/Δ MSPC, the LC3+ punctae are only marginally colocalized with either CDC42-GTP or F-actin compared with the MSPCs from control mice (Figure 5D-E), indicating diminished anchoring of LC3+ autophagosomes to the F-actin-cytoskeleton in O5AΔ/Δ MSPCs.
CDC42 activation and autophagy defects in MSPCs from human BM failure states
We then studied whether MSPCs from patients sharing features of peripheral cytopenias and hypocellular BM with the twice- 5-FU-treated O5AΔ/Δ mice showed similar alterations in the cytoskeleton and autophagy. For this, MSPCs were cultured from BM samples from patients with different BM failure states, such as SDS, an inherited BM failure syndrome, hypocellular MDS, or severe aplastic anemia. We also included 3 samples from leukemia/lymphoma patients undergoing allogeneic HSCT. Two patients had previously been conditioned with total body irradiation, which may contribute to niche damage, and the third patient showed incomplete BM reconstitution HSCT (likely due to extensive pretreatment) (supplemental Table 2). Interrogation of published gene expression data in freshly isolated mesenchymal cells from MDS vs normal controls revealed that whereas all control BM samples (10 of 10) express WNT5A, it is not detected in 33 of 45 MDS BM samples, and some actin-regulating intermediates are reduced (supplemental Figure 6A).37 In IF experiments, we subsequently found that expression of WNT5A or CDC42-GTP and colocalization of LC3/LAMP1 is similar in MSPCs from aged and young BM controls (supplemental Figure 6B), indicating their altered expression or colocalization is not due to aging. In comparison, staining of MSPCs from patients with the common features of peripheral cytopenia and BM hypocellularity revealed a slight but significant reduction of WNT5A, a clear increase of activated CDC42, and reduced F-actin stress fiber orientation (Figure 5F-H). Furthermore, LAMP1 is elevated with reduced LC3/LAMP1 colocalization (Figure 5I), indicative of cytoskeleton and autophagy defects.
Pharmacological attenuation of CDC42-GTP levels in vitro restores autophagy in MSPCs
Our data imply that cytostatic stress causes sustained CDC42 activation associated with deregulation of cytoskeleton-associated autophagy in MSPCs from cytopenic mice (Figure 5B-E). We consequently reasoned that inhibiting CDC42 activation would directly improve MSPC function. To test this hypothesis, we treated mouse MSPCs in vitro with the RhoGDI/CDC42 activation inhibitor CASIN (also described as Pirl1),8,38-41 or the allosteric CDC42 inhibitor ML14142,43 (supplemental Figure 7A-B). Both inhibitors reduced the cellular level of CDC42-GTP in O5AΔ/Δ MSPCs equally well. However, only CASIN restored CDC42-GTP localization at the cell edge and F-actin stress fibers in MSPCs from 5-FU-treated O5AΔ/Δ mice (supplemental Figure 7C-D).
To test whether CASIN improved autophagy, we compared treatments with CASIN, rapamycin, a known autophagy inducer, or chloroquine, a lysosomal lysis inhibitor (supplemental Figure 7E). These experiments showed that both rapamycin and CASIN increase LC3+ autophagosome formation without affecting LAMP1 expression (supplemental Figure 7F-H). Importantly, both rapamycin and CASIN increase colocalization of LC3 and LAMP1 (supplemental Figure 7I), indicating CASIN directly stimulates autophagy in MSPCs.
To determine whether the extrinsic WNT5A signal similarly restores CDC42 activation, recombinant WNT5A was added to MSPC cultures. These experiments show that rWNT5A reduces both LC3 and LAMP1, as well as their colocalization (supplemental Figure S7J-M), indicating that rWNT5A also directly improves autophagy.
Pharmacological attenuation of CDC42-GTP levels in vivo protects against stress-induced mortality
To appraise whether attenuation of CDC42 activation also restores niche function and hematopoiesis in vivo, we serially treated O5AΔ/Δ and control mice with 5-FU on day 0 and day 8 and included an additional treatment with CASIN41 (Figure 6A; supplemental Figure 8A). Strikingly, the application of CASIN protected O5AΔ/Δ mice from mortality induced by the second 5-FU-treatment (Figure 6B). In addition, we did not detect PB nor BM cytopenia with the rescue of both mature and immature hematopoietic cells (Figure 6C-J), particularly of LT-HSCs and BM T-cells (Figure 6E-G), in the CASIN-treated O5AΔ/Δ mice.
![Attenuation of elevated CDC42 activity ensures survival. (A) Serial IP injection of 5-FU in the following genotypes: CTRL: O5A+/+ n = 6, 5Afl/fl n = 7 and O5AΔ/Δ n = 7 at day 0 and day 8. Additional in vivo IP injection of CDC42-GTP inhibitor CASIN at day 5 through day 8. Analysis of animal survival, hematopoiesis (BM from 4 flushed long bones), and compact bone-derived MSPCs at day 14. (B) Percentage of mice survival after serial 5-FU treatment. Graph shows the survival curve of CTRL and O5AΔ/Δ mice treated with CASIN (+) or vehicle (-). (C) Graph shows the total BM cell number (left) and relative number of LSK cells (middle) and MPs (right) at day 14 of CTRL mice with vehicle ([-]; O5A+/+ n = 6, 5Afl/fl n = 7), O5AΔ/Δ mice with vehicle ([-]; n = 5, mice analyzed shortly before death) and O5AΔ/Δ mice with CASIN ([+]; n = 5). (D,F,H,J) Representative contour plots from BM populations. Graphs show relative number of LT-HSCs and ST-HSCs (E-F), T cells and B cells (G-H), and Gr1+ myeloid cells (I-J). *P < .05 (nonparametric Mann-Whitney test: C,E,G,I). The results represent 2 to 3 independent experiments. Data are represented as mean ± SEM. Symbol legend shown in Figure 6A.](https://ash.silverchair-cdn.com/ash/content_public/journal/blood/139/5/10.1182_blood.2021011775/7/m_bloodbld2021011775f6.png?Expires=1769116449&Signature=m9oof1QnRd05TI7GNPC2Si4z5-rHpBPJLpoOa5x5Kh4OAd5IOpAQl3kEpKMek5p6B6fVjbcSEpfKyCCo1-hDulRqRVy7G8Lz7GYI6Pv3g5b3Lr6vnstOeu2eCO0ljW8iWRe8DOhn5hAwZhyozzZtuXQjeZ7pbgblWzXIvPuxIJZZRLlxzU0C91QuhwemVuxHKouFuy4HWsbOs3yVT-kXzaZRxaQNo6OCPyUZmdIcBcEKiLDvT~zhd4KNazv58B4AV4NDmQQKOcHUH3Fu9Xz1p2kdVVwqMa2CNo6SjVey36QVpaO3u5z4WVS9TmIzZdNs38k9X1~CaB5b0AxQYdY1qQ__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
Attenuation of elevated CDC42 activity ensures survival. (A) Serial IP injection of 5-FU in the following genotypes: CTRL: O5A+/+ n = 6, 5Afl/fl n = 7 and O5AΔ/Δ n = 7 at day 0 and day 8. Additional in vivo IP injection of CDC42-GTP inhibitor CASIN at day 5 through day 8. Analysis of animal survival, hematopoiesis (BM from 4 flushed long bones), and compact bone-derived MSPCs at day 14. (B) Percentage of mice survival after serial 5-FU treatment. Graph shows the survival curve of CTRL and O5AΔ/Δ mice treated with CASIN (+) or vehicle (-). (C) Graph shows the total BM cell number (left) and relative number of LSK cells (middle) and MPs (right) at day 14 of CTRL mice with vehicle ([-]; O5A+/+ n = 6, 5Afl/fl n = 7), O5AΔ/Δ mice with vehicle ([-]; n = 5, mice analyzed shortly before death) and O5AΔ/Δ mice with CASIN ([+]; n = 5). (D,F,H,J) Representative contour plots from BM populations. Graphs show relative number of LT-HSCs and ST-HSCs (E-F), T cells and B cells (G-H), and Gr1+ myeloid cells (I-J). *P < .05 (nonparametric Mann-Whitney test: C,E,G,I). The results represent 2 to 3 independent experiments. Data are represented as mean ± SEM. Symbol legend shown in Figure 6A.
Attenuation of elevated CDC42 activity ensures survival. (A) Serial IP injection of 5-FU in the following genotypes: CTRL: O5A+/+ n = 6, 5Afl/fl n = 7 and O5AΔ/Δ n = 7 at day 0 and day 8. Additional in vivo IP injection of CDC42-GTP inhibitor CASIN at day 5 through day 8. Analysis of animal survival, hematopoiesis (BM from 4 flushed long bones), and compact bone-derived MSPCs at day 14. (B) Percentage of mice survival after serial 5-FU treatment. Graph shows the survival curve of CTRL and O5AΔ/Δ mice treated with CASIN (+) or vehicle (-). (C) Graph shows the total BM cell number (left) and relative number of LSK cells (middle) and MPs (right) at day 14 of CTRL mice with vehicle ([-]; O5A+/+ n = 6, 5Afl/fl n = 7), O5AΔ/Δ mice with vehicle ([-]; n = 5, mice analyzed shortly before death) and O5AΔ/Δ mice with CASIN ([+]; n = 5). (D,F,H,J) Representative contour plots from BM populations. Graphs show relative number of LT-HSCs and ST-HSCs (E-F), T cells and B cells (G-H), and Gr1+ myeloid cells (I-J). *P < .05 (nonparametric Mann-Whitney test: C,E,G,I). The results represent 2 to 3 independent experiments. Data are represented as mean ± SEM. Symbol legend shown in Figure 6A.
To find out whether in vivo attenuation of CDC42 activation maintains BM niche cell numbers and function, we found that the relative number of MSPCs was reverted to control levels in MSPCs from CASIN- and twice-5-FU-treated O5AΔ/Δ mice (supplemental Figure 8B-C). In addition, we observed similar MSPC differentiation in vitro (supplemental Figure 8D). In MSPCs from O5AΔ/Δ mice treated twice with 5-FU, CASIN treatment restored both CDC42-GTP localization to the cell edge and F-actin stress fiber orientation (supplemental Figure 8E-F).
Rescue of actin-guided autophagy and mitochondrial clearance in MSPCs
In experiments to determine whether in vivo attenuation of CDC42 activation prevents the decline of the F-actin-anchored autophagy in MSPCs prior to the second 5-FU treatment, we found that in vivo CASIN treatment reduces the levels and perinuclear localization of LC3 in O5AΔ/Δ MSPCs (Figure 7A-B) compared with controls (Figure 3B-C). Moreover, LC3 colocalizes with both CDC42-GTP and F-actin fibers (Figure 7C-D), indicating normalized cytoskeletal deregulation and actin cytoskeleton anchoring in MSPCs from 5-FU-treated O5AΔ/Δ mice. In experiments to determine whether the autophagy defects noted earlier (Figure 3) were also normalized, we found that both LAMP1 content and lysosome diameters were restored (Figure 7E). More importantly, LC3 and LAMP1 also colocalized (Figure 7F), indicating restored autophagy in MSPCs from 5-FU-treated O5AΔ/Δ mice. Indeed, we found that autophagic flux was substantially increased in MSPCs by prior in vivo CASIN treatment (Figure 7G). In addition, both reduction in ROS and MitoTracker Red levels and colocalization of LC3 and TOMM20 (Figure 7H; supplemental Figure 9A-C) indicate improved mitophagy after in vivo CASIN treatment.
![Attenuation of elevated CDC42 activity during auto/mitophagy. (A) IP injection of 5-FU in O5AΔ/Δ mice at day 0. Additional in vivo IP injection of vehicle ([-], n = 5) or CASIN ([+], n = 4) at day 5 through day 8. Rescue and analysis of autophagy relevant mechanisms in compact bone-derived MSPCs, isolated at day 8 and cultured until passage 4. (A-G) Shown are the results of O5AΔ/Δ mice with vehicle (-) and CASIN (+) treatment. CASIN-treated mice show the same phenotype as the control groups 5Afl/fl and O5A+/+ (Figure 3). (B) Fluorescent microscopy images of LC3 (red) and DAPI (blue) staining. Graph shows the pixel intensity as measured by ImageJ software. (C) Fluorescent microscopy image of CDC42-GTP (green), LC3 (red), and DAPI (blue) staining. The graph shows the percentage of MSPCs with colocalization measured by ImageJ software (plugin colocalization) and visualized in white. (D) Fluorescent microscopy images of F-actin (Phalloidin/green) and LC3 (red) in MSPCs. Colocalization was measured by ImageJ software (plug-in colocalization) and visualized in white. The graph shows colocalization counted with ImageJ software. (E) Fluorescent microscopy images of LAMP1 (green) and DAPI (blue) staining. The pixel intensity (left graph) and feret’s diameter (right graph) were measured by ImageJ software. (F) Fluorescent microscopy images of LAMP1 (green), LC3 (red), and DAPI (blue) staining. Yellow arrows show colocalized vesicle staining, red arrows depict LC3+ vesicles that did not colocalize with green LAMP1+ vesicles. Perinuclear colocalization of LAMP1 and LC3 measured by ImageJ software (plugin colocalization, depicted in white). (G) Representative FACS plots and quantification (graph) of Cyto-ID dye levels (DMFI: Cyto-ID dye level chloroquine treated, Cyto-ID dye level w/o treatment). (H) Fluorescent microscopy images of TOMM20 (green), LC3 (red), and DAPI (blue) staining. Colocalization was measured by ImageJ software (plugin colocalization) and visualized in the bottom row in white. Scale bars, 10 µm. *P < .05 (2-sided parametric Student’s t test; B-H). The results represent 2 independent experiments. Data are represented as mean ± SEM. Symbol legend shown in Figure 7A.](https://ash.silverchair-cdn.com/ash/content_public/journal/blood/139/5/10.1182_blood.2021011775/7/m_bloodbld2021011775f7.png?Expires=1769116449&Signature=nKJihs~iWQxd8l6ednzSrsUao9Tx8xpMpUWGWUTL94l4CkIFzoJxk2fZs2XhZUiwqkCpwJKW2l0~9rz9GahbQajABhnCd4rVnEDC5FolngkW-6~eSQZg-Zr6B2TU6hyvocjoy8o3FsYmNlKjqRNFN1IgsHbtr7ITolayyBlYFdkojweK-gM8jYJPxmHYsEM1KSbeFl3XaxRKjE3dJ59CmRsnhO-Ouam~kj6q2S8-XwrpJ53k9RgjqviCrKPCIVTT8Fq~wVfcYRfrknxdOphakQouz-nR4GwXo10QDcgJWvJjtz1JZcgvPUiT2BcuXJY5dJHqNpegVRqTqeXv8tLMKQ__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
Attenuation of elevated CDC42 activity during auto/mitophagy. (A) IP injection of 5-FU in O5AΔ/Δ mice at day 0. Additional in vivo IP injection of vehicle ([-], n = 5) or CASIN ([+], n = 4) at day 5 through day 8. Rescue and analysis of autophagy relevant mechanisms in compact bone-derived MSPCs, isolated at day 8 and cultured until passage 4. (A-G) Shown are the results of O5AΔ/Δ mice with vehicle (-) and CASIN (+) treatment. CASIN-treated mice show the same phenotype as the control groups 5Afl/fl and O5A+/+ (Figure 3). (B) Fluorescent microscopy images of LC3 (red) and DAPI (blue) staining. Graph shows the pixel intensity as measured by ImageJ software. (C) Fluorescent microscopy image of CDC42-GTP (green), LC3 (red), and DAPI (blue) staining. The graph shows the percentage of MSPCs with colocalization measured by ImageJ software (plugin colocalization) and visualized in white. (D) Fluorescent microscopy images of F-actin (Phalloidin/green) and LC3 (red) in MSPCs. Colocalization was measured by ImageJ software (plug-in colocalization) and visualized in white. The graph shows colocalization counted with ImageJ software. (E) Fluorescent microscopy images of LAMP1 (green) and DAPI (blue) staining. The pixel intensity (left graph) and feret’s diameter (right graph) were measured by ImageJ software. (F) Fluorescent microscopy images of LAMP1 (green), LC3 (red), and DAPI (blue) staining. Yellow arrows show colocalized vesicle staining, red arrows depict LC3+ vesicles that did not colocalize with green LAMP1+ vesicles. Perinuclear colocalization of LAMP1 and LC3 measured by ImageJ software (plugin colocalization, depicted in white). (G) Representative FACS plots and quantification (graph) of Cyto-ID dye levels (DMFI: Cyto-ID dye level chloroquine treated, Cyto-ID dye level w/o treatment). (H) Fluorescent microscopy images of TOMM20 (green), LC3 (red), and DAPI (blue) staining. Colocalization was measured by ImageJ software (plugin colocalization) and visualized in the bottom row in white. Scale bars, 10 µm. *P < .05 (2-sided parametric Student’s t test; B-H). The results represent 2 independent experiments. Data are represented as mean ± SEM. Symbol legend shown in Figure 7A.
Attenuation of elevated CDC42 activity during auto/mitophagy. (A) IP injection of 5-FU in O5AΔ/Δ mice at day 0. Additional in vivo IP injection of vehicle ([-], n = 5) or CASIN ([+], n = 4) at day 5 through day 8. Rescue and analysis of autophagy relevant mechanisms in compact bone-derived MSPCs, isolated at day 8 and cultured until passage 4. (A-G) Shown are the results of O5AΔ/Δ mice with vehicle (-) and CASIN (+) treatment. CASIN-treated mice show the same phenotype as the control groups 5Afl/fl and O5A+/+ (Figure 3). (B) Fluorescent microscopy images of LC3 (red) and DAPI (blue) staining. Graph shows the pixel intensity as measured by ImageJ software. (C) Fluorescent microscopy image of CDC42-GTP (green), LC3 (red), and DAPI (blue) staining. The graph shows the percentage of MSPCs with colocalization measured by ImageJ software (plugin colocalization) and visualized in white. (D) Fluorescent microscopy images of F-actin (Phalloidin/green) and LC3 (red) in MSPCs. Colocalization was measured by ImageJ software (plug-in colocalization) and visualized in white. The graph shows colocalization counted with ImageJ software. (E) Fluorescent microscopy images of LAMP1 (green) and DAPI (blue) staining. The pixel intensity (left graph) and feret’s diameter (right graph) were measured by ImageJ software. (F) Fluorescent microscopy images of LAMP1 (green), LC3 (red), and DAPI (blue) staining. Yellow arrows show colocalized vesicle staining, red arrows depict LC3+ vesicles that did not colocalize with green LAMP1+ vesicles. Perinuclear colocalization of LAMP1 and LC3 measured by ImageJ software (plugin colocalization, depicted in white). (G) Representative FACS plots and quantification (graph) of Cyto-ID dye levels (DMFI: Cyto-ID dye level chloroquine treated, Cyto-ID dye level w/o treatment). (H) Fluorescent microscopy images of TOMM20 (green), LC3 (red), and DAPI (blue) staining. Colocalization was measured by ImageJ software (plugin colocalization) and visualized in the bottom row in white. Scale bars, 10 µm. *P < .05 (2-sided parametric Student’s t test; B-H). The results represent 2 independent experiments. Data are represented as mean ± SEM. Symbol legend shown in Figure 7A.
Discussion
Understanding mechanisms involved in the hematopoiesis-supportive function of MSPCs is crucial for developing new treatments for debilitating hematologic diseases. Our present study shows that autophagy controlled by CDC42 is a critical factor for maintaining hematopoietic support by BM niche cells during stress. Furthermore, without Wnt5a in osteoprogenitors, CDC42 is constitutively active in MSPCs, which leads to secondary ineffective hematopoiesis and increased mortality after severe stress.
Our experiments show that regulation of CDC42 directly modulates F-actin and associated autophagic flux in niche cells. We found that only the RhoGDI/CDC42 association inhibitor CASIN, but not the allosteric inhibitor ML-141, restores cytoskeletal defects and CDC42-GTP localization. Thus, only RhoGDI/CDC42 attenuation effectively improves cellular processes resulting in healthier mice. Indeed, a similar 4-day treatment of aged mice with CASIN shows not only improved mouse health, it also extended their lifespan significantly.44
Interestingly, we found that similar changes in F-actin and autophagy also occur in MSPCs from patients sharing features of peripheral cytopenia and hypocellular BM, such as in different BM failure states, suggesting a possible role in their pathogenesis. Our findings add to reports showing that MSPCs from these diseases are defective in CFU-F content, show deregulated differentiation,3,37,45,46 and demonstrate defects in maintaining HSCs and progenitor cells.44,47-49 However, it remains elusive why and how CDC42 signaling, the cytoskeleton, or autophagy in MSPCs causes secondary ineffective hematopoiesis. Findings from SDS and like diseases showed that mutated SBDS and SRP54, which cause these syndromes, are both not only expressed by niche cells,50 but these proteins also bind to F-actin and deregulate small GTPase activation.51,52 Similar deregulation of these pathways may occur in MSPCs, also from other BM failure states. Alternatively, the processes mentioned may be linked indirectly through reduced mitophagy, causing oxidative injury33 or deregulation of autophagy-dependent secretion of cytokines.53,54
In summary, autophagy, cytoskeletal orientation, and CDC42 activation in niche cells are possible targets for improving inefficient hematopoiesis after severe stress. Furthermore, we provide a rationale for extending our findings in the O5AΔ/Δ mouse model to human diseases featuring peripheral cytopenia and hypocellular BM. In addition, our work offers attenuation of CDC42 activation in vivo as a therapeutic strategy to improve the homeostasis of the hematopoietic BM niche.
Acknowledgments
The authors thank Hannah Kitzberger and Tommaso di Genio (Department of Internal Medicine III - Hematology/Oncology, Technical University of Munich) for excellent help in some of the experiments. The authors thank Christian Landspersky for graphical illustration of animals. The authors also thank L. Henkel, I. Andrä, C. Angerpointner, and S. Dürr (Institute of Microbiology, Immunology, and Hygiene, Technical University of Munich) for cell sorting and help with fluorescent microscopy. Furthermore, the authors thank H. Harz at the Center for Advanced Light Microscopy (CALM, Ludwig-Maximilian University) and Ritu Mishra (Cell Analysis Core Facility, TranslaTUM), as well as L. Chen and M.H.G.P. Raaijmakers (Department of Hematology, Erasmus University, Rotterdam, Netherlands) for sharing data. Special thanks to C. Peschel (Clinic and Policlinic for Internal Medicine III, Klinikum rechts der Isar, Technical University of Munich) for support of this project.
This work was funded by the German Research Foundation (DFG) grants FOR 2033 (A2 and B3), SFB 1243 (A01, A04, and A09), and OO 8/16 and 8/18.
Authorship
T.L., C.S., J.S.H., K.S.G., H.G., and R.A.J.O. designed research; T.L., C.S., M.S., E.H., K.B., F.H., R.I., S.R.M., J.R., J.S.H., R.L., M.G., M.B. and M.W. performed research and collected data; M.S., J.R., D.S., A.S., K.M., T.P.Y., M.K., H.L., F.B., K.S.G and H.G. contributed vital reagents and analyticaltools; T.L., M.S., C.S., J.R., J.S.H., K.S.G., and R.A.J.O. analyzed and interpreted data and performed statistical analyses; and T.L., C.S., and R.A.J.O. wrote the manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
The current affiliation for R.I. is Technical University of Munich (TUM), School of Medicine, Department of Surgery, Munich, Germany.
Correspondence: Christina Schreck, Technical University of Munich, Klinikum rechts der Isar, Department of Internal Medicine III, Ismaningerstrasse 22, 81675 München, Germany; e-mail: christina.schreck@tum.de; and Robert A. J. Oostendorp, Technical University of Munich, Klinikum rechts der Isar, Department of Internal Medicine III, Ismaningerstrasse 22, 81675 München, Germany; e-mail: robert.oostendorp@tum.de.
The online version of this article contains a data supplement.
There is a Blood Commentary on this article in this issue.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal