

In this issue of Blood, reveal an intriguing link between cytoskeletal polarity as a regulator of autophagy in mesenchymal stem and progenitor cells (MSPCs) and the hematopoietic stress response.1
Microenvironmental niches, composed of hematopoietic stem cells (HSCs) and stromal elements, are the core units of hematopoiesis. Within these niches, HSCs have evolved a range of mechanisms to cope with stress, including autophagy, as a way to sustain nutrient and metabolic pools and remove damaged organelles such as mitochondria (mitophagy).2 An intricate relationship between autophagy, stemness, and self-renewal also exists for MSPCs.3
Landspersky et al demonstrate the unexpected impact of Wnt5a-CDC42–dependent autophagy in MSPCs on HSC self-renewal. Prior work by the group revealed that loss of Wnt5a signaling in the bone marrow niche deregulates cytoskeletal polarity through activation of the small GTPase CDC42 and thereby impairs homing and repopulation by adoptively transferred HSCs.4 The investigators discovered that F-actin polymerization also directs autophagosome-lysosome fusion and mitophagy in niche cells. In the current study, they test the impact of dysregulated Wnt5a-CDC42 in niche MSPCs on endogenous HSC function at homeostasis and under stress. In a series of experiments using a mouse model with Wnt5a loss restricted to Osterix+ stromal cells, they show that 5-fluorouracil (5-FU) treatment prompts CDC42 hyperactivation and leads to several predictable outcomes: a disorganized MSPC cytoskeleton with autophagosome accumulation, high levels of reactive oxygen species (ROS), and defective mitochondrial metabolism. Most striking are the phenotypic outcomes under stress, whereby repeated 5-FU administration depresses and ultimately exhausts the HSC pool. Remarkably, pharmacologic inhibition of CDC42 using CASIN reverses most of these end points and restores hematopoietic resilience under stress. Finally, the authors were able to test a small, heterogeneous population of patient samples to reproduce some of their findings.
The study raises a series of intriguing questions. Evidently, actin dynamics and cytoskeletal scaffolds are critical factors in multiple stages of autophagy, but how do metabolic impairment and loss of stromal cell polarity ultimately deplete neighboring HSCs?
Collective data support the importance of contact-dependent signals in determining asymmetric/polar and symmetric/nonpolar HSC division, thus guiding cell fates of self-renewal and exhaustion (see figure). Accordingly, loss of MSPC polarity and the corresponding HSC membrane domain organization may prevent proper attachment and orientation in the niche, shifting divisional symmetry and HSC anchorage in the niche under stress.5
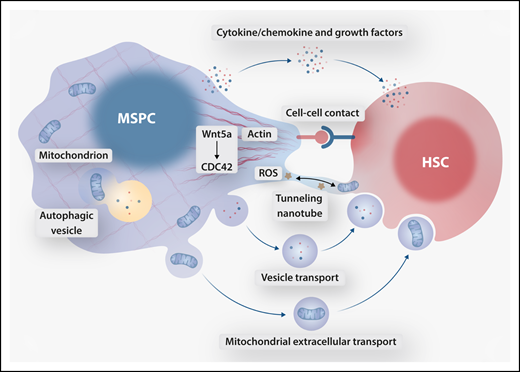
Proposed mechanisms of crosstalk between MSPCs and HSCs downstream of Wnt5a-CDC42–dependent actin organization. MSPC cytoskeletal polarity facilitates cell contact–dependent HSC orientation and bidirectional tunneling nanotube-mediated transfer of ROS and mitochondria. Cytoskeleton-dependent secretory autophagy contributes to the MSPC secretome, in part regulating release of freely secreted HSC-supportive factors, extracellular vesicles, and vesicle-bound mitochondria. Professional illustration by Somersault 18:24.
Proposed mechanisms of crosstalk between MSPCs and HSCs downstream of Wnt5a-CDC42–dependent actin organization. MSPC cytoskeletal polarity facilitates cell contact–dependent HSC orientation and bidirectional tunneling nanotube-mediated transfer of ROS and mitochondria. Cytoskeleton-dependent secretory autophagy contributes to the MSPC secretome, in part regulating release of freely secreted HSC-supportive factors, extracellular vesicles, and vesicle-bound mitochondria. Professional illustration by Somersault 18:24.
Several other possibilities exist that do not rely on cell-cell contact (see figure). HSC stress response is likely dependent upon supportive cytokines, chemokines, or angiocrine factors secreted by MSPCs; disruption to the MSPC secretome may spell trouble for retaining the regenerative reserve. Furthermore, existence of lysosomal exocytosis and secretory autophagy illustrates the intricate connection between degradative and secretory functions that likely play crucial roles in endogenous (ie, MSPC) cell maintenance as well as intercellular crosstalk with HSCs. Does disrupted cytoskeletal scaffolding promote the release of a toxic MSPC secretome or alternatively mediate the loss of HSC-supportive secreted factors? Such a sequence of events is supported by the canonical requirement of actin cytoskeleton in compartmental (autophagic and endolysosomal) recycling and extracellular release of metabolites.6 It is conceivable that disruption of normal secretory autophagy in MSPCs alters extrinsic stimuli delivered to HSCs in the niche.
Across different tissues, senescence is a common stress response, and a dual role for autophagy as a positive or negative regulator of MSPC senescence has been described. In steady-state conditions, autophagy may suppress MSPC senescence by clearing impaired organelles and reducing ROS production. On the other hand, replicative or genotoxic stress leading to DNA damage provides cues for MSPCs to initiate a senescence trajectory. In this setting, autophagic processes sustain cell viability by counteracting proteostasis disruption and eliminating toxic byproducts, resulting in a senescence-associated secretory phenotype (SASP).3 Although chronic exposure to the MSPC SASP secretome ultimately impairs HSC clonogenicity,7 these proinflammatory factors may be necessary to support canonical HSC response to acute stress, as evaluated in the current study.
Finally, recent observations of mitochondrial organelle transport raise the possibility of metabolic dependence between neighboring cells in the niche during the HSC stress response. Transfer of viable mitochondria within secreted microvesicles from MSPCs restores aerobic respiration-deficient cells in culture,8 while healthy HSCs have been demonstrated to transfer functional mitochondria to mesenchymal stromal cells in a post-irradiation setting.9 Tunneling nanotubes also mediate transfer of ROS from HSCs to stromal cells, preventing HSC stress-induced senescence.10 Bidirectional transfer of mitochondria and ROS in the niche may be an unconventional but plausible coping mechanism during stress.
Beyond a compelling mechanism that links the structural and metabolic capacities of MSPCs with HSC function, the current study leaves open questions surrounding the broader translational significance and application to clinically relevant disease states or regenerative conditions. For instance, could the Wnt5a-CDC42 axis be generally exploited in bone marrow failure states, including inflammatory stress, aging, or recovery from myelotoxic treatments? With CASIN likely to have more pleiotropic effects, can other regulators of Wnt5a-CDC42 restore actin polarity and subsequently reestablish controlled autophagy in MSPCs? Broader appreciation of actin-dependent regulation of MSPC autophagy may provide key biologic insights and clinically relevant approaches to restore regenerative hematopoiesis after bone marrow injury.
Conflict-of-interest disclosure: The authors declare no competing financial interests.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal