

In this issue of Blood, identify a cascade of protein tyrosine phosphatases, including SHP2, that contribute to the clinical challenge of ALK inhibitor resistance in ALK+ anaplastic large-cell lymphoma (ALCL).1 In stark contrast to the persistent challenges posed by the more prevalent peripheral T-cell lymphomas, the current standard of care (ie, brentuximab vedotin, cyclophosphamide, doxorubicin, and prednisone) is curative in ∼90% of adult patients with ALK+ ALCL.2
Given ALK’s primary oncogenic role in these lymphomas, ∼90% of patients with recurrent disease may anticipate achieving a response to the ALK inhibitor (ALKi) crizotinib, some of which are durable.3 However, ALK inhibition is not curative; recurrences are observed upon treatment discontinuation4 and both primary and acquired resistance to ALKi pose a significant challenge. ALK mutations,5 amplifications,6 and the activation of alternative pathways,5,7 including cytokine (ie, interleukin-10)-dependent activation of STAT3,8 all contribute to this challenge. Furthermore, each mechanism of ALKi resistance demands an alternative response, ranging from the use of an alternative ALKi to cessation of ALKi therapy or the initiation of an alternative targeted agent altogether.
In their study, Karaca-Ataby et al contribute to our understanding of crizotinib resistance by performing an unbiased, genome-scale CRISPR-Cas9 screen in crizotinib-treated cells. In addition to identifying known dependencies (eg, c-myc, STAT3) beyond ALK, they identified 2 protein tyrosine phosphatases (PTPN1 and PTPN2), loss of which culminate in crizotinib resistance. These protein tyrosine phosphatases negatively regulate multiple kinases and pathways with a pathogenic role across the spectrum of the peripheral T-cell lymphomas, including the Src family kinases and the JAK/STAT pathway. Mechanistically, PTPN1/PTPN2 were shown to bind and dephosphorylate NPM-ALK, and loss of these protein tyrosine phosphatases further led to a substantial increase in STAT3, ERK1/2, and SHP2 phosphorylation. Furthermore, STAT3, a downstream target of oncogenic NPM-ALK signaling, was shown to bind the PTPN1 promoter and transcriptionally repress its expression. In addition, SHP2 (PTPN11), dephosphorylation of which was predominantly regulated by PTPN1, was further scrutinized because this oncogenic phosphatase cooperates with tyrosine-kinase driven malignancies, and allosteric SHP2 inhibitors are in clinical development and early-phase clinical trials.9 Importantly, they showed that SHP2 activation not only contributes to the challenge of ALKi resistance, but further demonstrate significant synergy between allosteric SHP2 and ALK inhibitors. Given the diverse kinases regulated by these protein tyrosine phosphatases, Karaca-Ataby et al correctly suggest that their findings may be the tip of the iceberg, and that future studies, including those in patients with ALKi-resistant disease, are warranted. Nonetheless, given the availability of both SHP2 inhibitors and STAT3 degraders with activity in ALK+ ALCL,10 their findings have significant therapeutic implications (see figure).
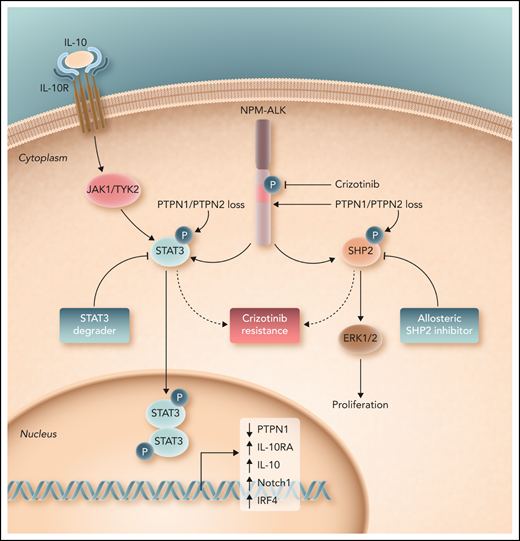
Loss of the protein tyrosine phosphatases PTPN1 and PTPN2 promote ALK-dependent phosphorylation and activation of STAT3 and SHP2, both of which promote resistance to crizotinib in ALK+ ALCL. Consequently, as demonstrated by Karaca-Ataby et al, allosteric SHP2 inhibitors may be rationally combined with crizotinib in ALK+ ALCL. Professional illustration by Somersault 18:24.
Loss of the protein tyrosine phosphatases PTPN1 and PTPN2 promote ALK-dependent phosphorylation and activation of STAT3 and SHP2, both of which promote resistance to crizotinib in ALK+ ALCL. Consequently, as demonstrated by Karaca-Ataby et al, allosteric SHP2 inhibitors may be rationally combined with crizotinib in ALK+ ALCL. Professional illustration by Somersault 18:24.
Conflict-of-interest disclosure: The author declares no competing financial interests.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal