

Key Points
Systems-level immune profiling reveals perturbed NK-cell homeostasis in a subgroup of patients with chronic severe-grade neutropenia.
Coordinated transcriptional and proteomic changes in apoptotic pathways and cell turnover underlie defective NK-cell homeostasis.

Abstract
Neutrophils have been thought to play a critical role in terminal differentiation of NK cells. Whether this effect is direct or a consequence of global immune changes with effects on NK-cell homeostasis remains unknown. In this study, we used high-resolution flow and mass cytometry to examine NK-cell repertoires in 64 patients with neutropenia and 27 healthy age- and sex-matched donors. A subgroup of patients with chronic neutropenia showed severely disrupted NK-cell homeostasis manifesting as increased frequencies of CD56bright NK cells and a lack of mature CD56dim NK cells. These immature NK-cell repertoires were characterized by expression of the proliferation/exhaustion markers Ki-67, Tim-3, and TIGIT and displayed blunted tumor target cell responses. Systems-level immune mapping revealed that the changes in immunophenotypes were confined to NK cells, leaving T-cell differentiation intact. RNA sequencing of NK cells from these patients showed upregulation of a network of genes, including TNFSF9, CENPF, MKI67, and TOP2A, associated with apoptosis and the cell cycle, but different from the conventional CD56bright signatures. Profiling of 249 plasma proteins showed a coordinated enrichment of pathways related to apoptosis and cell turnover, which correlated with immature NK-cell repertoires. Notably, most of these patients exhibited severe-grade neutropenia, suggesting that the profoundly altered NK-cell homeostasis was connected to the severity of their underlying etiology. Hence, although our data suggest that neutrophils are dispensable for NK-cell development and differentiation, some patients displayed a specific gap in the NK repertoire, associated with poor cytotoxic function and more severe disease manifestations.
Introduction
NK cells are a diverse population of innate leukocytes that are important in the control of infected and transformed cells. The differentiation spectrum of peripheral NK cells spans from CD56bright cells to adaptive CD56dim cells induced by cytomegalovirus (CMV) infection.1-4 Immunoregulatory CD56bright NK cells are highly responsive to cytokines, whereas cytotoxic CD56dim NK cells favor receptor ligation input, although the response is tissue and context dependent.5 CD56dim NK cells can be subdivided according to the expression of the inhibitory receptor NKG2A, killer immunoglobulin-like receptors (KIRs), and the carbohydrate epitope CD57, which associates with changes in functionality and longevity.6-8 Recent studies have revealed a set of regulatory gene circuits that reciprocally regulate NK-cell differentiation, including TCF7-LEF1, MYC, and RUNX2, associated with the early CD56bright stage, and PRDM1-ZEB2 and BCL11B, associated with the CD56dim stage and CMV-induced adaptive NK-cell expansions.9,10
Neutrophils are the most abundant leukocytes with functions including phagocytosis, production of reactive oxygen species and chemokines, and release of neutrophil extracellular traps.11 They are involved in innate and adaptive immunoregulatory networks that may occur through direct effects on cell activation by the release of effector molecules or in the support of cellular differentiation.12 Human neutrophil deficiencies (neutropenias), are characterized by a consistently low or fluctuating number of blood neutrophils and functional deficiencies, such as low levels of antibacterial peptides (eg, hCAP18/pro-LL-37), which may contribute to enhanced risk for infection.13 Neutropenia can be acquired or hereditary, where the hereditary forms have been linked to mutations in a high number of genes, resulting in a heterogenous disease spectrum.14 The clinical presentations vary, and the underlying mechanisms are not fully understood, but may all lead to enhanced apoptosis of neutrophil precursors or mature neutrophils. Depending on the underlying mutation, patients may also show alterations in the number of lymphocytes or functions, and have an increased risk of disease progression to myelodysplastic syndrome and/or acute myeloid leukemia.15
NK cells and neutrophils have been thought to constitute a new “defensive alliance.”16 Neutrophil-derived factors are reported to have both positive and negative effects on NK cells in terms of cytolysis, interferon-γ release, proliferation, and survival.12,17-20 A study of patients with neutropenia and of mice carrying an induced point mutation leading to neutrophil deficiency showed that NK-cell function and differentiation is dependent on interactions with neutrophils.21 To obtain a global view of the relationship between neutropenia and NK-cell homeostasis in the context of the immune environment, we performed comprehensive flow and mass cytometry analyses, as well as plasma protein analyses and RNA sequencing of the NK-cell compartment in 64 patients with different forms of neutropenia. We found a profound skewing of NK-cell homeostasis that manifested as a predominance of CD56bright NK cells and selective loss of CD56dim NK cells leading to NK lymphopenia, but only in a subgroup of patients with chronic neutropenia, suggesting a nonubiquitous role for neutrophils in NK-cell differentiation. The results provide insights into the cell-intrinsic and -extrinsic events that regulate NK-cell homeostasis and suggest common underlying dysregulation in neutrophils and NK cells in some patients with severe disease.
Methods
Patient samples and clinical data
The study was approved by the regional ethics committee (Stockholm, Sweden, Dnr 2014/988-31 and 2016/1105-32) and was conducted in accordance with the Declaration of Helsinki. Patients (n = 64) with various forms of neutropenia were recruited from the Hematology Unit at the Karolinska University Hospital in Huddinge, Sweden; Astrid Lindgren Children’s Hospital, Huddinge, Sweden; and Sunderbyn Hospital, Luleå, Sweden. The median age of the cohort was 40 years (range, 10 months to 89 years). Twenty adult, healthy, age- and sex-matched donors (HDs) were recruited, along with 7 control healthy siblings of the pediatric patients with neutropenia. A full description of the cohort is available in Table 1. The workup for neutropenia subtypes in adults and children followed published algorithms.22,23
Study cohort
| Neutropenia/diagnosis | n | Age, median y (range) | Sex, F/M | G-CSF, Y/N | ANC, median n (range) | Genetics Y/N |
|---|---|---|---|---|---|---|
| Adult neutropenia | 53 | 51 (19-89) | 38/15 | 14/38 | 1.1 (0.1-15) | — |
| AIN | 20 | 53 (21-8) | 18/2 | 7/12 | 1.2 (0.3-5.8) | — |
| SCN | 5 | 28 (19-68) | 4/1 | 3/2 | 0.4 (0.3-2.8) | 3/2 (ELANE, HAX1) |
| FN | 4 | 37 (29-40) | 2/2 | 0/4 | 1.1 (0.2-1.8) | — |
| EN | 7 | 52 (21-89) | 1/6 | 0/7 | 0.9 (0.7-1.2) | 7/7 (DARC/ACKR1) |
| IN | 13 | 59 (33-80) | 13/0 | 2/11 | 1.1 (0.1-3.2) | — |
| T-LGL | 4 | 52 (39-82) | 4/0 | 2/2 | 1.5 (0.2-15) | — |
| Pediatric neutropenia | 11 | 2 (0.9-14) | 2/9 | 3/8 | 1.0 (0.1-5.4) | |
| AIN | 5 | 2 (0.9-4) | 0/5 | 0/5 | 0.5 (0.1-1.1) | — |
| SCN | 3 | 4 (1.5-8) | 1/2 | 3/0 | 2.6 (1.4-5.4) | 2/1 (ELANE, JAGN1) |
| IN | 3 | 2 (1.5-14) | 1/2 | 0/3 | 1 (0,9-1.2) | — |
| Adult HD | 20 | 45 (24-79) | 17/3 | — | — | — |
| Pediatric HD | 7 | 10 (1-16) | 1/6 | — | — | — |
| Neutropenia/diagnosis | n | Age, median y (range) | Sex, F/M | G-CSF, Y/N | ANC, median n (range) | Genetics Y/N |
|---|---|---|---|---|---|---|
| Adult neutropenia | 53 | 51 (19-89) | 38/15 | 14/38 | 1.1 (0.1-15) | — |
| AIN | 20 | 53 (21-8) | 18/2 | 7/12 | 1.2 (0.3-5.8) | — |
| SCN | 5 | 28 (19-68) | 4/1 | 3/2 | 0.4 (0.3-2.8) | 3/2 (ELANE, HAX1) |
| FN | 4 | 37 (29-40) | 2/2 | 0/4 | 1.1 (0.2-1.8) | — |
| EN | 7 | 52 (21-89) | 1/6 | 0/7 | 0.9 (0.7-1.2) | 7/7 (DARC/ACKR1) |
| IN | 13 | 59 (33-80) | 13/0 | 2/11 | 1.1 (0.1-3.2) | — |
| T-LGL | 4 | 52 (39-82) | 4/0 | 2/2 | 1.5 (0.2-15) | — |
| Pediatric neutropenia | 11 | 2 (0.9-14) | 2/9 | 3/8 | 1.0 (0.1-5.4) | |
| AIN | 5 | 2 (0.9-4) | 0/5 | 0/5 | 0.5 (0.1-1.1) | — |
| SCN | 3 | 4 (1.5-8) | 1/2 | 3/0 | 2.6 (1.4-5.4) | 2/1 (ELANE, JAGN1) |
| IN | 3 | 2 (1.5-14) | 1/2 | 0/3 | 1 (0,9-1.2) | — |
| Adult HD | 20 | 45 (24-79) | 17/3 | — | — | — |
| Pediatric HD | 7 | 10 (1-16) | 1/6 | — | — | — |
AIN, autoimmune neutropenia; ANC, absolute neutrophil count; EN, ethnic neutropenia; F/M, female/male; FN, familial neutropenia other than SCN and EN; HD, healthy donors; IN, idiopathic neutropenia; SCN, severe congenital neutropenia; T-LGL, T-cell large granular lymphoma associated neutropenia; Y/N, yes/no.
Cell processing
Peripheral blood mononuclear cells (PBMCs) from healthy controls and patients were isolated by density gradient centrifugation (Ficoll-Hypaque; GE Healthcare) and cryopreserved in 10% dimethyl sulfoxide and 90% heat-inactivated fetal calf serum (Sigma-Aldrich, St Louis, MO). EDTA plasma was collected by centrifugation and cryopreserved.
Flow and mass cytometry analysis
Fluorochrome-conjugated antibodies used for flow cytometry are listed in the supplemental Methods available on the Blood Web site. Data were acquired in FACSDiva on an LSR Fortessa or LSR II (BD Biosciences). Acquired data were analyzed in FlowJo (Ashland, OR). For mass cytometry, a panel of 36 parameters was established (supplemental Table 1). For t-distributed stochastic neighbor analysis, all events were then pooled and clustered, using the Rtsne R package (version 0.15). See further details in the supplemental Methods and supplemental Figure 1, available on the Blood Web site.
Functional assays
NK-cell function was evaluated by mixing PBMCs at a 10:1 ratio with K562 target cells and incubating in a V-bottom 96-well plate for 4 hours at 37°C in 5% CO2. For measurements of NK-cell degranulation and interferon-γ production, monensin (Golgi Stop, 1:1500; BD Biosciences), brefeldin A (GolgiPlug, 1:1000; BD Biosciences), and anti-CD107a PE were added at the start of the 4-hour coincubation. Further details are provided in supplemental Methods.
Plasma protein analysis
Plasma samples were analyzed externally in a proximity extension assay (Olink Proteomics). A total of 263 plasma proteins were analyzed in each sample (supplemental Table 2). Of these, 14 proteins were excluded for >50% missing values. Missing values for 12 of the remaining 249 proteins were replaced with limit of detection values. The log2-transformed, arbitrary unit–normalized protein expression was used for all calculations and visualizations. Plasma levels of the human hCAP-18/pro-LL-37 were analyzed with a recently described in-house enzyme-linked immunosorbent assay.24
KIR ligand typing
Genomic DNA was isolated from PBMCs with a commercial kit (DNeasy, Qiagen). KIR ligands were determined with the KIR HLA ligand kit (Olerup SSP; Qiagen) for detection of the HLA-C1 and -C2 motifs.
RNA sequencing
NK cells were isolated from cryopreserved PBMCs (MACS columns and reagents; Miltenyi BioTech). Total RNA was isolated from samples containing 75 × 103 to 200 × 103 cells per sample with the miRNeasy Micro Kit (Qiagen) for small amounts of cells. RNA quality control and library preparation are described in the supplemental Methods. Paired-end with 75-bp read length (2 × 75) was performed on the HiSeq4000 system. Sequencing data were demultiplexed with bcl2fastq v2 software from Illumina. Resulting fastq-files were further processed with STARaligner25 (v 2.5.0b) and the Homo sapiens reference genome and annotation (UCSC hg19). For transcript assembly Cufflinks 2.2.1, was used, and for differential gene expression, Cuffdiff 2.2.0 was used.26
Network analyses
For network analyses, the DifferentialNet database27 (integrated data from experimentally detected protein–protein interactions [PPIs], and RNA sequencing gathered by the Genotype-Tissue Expression consortium [GTEx]) was used to generate tissue-specific (whole blood) PPI networks that were visualized with NetworkAnalyst 3.0. Enrichment analyses were performed in NWA3.0 with the Gene Ontology (Biological Processes) database. Venn diagrams and related tables were created with BioVenn.28
Statistical analyses
Statistical analyses and visualization were performed in Prism 9 (GraphPad, La Jolla, CA) and R with the ggplot2 and dunn.test packages. For unpaired comparison of 3 groups, the Kruskal-Wallis rank sum test was used along with the 2-sided Mann-Whitney post hoc test, Wilcoxon rank sum exact test or Dunn’s test when the initial test gave P ≤ .05. The 2-sided Wilcoxon matched-pairs signed rank test was used for paired comparisons. Where indicated, P-values were corrected with the Benjamini-Hochberg false discovery rate. Nonparametric Spearman rank correlations were performed. Binomial distribution was used to compare the observed outcome relative to expected frequencies of donors with adaptive NK cells. Fisher’s exact test and the χ2 test were used to determine differences in proportions in the clinical data.
Results
Perturbed NK-cell homeostasis in neutropenia
To understand the impact of neutropenia on immune homeostasis, and in particular NK cells, a cohort of 53 adults and 11 children with neutropenia were recruited, along with 27 sex- and age-matched HDs and siblings (Figure 1A; Table 1). Using the reported shift in the CD56bright/CD56dim ratio in patients with neutropenia,21 as a point of departure, we identified 14 of 53 adult patients with neutropenia as having immature NK-cell repertoires with elevated frequencies of CD56bright NK cells (defined as >3 standard deviations [SD] above the HD mean and subsequently relabeled the NKimmgroup), a phenotype that was consistent over time (Figure 1B-D). This subgroup of patients displayed decreased frequencies of NK cells among PBMCs, as well as absolute counts of NK cells, with several patients presenting severe NK lymphopenia (<50 cells per microliter of blood), with a selective loss of CD56dim cells (Figure 1E-F). Similar results were observed in children with neutropenia (supplemental Figure 2A-B). Further delineation of the CD56dim NK-cell phenotype revealed increased frequencies of NKG2A+KIR−CD57− cells and decreased differentiated NKG2A−KIR+CD57+ cells (Figure 1G-H). Acquisition of a first KIR was significantly lower in CD56dim cells from the NKimm group (supplemental Figure 2C-D). Adaptive NK cells, as defined by >10% NKG2C+CD57+ or Fcerg−CD2+CD7lowCD57+ of CD56dimcells, were present in ∼25% of CMV+ individuals in the HD and NP groups, but not in the CMV+ NKimm group (Figure 1I; supplemental Figure 2E). This finding shows that a subgroup of patients with neutropenia had severe NK lymphopenia with a stable imprint toward NKimm-cell repertoires with selective lack of mature CD56dim NK cells.
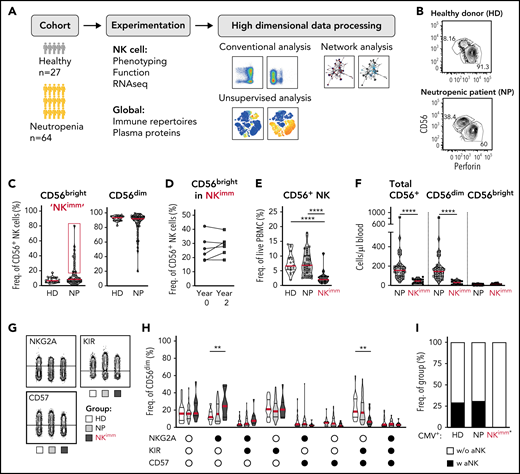
Altered NK-cell homeostasis in a subset of patients with neutropenia. (A) Study design. (B-C) Flow cytometry analysis of frequencies of CD56bright/dim among total NK cells. A subset of 14 patients with neutropenia (NP) with elevated levels of CD56bright cells (defined as >HD mean +3 standard deviations, were relabeled NKimm. (D) Frequencies of CD56bright cells in NKimm patients sampled 2 years apart. (E) Frequency of total CD56+ NK among PBMCs. (F) Cell counts of CD56+ NK cells. (G-H) Subset level phenotyping of CD56dim NK cells based on NKG2A, KIR, and CD57 expression. (I) The frequency of CMV+ donors with an adaptive NK-cell population (aNK) within each group. HD (n = 20), NP (n = 39), and NKimm (n = 14). *P < .05; **P < .01; ***P <.001; ****P < .001 by nonparametric test, and binomial distribution (I).
Altered NK-cell homeostasis in a subset of patients with neutropenia. (A) Study design. (B-C) Flow cytometry analysis of frequencies of CD56bright/dim among total NK cells. A subset of 14 patients with neutropenia (NP) with elevated levels of CD56bright cells (defined as >HD mean +3 standard deviations, were relabeled NKimm. (D) Frequencies of CD56bright cells in NKimm patients sampled 2 years apart. (E) Frequency of total CD56+ NK among PBMCs. (F) Cell counts of CD56+ NK cells. (G-H) Subset level phenotyping of CD56dim NK cells based on NKG2A, KIR, and CD57 expression. (I) The frequency of CMV+ donors with an adaptive NK-cell population (aNK) within each group. HD (n = 20), NP (n = 39), and NKimm (n = 14). *P < .05; **P < .01; ***P <.001; ****P < .001 by nonparametric test, and binomial distribution (I).
Perturbed homeostasis is confined to NK cells
Next, we performed systems-level immune mapping, using 44-parameter mass cytometry, which revealed distinct topological regions corresponding to CD4+ and CD8+ T cells, B cells, monocytes, and NK cells (Figure 2A-C). Apart from NK cells, no significant influence on the broader composition of immune cells was found in the aggregated analysis of the NKimm group (Figure 2D). On the subset level, the B-cell compartment was tilted in favor of both unswitched and class-switched memory B cells in all patients with neutropenia (Figure 2E). For CD4+/CD8+ T-cell subsets (naive, Ttm, Tcm, Tem, Temra, and Treg) and γδ T cells; ILC1, -2, and -3; and myeloid cell subsets (classical, intermediate, and nonclassical monocytes, as well as pDCs and mDCs), differences were small and nonsignificant between groups (Figure 2F-G). Hence, the immature profile and disrupted homeostasis defining the NKimm phenotype was isolated in the NK-cell compartment.
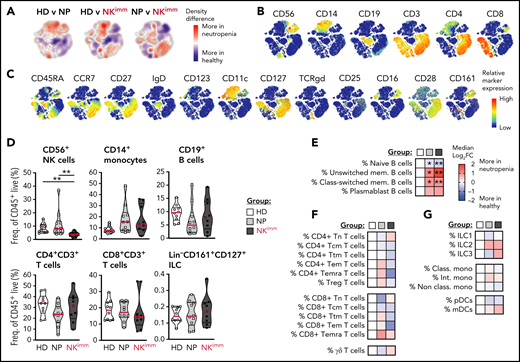
The perturbed homeostasis observed in the NKimm group is confined to NK cells. Systems level immune mapping by flow and mass cytometry. (A) Differences in the density of cells from each group visualized in a tSNE map. (B) Relative expression of selected markers for main immune cell lineages and (C) other assessed markers shown on a t-SNE clustering of sampled events from all individuals (n = 29). Aggregated group level analysis of major immune cell subsets (D) and subset-level (E-G) analyses. Data shown in heat maps are depicted as median log2-fold change as compared with HD. Frequencies were drawn from the parent population. HD (n = 10), NP (n = 10), and NKimm (n = 9). *P < .05; **P < .01, by nonparametric test. t-SNE, t-distributed stochastic neighbor embedding.
The perturbed homeostasis observed in the NKimm group is confined to NK cells. Systems level immune mapping by flow and mass cytometry. (A) Differences in the density of cells from each group visualized in a tSNE map. (B) Relative expression of selected markers for main immune cell lineages and (C) other assessed markers shown on a t-SNE clustering of sampled events from all individuals (n = 29). Aggregated group level analysis of major immune cell subsets (D) and subset-level (E-G) analyses. Data shown in heat maps are depicted as median log2-fold change as compared with HD. Frequencies were drawn from the parent population. HD (n = 10), NP (n = 10), and NKimm (n = 9). *P < .05; **P < .01, by nonparametric test. t-SNE, t-distributed stochastic neighbor embedding.
Activation, proliferation, and exhaustion of the CD56dim NK compartment
Given the selective loss of mature NK cells, we performed an extended phenotyping of NK cells by flow and mass cytometry. We found that CD56dim, but not CD56bright, NK cells had increased Ki-67+, Tim-3+, and TIGIT+ fractions (Figure 3A; supplemental Figure 3A-B), suggesting an activated state associated with proliferation and/or exhaustion. No significant differences were found between groups in expression of CD127, CD16, CD38, CD161, NKp30, LILRB1, CD2, CD98, CD71, or CD25 (supplemental Figure 3B). Further CD56dim NK cells displayed decreased levels of Bcl-2, leading to a lower BIM/Bcl-2 ratio and indicating a shift toward proapoptotic pathways (Figure 3B; supplemental Figure 3C). Levels of proapoptotic and antiapoptotic proteins Bcl-xL, Mcl-1, NOXA, and BIM in NK cells were similar in all groups (supplemental Figure 3C). Although levels of Bcl-2 were overall lower in Ki-67+ cells, low Bcl-2/BIM ratios were particularly pronounced in proliferating CD56dim cells within the NK compartment (Figure 3C).
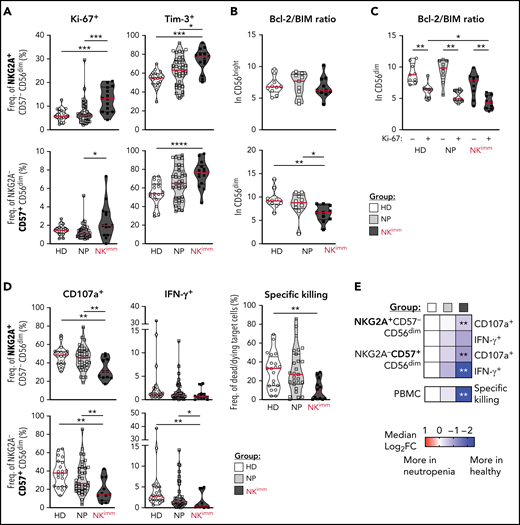
Activation, proliferation, and exhaustion of the CD56dim NK compartment in the NKimm group. Extended NK-cell phenotype analysis by flow and mass cytometry. (A) Ki-67+ and Tim-3+ cells in NKG2A+/−CD57+/− CD56dim NK-cell subsets. (B) Bcl-2 to BIM ratio in CD56bright and CD56dim NK cells and in panel C, based on Ki-67 expression in CD56dim cells. (D-E) NK-cell subset responses to K562 tumor target cells in terms of degranulation (CD107a), interferon-γ production, and specific killing of target cells by NK cells among PBMCs, as judged by Dead Cell Marker+ caspase-3+ target cells. Data shown in heat maps are depicted as median log2-fold change, compared with HD cells. HD (n = 10), NP (n = 10), and NKimm (n = 9). *P < .05; **P < .01; ***P < .001; ****P < .0001, by nonparametric test. HD (n = 10-20), NP (n = 10-39), and NKimm (n = 10-14).
Activation, proliferation, and exhaustion of the CD56dim NK compartment in the NKimm group. Extended NK-cell phenotype analysis by flow and mass cytometry. (A) Ki-67+ and Tim-3+ cells in NKG2A+/−CD57+/− CD56dim NK-cell subsets. (B) Bcl-2 to BIM ratio in CD56bright and CD56dim NK cells and in panel C, based on Ki-67 expression in CD56dim cells. (D-E) NK-cell subset responses to K562 tumor target cells in terms of degranulation (CD107a), interferon-γ production, and specific killing of target cells by NK cells among PBMCs, as judged by Dead Cell Marker+ caspase-3+ target cells. Data shown in heat maps are depicted as median log2-fold change, compared with HD cells. HD (n = 10), NP (n = 10), and NKimm (n = 9). *P < .05; **P < .01; ***P < .001; ****P < .0001, by nonparametric test. HD (n = 10-20), NP (n = 10-39), and NKimm (n = 10-14).
We next interrogated the functional consequences of the immature NK-cell repertoires in affected patients (Figure 3D-E; supplemental Figure 3D). Levels of granzyme B and perforin in CD56dim NK cells were similar to those with intact NK-cell repertoires (supplemental Figure 3E). Likewise, NK-cell education of self-KIR–expressing NK cells was normal (supplemental Figure 3F). However, patients in the NKimm group had significantly lower global target cell responses and diminished target cell killing (Figure 3D-E), because of the lower presence of mature CD56dim NK cells among PBMCs (supplemental Figure 3G). Taken together, these results revealed that CD56dim NK cells in the NKimm group showed signs of activation and proliferation with impaired target cell responses, irrespective of differentiation and education status.
Transcriptional profiling and network analysis reveals intrinsic defects in NK-cell homeostasis
To decipher the transcriptional programs underlying the altered NK-cell homeostasis, we performed bulk RNA sequencing on isolated NK cells from patients and controls, including sorted CD56bright NK cells. We identified 105 upregulated and 47 downregulated genes between NK cells from the HD and NKimmgroups, and 62 upregulated and 45 downregulated genes between the NP and NKimm groups (Figure 4A). Forty upregulated and 28 downregulated genes were significant in both comparisons (Figure 4B; supplemental Table 3). PPI analysis with the shared differentially expressed genes as a base resulted in a network of 511 elements (nodes; Fig. 4C). Cross-reference to HD CD56bright gene signatures revealed a small set of 7 upregulated and 4 downregulated genes that were unique to NK cells from the NKimm group (Figure 4D) and therefore were unrelated to the normal CD56bright signature. To delineate the cell-intrinsic processes specific to the NKimm phenotype, the signature genes (CENPF, DTX1, MKI67, TNFSF9, PTGDS, and TOP2A) and related nodes within the network were queried for pathway enrichment (Figure 4E). Corroborating the flow cytometry–based phenotyping data, cell cycle regulation, as well as proliferation, programmed cell death, and the MAPK cascade dominated the NKimm-instructed gene network (Figure 4E; supplemental Table 4).
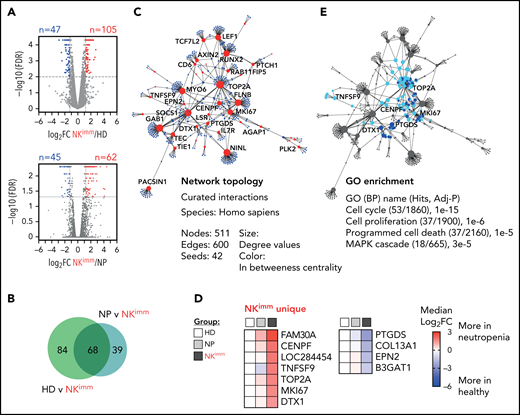
Transcriptional profile of the NKimm-cell repertoire: dysregulation of the proliferation and apoptosis machinery. RNA sequencing of the NK-cell compartment. (A) There were 152 DE genes in the HD and NKimm groups combined (105 upregulated and 47 downregulated) and 107 DE genes in the NP and NKimm groups combined (62 upregulated and 45 downregulated) after filtering for log2 FC > 1.2. (B) Venn diagram displaying the shared DE genes between NKimm/HD and NKimm/NP groups. (C) Topology and content of the protein-protein interaction network driven by shared DE genes in CD56+ NK cells of the NKimm group. (D) Heat maps illustrating median log2-fold change in expression of NKimm-unique genes identified by cross-referencing shared DE genes in panel B to DE genes in HD NK cell/CD56bright NK-cell controls. (E) Query of NKimm-unique seeds and related nodes (blue) in the PPI network that, when identified in GO biological processes are highlighted (teal). HD (n = 4), NP (n = 8), and NKimm (n = 7). DE, differentially expressed.
Transcriptional profile of the NKimm-cell repertoire: dysregulation of the proliferation and apoptosis machinery. RNA sequencing of the NK-cell compartment. (A) There were 152 DE genes in the HD and NKimm groups combined (105 upregulated and 47 downregulated) and 107 DE genes in the NP and NKimm groups combined (62 upregulated and 45 downregulated) after filtering for log2 FC > 1.2. (B) Venn diagram displaying the shared DE genes between NKimm/HD and NKimm/NP groups. (C) Topology and content of the protein-protein interaction network driven by shared DE genes in CD56+ NK cells of the NKimm group. (D) Heat maps illustrating median log2-fold change in expression of NKimm-unique genes identified by cross-referencing shared DE genes in panel B to DE genes in HD NK cell/CD56bright NK-cell controls. (E) Query of NKimm-unique seeds and related nodes (blue) in the PPI network that, when identified in GO biological processes are highlighted (teal). HD (n = 4), NP (n = 8), and NKimm (n = 7). DE, differentially expressed.
Systemic protein profiling reveals altered regulation of cell turnover, apoptosis, and immune regulation
Our multilayered analysis of NK cells in patients with neutropenia suggested a cell-intrinsic perturbation in homeostasis related to regulation of cell cycling and apoptosis. To decipher the contribution of, and interplay with, the global immune environment, we used the proximity extension assay, a high-throughput proteomics method, to quantify 249 proteins in plasma of patients and HDs. We found 39 proteins with significantly different levels in the NKimm group compared with that in the HD group, of which 17 were shared with the NP group (Figure 5A-D). Children with neutropenia displayed no alterations in plasma profiles, compared with their healthy siblings (supplemental Figure 4A). Among the abundant plasma proteins, we found CCL3 (MIP-1α), interleukin-18 (IL-18) and IL-27, MARCO, and OSCAR, indicating myeloid cell activation. Levels of proteins associated with regulation of cell death and differentiation, such as FADD, TNFSF12, OLR1, DLL1, and CEACAM5, were also altered in the NKimm group. Among the 22 plasma proteins that were unique to patients with the NKimm phenotype, many correlated with each other (supplemental Figure 4B-C), suggesting coordinated activation of specific programs. The dominating pathways included regulation of MAPK cascades, proliferation, and programmed cell death (Figure 5E-F; supplemental Table 5).
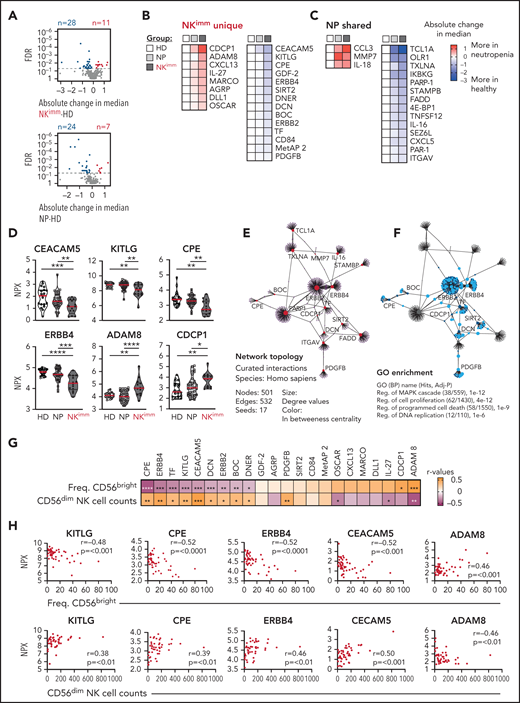
Plasma protein profile in NKimm group suggests cell turnover, apoptosis, and immune regulation. Analysis of 249 plasma proteins by proximity extension assay (Olink). (A-C) Levels of 39 proteins differed significantly in the NKimm group compared with the HD group (nonparametric testing, Benjamini-Hochberg false discovery rate <.05) of which 22 were NKimm unique and 17 were shared with the NP group. (D) Raw normalized protein expression values for selected proteins. (E) Topology and content of the protein-protein interaction network driven by significant plasma proteins (seeds) in the NKimm group. (F) Query of NKimm-unique seeds and related nodes (blue) in the PPI network, that, when identified in Gene Ontology (Biological Processes), are highlighted (teal). (G) Spearman rank correlations for NKimm-defining parameters and plasma protein data for the 22 uniquely altered proteins in this group. (H) Raw plots for top correlating proteins. HD (n = 19-20), NP (n = 36-38), and NKimm (n = 14). Data in the heat maps in panels B and C are depicted as absolute change in median compared with the HD median. *P < .05; **P < .01; ***P < .001; ****P < .0001, by nonparametric test.
Plasma protein profile in NKimm group suggests cell turnover, apoptosis, and immune regulation. Analysis of 249 plasma proteins by proximity extension assay (Olink). (A-C) Levels of 39 proteins differed significantly in the NKimm group compared with the HD group (nonparametric testing, Benjamini-Hochberg false discovery rate <.05) of which 22 were NKimm unique and 17 were shared with the NP group. (D) Raw normalized protein expression values for selected proteins. (E) Topology and content of the protein-protein interaction network driven by significant plasma proteins (seeds) in the NKimm group. (F) Query of NKimm-unique seeds and related nodes (blue) in the PPI network, that, when identified in Gene Ontology (Biological Processes), are highlighted (teal). (G) Spearman rank correlations for NKimm-defining parameters and plasma protein data for the 22 uniquely altered proteins in this group. (H) Raw plots for top correlating proteins. HD (n = 19-20), NP (n = 36-38), and NKimm (n = 14). Data in the heat maps in panels B and C are depicted as absolute change in median compared with the HD median. *P < .05; **P < .01; ***P < .001; ****P < .0001, by nonparametric test.
Notably, the alterations in protein profiles in patients with the NKimm phenotype correlated with frequencies of CD56bright and absolute CD56dim NK-cell counts (Figure 5G-H). Of these, KITLG, CPE, ERBB4, CEACAM5, and ADAM8 showed the strongest correlations to both parameters. Together, these data reveal a coordinated systemic plasma protein profile connected to dysregulated NK-cell homeostasis.
Severe-grade neutropenia is associated with the NKimm phenotype
We next examined the relationship between the observed perturbation in NK-cell homeostasis and clinical parameters. We found no significant differences in age, sex, or CMV seropositivity between the NP and NKimm groups (Figure 6A). Patients receiving regular granulocyte colony-stimulating factor (G-CSF) to increase peripheral neutrophil counts were asked to make a scheduled pause in treatment for 1 week before sampling, to enable neutrophil levels to return to steady state. G-CSF treatment at baseline was overrepresented within the NKimm group (Figure 6A), but was not associated with increased CD56bright NK cells in all G-CSF–treated patients (Figure 6B). Furthermore, we found no correlation between G-CSF treatment dose and NK-cell parameters (supplemental Figure 4D). Patients with severe congenital neutropenia (SCN), who had a maturational block of neutrophils in the bone marrow, and patients with autoimmune neutropenia (AIN), with supposed augmented neutrophil destruction in the periphery, were found in the regular NP group and in the NKimm group (Figure 6C). Notably, all patients with T-cell large granulocyte leukemia were found in the NKimm subgroup.
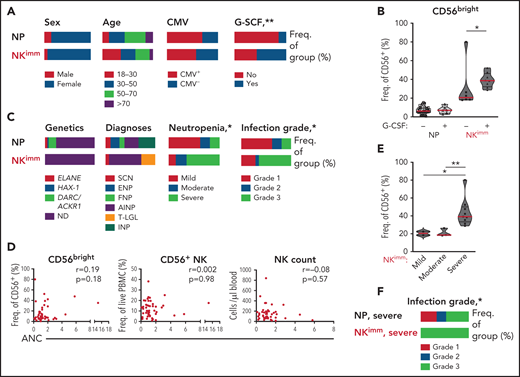
Severe-grade neutropenia is associated with the NKimm phenotype. (A,C) Clinical data for the NP and NKimm groups, presented as frequency of total group. (B) Frequency of CD56bright NK cells for NP and NKimm groups divided by G-CSF treatment (−, no treatment; +, patients on G-CSF, but with scheduled stop in treatment). (D) Spearman rank correlations for NKimm-defining parameters and ANCs. (E) Frequency of CD56bright NK cells divided by severity of neutropenia within the NKimm group. (F) Documented grade 1 to 3 infection history in patients in the NP and NKimm groups with severe-grade neutropenia. HD (n = 20), NP (n = 39), and NKimm (n = 14). *P < .05; **P < .01 by nonparametric test and χ2 test for trend.
Severe-grade neutropenia is associated with the NKimm phenotype. (A,C) Clinical data for the NP and NKimm groups, presented as frequency of total group. (B) Frequency of CD56bright NK cells for NP and NKimm groups divided by G-CSF treatment (−, no treatment; +, patients on G-CSF, but with scheduled stop in treatment). (D) Spearman rank correlations for NKimm-defining parameters and ANCs. (E) Frequency of CD56bright NK cells divided by severity of neutropenia within the NKimm group. (F) Documented grade 1 to 3 infection history in patients in the NP and NKimm groups with severe-grade neutropenia. HD (n = 20), NP (n = 39), and NKimm (n = 14). *P < .05; **P < .01 by nonparametric test and χ2 test for trend.
We found no association between absolute neutrophil counts (ANCs) and frequency of CD56bright cells or frequency of total NK cells or absolute counts of NK cells (Figure 6D). However, significantly more patients with NKimm displayed severe-grade neutropenia (defined as ANC < 500 cells per microliter untreated, with increased propensity for infection), which correlated with the highest frequencies of CD56bright NK cells within this group (Figure 6C,E). Interestingly, altered plasma proteins with strong correlation to CD56bright frequencies were also significantly associated with severe neutropenia (supplemental Figure 4E). To investigate whether the maturation status of neutrophils was related to perturbed NK-cell homeostasis, plasma levels of the peptide hCAP-18/pro-LL-37, previously shown to distinguish neutropenia caused by perturbed neutrophil maturation,29 were measured. Although we could confirm low levels in SCN, hCAP-18 levels varied greatly in the NKimm group and were not significantly different from other patients with neutropenia (supplemental Figure 4F).
Severe neutropenia is generally associated with increased infectious complications; the lower the ANC, the greater the risk for bacterial and fungal infections.30 We compared the incidence of infections requiring treatment with antibiotics for the past 10 years among the patients with neutropenias (grade 1 is <2 antibiotic regimens per 10 years, grade 2 is >2 antibiotic-regimens per 10 years, and grade 3 is hospitalized for treatment with IV antibiotics). No data were available on viral infections. Grade 3 infections requiring hospital care with IV antibiotics were overrepresented in the NKimm group (Figure 6C), as was the case when the analysis was limited to patients with severe-grade neutropenia only (Figure 6F). Thus, disrupted NK-cell homeostasis can be found across a spectrum of clinical subtypes of chronic neutropenia and is associated with severe-grade neutropenia and a more complex infectious history.
Discussion
Neutrophils have been suggested to be essential for proper NK-cell maturation,21 but how this relationship is regulated at a cellular and molecular level remains unknown. Using high-dimensional flow and mass cytometry, soluble factor analysis, and RNA sequencing we characterized immunophenotypes in patients with distinct clinical subtypes of neutropenia. Whereas most patients with neutropenia had seemingly normal levels of NK cells, a subgroup of patients displayed a profoundly disturbed NK-cell differentiation, manifesting as moderate to severe NK lymphopenia with a marked overrepresentation of the CD56bright subset. Comprehensive profiling of CD56dim NK cells in these patients showed loss of more differentiated subsets, increased proliferation, and activation of proapoptotic pathways. RNA sequencing and plasma protein profiles revealed dysregulation of the cell cycle and apoptotic pathways associated with the inability to maintain homeostasis in the NK-cell compartment.
An increase in CD56bright NK cells and diminished NK functional responses was previously shown for patients with AIN and SCN, but the total NK-cell number was within the normal range.21 In a more recent study, patients with DARC/ACKR1-null neutropenia had lower frequencies of mature NK cells related to ANC, yet no functional impairments.31 The NK-cell defect described herein presents with loss of functionality and a reduction in the number of NK-cell subsets, reminiscent of a classic NK-cell deficiency (NKD), yet only as a contributing factor in the overall immunodeficiency.32 In our cohort, patients carrying ELANE and HAX1 mutations, known to severely affect the neutrophil population,33,34 had normal NK-cell repertoires. We found no correlations between the number of ANC and NK cells, although the reliability of ANC as an estimation of neutrophil presence can be variable. Instead, the strongest common denominator for the NKimm group was severe-grade neutropenia, and we considered that the quality of neutrophils egressing from the bone marrow may have differed between patients. In this context, neutrophils induced by G-CSF can be more immature.35 Baseline G-CSF treatment (before treatment interruption) was associated with the NKimm phenotype, but patients ordinarily treated with high-dose G-CSF were also found among those with normal NK-cell repertoires. Low levels of hCAP-18, a granule propeptide synthesized by neutrophil precursors during differentiation in the bone marrow, distinguishes a stop in the neutrophil maturation process,29 which was apparent in patients with SCN. However, we found no difference in levels between the study-defined groups. Altogether, these results suggest that direct or indirect interactions with mature neutrophils in the periphery or in the bone marrow are not strictly needed for proper NK-cell development and maturation, as previously suggested.21
NK cells of patients in the NKimm group displayed blunted target cell responses and higher fractions of Ki-67+ cells. It is possible that a switch from more functional to proliferative programs, unrelated to the differentiation process, made the NK cells hyporesponsive.36 The ability to proliferate is central in maintaining homeostasis and in the progression of NK cells through differentiation stages.32,37 However, increased turnover accompanied by decreased survival of proliferating cells may lead to disturbances in population maintenance. Low proapoptotic-to-antiapoptotic ratios were particularly pronounced in Ki-67+ CD56dim cells within the NK compartment in the NKimmgroup, implying that they could be more prone to undergoing apoptosis, thus effectively preventing progression through differentiation stages.
Next, we sought a transcriptional basis for the observed alteration in NK homeostasis. Several recent publications have described the transcriptome of NK cells related to differentiation stages by using bulk or single-cell RNA sequencing that showed a distinct profile of the immature CD56bright NK-cell subset.9,10,38 Given the relative increase in CD56bright NK cells in patients with perturbed NK-cell homeostasis, the transcriptional profiles were compared with both bulk and sorted CD56bright NK cells from healthy donors. Many of the identified differentially expressed genes for NK cells of the NKimm group were clearly associated with the CD56bright transcriptome, suggesting an exaggerated immature gene profile in NK cells of the NKimmgroup. Interestingly, we found several unique genes in the NKimm group, including CENPF, MKI67, and TOP2A, all of which have been connected to various cancer forms because of their participation in cell proliferation.39,40 The expression of CENPF, TOP2A, MKI67, and the Ki-67 protein, show a cell-cycle–dependent regulation of abundance,41-43 suggesting that dysregulation in cell cycle progression is a contributing factor to the NKimm NK phenotype. Indeed, enrichment analysis based on the NKimm dominant seed genes revealed pathways connected to cell cycle and programmed cell death/apoptosis.
We speculated that alterations to the plasma protein environment in patients with neutropenia contributes to increased turnover and activation of some of these transcriptional programs, which was also suggested by the protein-protein networks built on significantly altered proteins. A core set of plasma proteins had diminished levels in cells in the NKimm group and correlated with the frequency of CD56bright NK cells and NK-cell counts, suggesting a link to homeostasis in the NK-cell population. The KIT ligand (SCF/CD117) can suppress NK apoptosis by upregulation of Bcl-2,44 and thus the relatively lower levels observed could affect the balance of proapoptotic and antiapoptotic signals in the NK-cell population. ADAM-8 is highly expressed by neutrophils and can be released to the extracellular milieu by metalloprotease-dependent proteolysis, and this release is increased in pathophysiological processes.45 ADAM-8, as well as closely correlated downregulated proteins, were associated with severe-grade disease, reinforcing the connection to the severity of the underlying disease and illuminating a complex relationship between these factors and the NK-cell population.
The NKimm phenotype was found in patients with AIN, SCN, and T-LGL-associated neutropenia. The underlying mechanism for neutropenia in LGL is not fully elucidated, but an apoptosis mechanism via FAS/FASLG is implicated in the pathogenesis.46 A STAT3 mutation is the most common one in T-LGL, with STAT5B in rarer cases.47,48 Interestingly, STAT5B mutations have effects on NK cells similar to those described herein.49 Whether affected NK homeostasis is a common feature of T-LGL and is associated with any particular genetic background should be explored in a larger patient cohort, as well as in patients with T-LGL who do not display neutropenia.
Our systems-level immune mapping in patients with neutropenia demonstrated that no other immune cell subset was affected that was akin to NK cells, suggesting that neutrophils and NK cells may be uniquely affected by common upstream mechanisms. The theme of cell cycle regulation and proliferation is emerging as a requirement for maintained NK homeostasis and, when dysregulated, as an origin for NKDs, as recently reviewed by Mace and Orange.32 Several bona fide NKDs have been reported with mutations in genes relating to the DNA replication machinery. IRF8, MCM4, MCM10, and GINS1 deficiency all lead to selective loss of the CD56dim subset and concomitant overrepresentation of the CD56bright subset.50-53 Especially the GINS1 mutation with selective effects on neutrophils and NK cells in terms of proliferation is relevant in this context.52,54
The outstanding question is at what point the NK-cell maturational trajectory is interrupted in these patients. NK-cell progenitors are functionally impaired in G-CSF–mobilized stem cell products, although the peripheral CD56bright/dim ratios appear to be unaffected.55,56 We found normal absolute CD56bright cell counts with nonaberrant phenotypes, suggesting that the homeostatic defect is downstream of progenitor stages, primarily affecting the CD56dim NK-cell compartment. Thus, our data imply that, rather than a block in CD56bright to CD56dim transition, CD56dim expansion, survival and subsequent maturation is disrupted. This interpretation was further supported by monitoring adaptive NK cells in patients with perturbed NK-cell homeostasis. Although the number of CMV+ patients in the NKimm group was low, there was a significant lack of adaptive NK cells, compared with expectations. Adaptive NK cells are generated in a proliferative burst in response to CMV infection,2,3 and their absence corroborates the inability of CD56dim NK cells in the NKimmgroup to undergo regulated expansion. It will be of interest to assess the NK-cell status in relevant tissues, especially bone marrow, to investigate whether hematopoiesis/early lineage differentiation is affected, to gain a further insight into the depth of the NK-cell dysfunction.
In summary, we identified a subgroup of patients with chronic neutropenia associated with a selective loss of mature CD56dim NK cells. This perturbed homeostasis was unique to NK cells and correlated with disease grade. Although the phenotype was profound in these patients, there did not seem to be a ubiquitous role for neutrophils in NK development, but rather a uniting theme of accelerated cell cycle progression and dysregulated apoptosis, associated with both neutropenia and defective NK-cell homeostasis.
Acknowledgments
The authors thank all the patients with neutropenia, their siblings, and the healthy control subjects for their participation in the study; the research nurses and doctors for kind assistance with sample collection; and Marie Schaffer, codirector of the Tissue-typing Laboratory at the Clinical Immunology Department at Karolinska University Hospital, for KIR-L typing. BioRender.com was used for creation of some figure elements.
This work was supported by grants from the Swedish Research Council, the Swedish Children’s Cancer Society, the Swedish Cancer Society, the Karolinska Institutet, Oslo University Hospital, the Norwegian Research Council, the KG Jebsen Foundation, the South-Eastern Norway Regional Health Authority, the Norwegian Cancer Society, and Knut and Alice Wallenberg Foundation.
Authorship
Contribution: E.S., A.P., E.H.A., A.T.-P., B.J., H.N., S.L., M.K., M.K.-K., and A.M. conducted experiments and analyzed data; M.S., G.C., and J.P. provided clinical material and scientific input; S.M. and P.H. aided in sample collection and provided scientific input; and E.S. and K.-J.M. designed the research and wrote the manuscript.
Conflict-of-interest disclosure: K.-J.M. is a consultant and has ownership interests in Fate Therapeutics and Vycellix; and has received research funding from Fate Therapeutics, Oncopeptides, and Merck and honoraria from Oncopeptides and Cytovia. E.S. is a consultant to Fate Therapeutics. All relationships have been reviewed and managed by Oslo University Hospital and Karolinska Institute, in accordance with its conflict-of-interest polices. The remaining authors declare no competing financial interests.
Correspondence: Karl-Johan Malmberg, Department of Medicine, Karolinska Institutet, Karolinska University Hospital, Stockholm, SE-171 76, Sweden; e-mail: kalle.malmberg@ki.se; and Ebba Sohlberg, Department of Medicine, Karolinska Institutet, Karolinska University Hospital, Stockholm, SE-171 76, Sweden; e-mail: ebba.sohlberg@ki.se.
The Proximity Extension Assay data used in this publication will be deposited in Mendeley Data (http://dx.doi.org/10.17632/m4zv2p4tsy.1). Mass cytometry data will be made available at FlowRepository.org using an accession code (http://flowrepository.org/id/FR-FCM-Z4L3). RNA Sequencing data will be deposited at the EMBL-EBI European Nucleotide Archive (EGAS000010-05666). Any requests should be directed to the corresponding authors.
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal