

Key Points
In patients with AA, somatic mutations in HLA genes correlated with functionality, as measured by cell surface expression.
Loss of HLA expression and specific HLA genotypes correlated with clinical manifestations, especially age of onset and clonal evolution.

Abstract
Immune aplastic anemia (AA) features somatic loss of HLA class I allele expression on bone marrow cells, consistent with a mechanism of escape from T-cell–mediated destruction of hematopoietic stem and progenitor cells. The clinical significance of HLA abnormalities has not been well characterized. We examined the somatic loss of HLA class I alleles and correlated HLA loss and mutation-associated HLA genotypes with clinical presentation and outcomes after immunosuppressive therapy in 544 AA patients. HLA class I allele loss was detected in 92 (22%) of the 412 patients tested, in whom there were 393 somatic HLA gene mutations and 40 instances of loss of heterozygosity. Most frequently affected was HLA-B*14:02, followed by HLA-A*02:01, HLA-B*40:02, HLA-B*08:01, and HLA-B*07:02. HLA-B*14:02, HLA-B*40:02, and HLA-B*07:02 were also overrepresented in AA. High-risk clonal evolution was correlated with HLA loss, HLA-B*14:02 genotype, and older age, which yielded a valid prediction model. In 2 patients, we traced monosomy 7 clonal evolution from preexisting clones harboring somatic mutations in HLA-A*02:01 and HLA-B*40:02. Loss of HLA-B*40:02 correlated with higher blood counts. HLA-B*07:02 and HLA-B*40:01 genotypes and their loss correlated with late-onset of AA. Our results suggest the presence of specific immune mechanisms of molecular pathogenesis with clinical implications. HLA genotyping and screening for HLA loss may be of value in the management of immune AA. This study was registered at clinicaltrials.gov as NCT00001964, NCT00061360, NCT00195624, NCT00260689, NCT00944749, NCT01193283, and NCT01623167.
Medscape Continuing Medical Education online
In support of improving patient care, this activity has been planned and implemented by Medscape, LLC and the American Society of Hematology. Medscape, LLC is jointly accredited by the Accreditation Council for Continuing Medical Education (ACCME), the Accreditation Council for Pharmacy Education (ACPE), and the American Nurses Credentialing Center (ANCC), to provide continuing education for the healthcare team.
Medscape, LLC designates this Journal-based CME activity for a maximum of 1.00 AMA PRA Category 1 Credit(s)™. Physicians should claim only the credit commensurate with the extent of their participation in the activity.
Successful completion of this CME activity, which includes participation in the evaluation component, enables the participant to earn up to 1.0 MOC points in the American Board of Internal Medicine's (ABIM) Maintenance of Certification (MOC) program. Participants will earn MOC points equivalent to the amount of CME credits claimed for the activity. It is the CME activity provider's responsibility to submit participant completion information to ACCME for the purpose of granting ABIM MOC credit.
All other clinicians completing this activity will be issued a certificate of participation. To participate in this journal CME activity: (1) review the learning objectives and author disclosures; (2) study the education content; (3) take the post-test with a 75% minimum passing score and complete the evaluation at http://www.medscape.org/journal/blood; and (4) view/print certificate. For CME questions, see page 2891.
Disclosures
Associate Editor Jeanne Hendrickson, CME questions author Laurie Barclay, freelance writer and reviewer, Medscape, LLC, and the authors declare no competing financial interests.
Learning objectives
Upon completion of this activity, participants will:
- 1.
Determine somatic loss of HLA class I alleles in a study of 544 patients with immune aplastic anemia (AA)
- 2.
Describe allele frequencies of HLA-A and HLA-B associated with somatic loss in a study of 544 patients with immune AA
- 3.
Identify correlations of HLA alleles and HLA loss with clinical presentation and outcome after IST in a study of 544 patients with immune AA
Release date: December 30, 2021; Expiration date: December 30, 2022
Introduction
Immune aplastic anemia (AA) is caused by T cells that destroy hematopoietic stem cells (HSCs), and marrow failure is successfully treated with hematopoietic cell transplantation (HCT) or immunosuppressive therapy (IST).1 Eltrombopag (EPAG) combined with IST yielded higher hematologic responses and survival compared with IST alone,2 but long-term outcomes such as relapse and clonal evolution remain clinically problematic and biologically not well understood.
The immune pathophysiology of AA has been, in part, inferred from frequent somatic loss of HLA class I alleles. Increased frequency of some HLA alleles has been reported in AA patients of various ethnicities,3-14 and has been confirmed in a recent genome-wide association study.15 Somatic loss of HLA class I alleles may result from copy-neutral chromosome 6p loss of heterozygosity (6p LOH)11,16,17 or acquired inactivating HLA gene mutations.18,19 A limited set of HLA-A and HLA-B alleles are more likely to acquire somatic mutations18,19; a T-cell line specific for a missing HLA class I allele has been isolated from an AA patient.20 Loss in a recurrently mutated HLA allele may characterize a specific immune pathogenesis and associated clinical manifestations. Previous studies suggested better IST responses and survival in patients with HLA loss11,18,21 and poor outcomes, including frequent clonal evolution, in patients who harbored HLA alleles related to somatic mutations, irrespective of somatic loss of an HLA allele.19
Clonality is common in AA.22 Paroxysmal nocturnal hemoglobinuria (PNH)-clones, cells deficient in glycosylphosphatidylinositol (GPI)-anchored proteins due to acquired PIGA mutations, are most frequent. PNH is closely related to immune marrow failure, and the GPI anchor itself may be a target of immune attack.23,24 Somatic mutations are also present in genes recurrently mutated in myelodysplastic syndromes or acute myeloid leukemia (AML), especially DNMT3A, ASXL1, and BCOR.17 However, the allelic burden of these clones in AA are usually small, clones remain stable for years, and affected cells infrequently drive evolution to myeloid neoplasms, which are usually characterized by complete or partial loss of chromosome 7.1,25,26 Of all clonal associations, the mechanism of HLA loss is most clearly related to escape from immune cell destruction.
Our aim was to clarify and enlarge on the clinical significance of HLA class I allele loss and recurrently mutated genotypes in a large cohort of patients with immune AA.
Methods
Study design and participants
A total of 544 patients with AA, aged 2 years or older, treated at the NIH Clinical Center, Bethesda, MD, between May 1999 and August 2019 were enrolled in this retrospective study. Patients were enrolled in various IST protocols approved by the Institutional Review Board of the National Heart, Lung, and Blood Institute (clinicaltrials.gov: NCT00001964, NCT00061360, NCT00195624, NCT00260689, NCT00944749, NCT01193283, and NCT01623167), which entailed informed consent. All horse antithymocyte globulin (hATG)-based IST protocols have been published2,27-29 and briefly described in the supplemental Methods (available on the Blood Web site).
Definitions
All definitions have been consistent across protocols. Severe AA was diagnosed when at least 2 of the following 3 criteria were met: a neutrophil count <0.5 × 109/L, a reticulocyte count <60 × 109/L, and a platelet count <20 × 109/L. Hematologic responses were assessed 6 months after institution of hATG: an overall response was defined as blood counts no longer meeting the above stated criteria for severe AA; a complete response required a neutrophil count of at least 1.0 × 109 cells/L, a hemoglobin level of at least 10 g/dL, and a platelet count of at least 100 × 109/L. High-risk clonal evolution was defined as the acquisition of either chromosome 7 abnormalities, complex cytogenetics, myelodysplastic syndrome, or AML, and low-risk clonal evolution was the other cytogenetic change, as previously published.30
Detection of HLA class I loss
Detailed methods for the detection of HLA loss are described in the supplemental Methods. In brief, HLA loss was assessed by either HLA flow cytometry plus deep nucleotide sequencing or deep sequencing alone. HLA flow cytometry was performed using cryopreserved peripheral blood mononuclear cells (PBMCs) stained with HLA-A and HLA-B allele-specific monoclonal antibodies and assessed in CD33hi monocytes. Deep sequencing of HLA class I genes was performed using sorted peripheral blood cell subpopulations, including HLA allele-lacking and expressing monocytes or using whole blood for the deep sequencing-only samples. HLA class I genes were enriched by locus-specific long-range PCR, as previously described.31 To precisely detect low-frequency mutations, we used hla-mapper (version 2.3),32 and used thresholds defined by base-position error rates in T-cell samples, as previously published for non-HLA genes.33,34 We inferred 6p LOH using read counts of allele-specific single-nucleotide polymorphisms in HLA genes.
Statistics
Statistical analyses were performed using R (version 4.0.3) with EZR package (version 1.54)35; individual statistical methods are described in the supplemental Methods. HLA allele frequencies in patients were compared with those in in the National Marrow Donor Program (NMDP) datasets according to 4 wide ethnic groups including Whites, African Americans, Hispanics, and Asians,36 and the results were combined by fixed-effect meta-analysis to elucidate consistent impacts across ethnic groups.
Data sharing statement
All somatic HLA gene mutation information is available in the supplemental Data.
Results
Patients
All 544 patients had HLA allele genotyping performed during evaluation on clinical protocols; 515 of the 544 were treatment-naïve severe AA, and 416 of 515 were initially treated with hATG-based standard IST (Figure 1). Clinical characteristics of the patients are summarized in Table 1. The median time to clonal evolution and observation period after hATG treatment was 25 (interquartile range [IQR], 6 to 41) months, and 41 (IQR, 22 to 61) months, respectively. Of the 544 total subjects, 412 were tested for the presence of HLA allele loss.
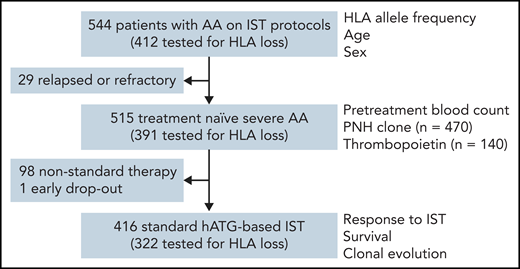
Patients and clinical parameters studied. Correlations of clinical parameters with HLA genotype and HLA loss were tested in subgroups based on the availability of data.
Patients and clinical parameters studied. Correlations of clinical parameters with HLA genotype and HLA loss were tested in subgroups based on the availability of data.
Clinical characteristics of patients
| n | Value, n (%) | |
|---|---|---|
| Age, y | 544 | 29 (18-54) |
| Range | 2-82 | |
| Male sex | 544 | 304 (56) |
| Ethnicity | 544 | |
| White | 275 (51) | |
| African American | 120 (22) | |
| Hispanic | 109 (20) | |
| Asian | 35 (6) | |
| Other | 5 (1) | |
| Disease status | 544 | |
| Treatment naïve severe AA | 515 (95) | |
| Relapsed or refractory AA | 29 (5) | |
| Pretreatment blood values | ||
| Neutrophil count, ×109/L | 515 | 0.29 (0.09-0.51) |
| Lymphocyte count, ×109/L | 515 | 1.27 (0.92-1.63) |
| Reticulocyte count, ×109/L | 515 | 15.3 (6.5-32.0) |
| Platelet count, ×109/L | 515 | 9 (5-13) |
| Thrombopoietin, ng/mL | 140 | 2610 (2220-3080) |
| PNH clone ≥1% | 470 | 177 (38) |
| hATG-based IST | 416 | |
| hATG and CsA | 102 (25) | |
| hATG, CsA, and MMF | 103 (25) | |
| hATG, CsA, and rapamycin | 35 (8) | |
| hATG, CsA, and EPAG | 176 (42) | |
| Response to IST | 416 | |
| Overall response | 293 (70) | |
| Complete response | 101 (24) | |
| Clonal evolution | 416 | |
| High-risk clonal evolution | 31 (7) | |
| Low-risk clonal evolution | 26 (6) |
| n | Value, n (%) | |
|---|---|---|
| Age, y | 544 | 29 (18-54) |
| Range | 2-82 | |
| Male sex | 544 | 304 (56) |
| Ethnicity | 544 | |
| White | 275 (51) | |
| African American | 120 (22) | |
| Hispanic | 109 (20) | |
| Asian | 35 (6) | |
| Other | 5 (1) | |
| Disease status | 544 | |
| Treatment naïve severe AA | 515 (95) | |
| Relapsed or refractory AA | 29 (5) | |
| Pretreatment blood values | ||
| Neutrophil count, ×109/L | 515 | 0.29 (0.09-0.51) |
| Lymphocyte count, ×109/L | 515 | 1.27 (0.92-1.63) |
| Reticulocyte count, ×109/L | 515 | 15.3 (6.5-32.0) |
| Platelet count, ×109/L | 515 | 9 (5-13) |
| Thrombopoietin, ng/mL | 140 | 2610 (2220-3080) |
| PNH clone ≥1% | 470 | 177 (38) |
| hATG-based IST | 416 | |
| hATG and CsA | 102 (25) | |
| hATG, CsA, and MMF | 103 (25) | |
| hATG, CsA, and rapamycin | 35 (8) | |
| hATG, CsA, and EPAG | 176 (42) | |
| Response to IST | 416 | |
| Overall response | 293 (70) | |
| Complete response | 101 (24) | |
| Clonal evolution | 416 | |
| High-risk clonal evolution | 31 (7) | |
| Low-risk clonal evolution | 26 (6) |
Values are n (%) or median (IQR).
CsA, cyclosporine; MMF, mycophenolate mofetil.
Detection of HLA class I gene mutations
Our sequencing method allowed us to identify variant allele frequency as low as 0.3% in more than 50% of base positions of single nucleoide variants and 90% of indels (supplemental Figure 1). The accuracy of the method was supported by the following observations and experiments: HLA class I gene mutations were not detected from additional 16 control T-cell samples nor from monocytes of patients expressing respective HLA alleles as determined by flow cytometry (except for missense mutations and 1 intronic mutation); somatic mutations detected from patient samples were generally predicted to inactivate affected HLA alleles, irrespective of variant allele frequencies and samples (supplemental Figure 2), in contrast to pseudomutations created by PCR errors (supplemental Figure 3); serial mixing of HLA-lacking monocytes and HLA-expressing T cells showed a linear correlation between variant allele frequency and mixing ratio (supplemental Figure 4). Low-frequency mutations were confirmed by droplet digital PCR (supplemental Figure 5), and functional HLA loss was demonstrated in the large cohort of samples also studied by flow cytometry. Small cell populations deficient in surface HLA-A or HLA-B antigens were not detected in 41 healthy individuals by flow cytometry at a threshold of 0.1%, consistent with the very infrequent spontaneous inactivating mutations in leukocytes of healthy individuals.37,38 Deep sequencing could detect 6p LOH at a clone size of 15% (supplemental Figure 6).
Loss of HLA class I allele
HLA class I allele loss was detected in 92 (22%) of 412 patients (Figure 2A) by flow cytometry plus deep sequencing of HLA allele-expressing and -deficient cells (51 of 201) (supplemental Figures 7 and 8) or only whole blood deep sequencing (41 of 211). A total of 393 somatic HLA gene mutations (supplemental Data) were identified from 92 patients, in HLA-A (n = 124), HLA-B (n = 267), and HLA-C (n = 2); 29 of the 92 patients also had 6p LOH and a further 11 patients had 6p LOH only. Multiple mutant clones (median, 5 [range, 2 to 20]) with HLA gene mutations or with 6p LOH coexisted in 62 of the 92 patients, almost always restricted to a single HLA-A or HLA-B allele in individuals. HLA-B*14:02, HLA-A*02:01, HLA-B*40:02, HLA-B*07:02, and HLA-B*08:01 were frequently absent (Figure 2B). The median proportion of cells with HLA loss (combined size of all clones with HLA gene mutations and 6p LOH) was 16.3% (range, 0.2% to 100%; IQR, 2.4% to 62%) (Figure 2C).

HLA class I allele loss. (A) Number of patients with HLA loss due to HLA gene mutations, 6p LOH, or both in a total of 412 subjects tested. (B) The number and proportion of patients who lacked individual HLA alleles (left); the total and mean number of clones with HLA gene mutations and 6p LOH among patients who lost respective HLA alleles (right). The number of patients tested for the presence of HLA loss is shown in parenthesis. (C) Combined clone size of cells with HLA loss (upper) and the clone size of individual HLA gene mutations and 6p LOH among cells with HLA loss (lower). (D) Positions and types of somatic inactivating mutations in HLA-A (n = 123) and HLA-B (n = 267). Hotspot mutations are noted on the figure.
HLA class I allele loss. (A) Number of patients with HLA loss due to HLA gene mutations, 6p LOH, or both in a total of 412 subjects tested. (B) The number and proportion of patients who lacked individual HLA alleles (left); the total and mean number of clones with HLA gene mutations and 6p LOH among patients who lost respective HLA alleles (right). The number of patients tested for the presence of HLA loss is shown in parenthesis. (C) Combined clone size of cells with HLA loss (upper) and the clone size of individual HLA gene mutations and 6p LOH among cells with HLA loss (lower). (D) Positions and types of somatic inactivating mutations in HLA-A (n = 123) and HLA-B (n = 267). Hotspot mutations are noted on the figure.
Several “hotspots” were observed (Figure 2D; supplemental Figure 9): nonsense mutation R7X (n = 55) and frameshift mutation R7fs (n = 28) in both HLA-A and HLA-B; and start codon mutation (n = 15) and missense mutation T118P (n = 11) in HLA-B. Mutations in the start codon were detected only for HLA-B*40:02, HLA-B*27:05, and HLA-B*13:01, which lack the second start signal in the fourth codon (supplemental Figure 10).
When assessed by flow cytometry, all frameshift mutations (n = 76), nonsense mutations (n = 72), intronic mutations (n = 24), and even synonymous mutations reduced cell surface expression of affected HLA alleles (except for 1 mutation in HLA-A intron 7). Missense mutations were inconsistently associated with loss of expression: 21 of 69 expressed HLA antigens on the cell surface, and for 48, there was a loss of expression (supplemental Figure 11A); 6 of the 21 missense mutations that were expressed on cell surface located to the peptide-binding groove (S37F [HLA-B*08:01], F57L [HLA-B*40:01], L102Q [HLA-B*40:01], H117Y [HLA-A*02:01], T118P [HLA-B*40:02], and D126Y [HLA-B*14:02]) (supplemental Figure 11B), where they might interact with antigens presented by HLA alleles; another 13 missense mutations were located in the α-3 domain where they could associate with CD8 molecules on cytotoxic T cells (supplemental Figure 11C).18 Eleven missense mutations and 2 synonymous mutations were predicted to create alternative splice sites and generate truncated proteins (supplemental Table 1). Of note, 12 of 24 functionally confirmed intronic mutations and 7 unconfirmed mutations did not involve donor 5′-GT nor acceptor AG-3′ canonical splice sites (supplemental Table 2): 8 of these mutations proximate to the splice sites have been predicted to reduce splicing efficiency; 6 mutations at 19 to 21 nucleotides from the 3′ splice site were found to impair the consensus branchpoint sequences, CTGAC, CTCAC, or CTCAG, also critical for splicing39; the remaining 5 mutations in the middle of HLA-B intron 3 (c.619 + 123A>G and c.619 + 133C>G) have been reported to create alternative 5′ splice sites.18 One mutation in intron 7 of HLA-A and 2 mutations in cytoplasmic tail (exon 7) of HLA-C (S357A and E359del) were assumed to be passenger mutations, as these clones were small (1.0%, 3.0%, and 0.6%) and multiple clones with HLA-B mutations (14, 7, and 6) were also detected from the 3 patients.
The proportions of HLA allele-lacking cells in total monocytes were reduced in response to IST plus EPAG in all 18 patients sequentially tested, irrespective of hematological responses, and in 2 patients, HLA allele-lacking cells had disappeared at 6 months; they were thereafter stable over many years in 13 patients (supplemental Figure 12), as has been reported.18
HLA loss and other hematopoietic clones
HLA loss was relatively infrequent among patients with other types of clonal hematopoiesis compared with patients without clonal hematopoiesis, including PNH clones (18% [24 of 136] vs 24% [54 of 225], P = .19) (supplemental Figure 13A; supplemental Table 4) and somatic myeloid neoplasm gene (20% [4 of 20] vs 28% [33 of 119], P = .25) (supplemental Figure 14). When PNH clones and HLA loss were simultaneously analyzed by sensitive flow cytometry, PNH clones at a median frequency of 4.6% (range, 0.1% to 39%) in total monocytes were present in 27 of the 51 patients (53%) with concomitant HLA loss; such PNH clones always retained cell surface expression of HLA class I (supplemental Figure 13B-C). Neither PNH clone nor clones mutated in myeloid malignancy-related genes correlated with loss of specific HLA alleles.
Clinical significance of HLA class I allele loss and individual alleles
Recurrently inactivated class I alleles HLA-B*14:02, HLA-B*40:02, and HLA-B*07:02 were significantly overrepresented in patients with AA (Table 2; supplemental Table 3).
HLA allele frequencies in patients with AA and healthy individuals in the NMDP dataset
| HLA allele | Allele frequency, % (phenotype frequency, %) | Odds ratio of allele frequency (95% CI) | |||||||
|---|---|---|---|---|---|---|---|---|---|
| White | African American | Hispanic | Asian | ||||||
| AA n = 275 | Control n = 3 912 440 | AA n = 120 | Control n = 505 250 | AA n = 109 | Control n = 712 764 | AA n = 35 | Control n = 568 597 | ||
| HLA-B*14:02 | 7.3 (12.7) | 2.7 (5.4) | 5.0 (10.0) | 2.2 (4.3) | 7.8 (14.7) | 4.2 (8.3) | 0.0 (0) | 0.2 (0.3) | 2.44 (1.91-3.12) |
| HLA-B*40:02 | 2.4 (4.7) | 1.3 (2.7) | 0.4 (0.8) | 0.3 (0.7) | 7.3 (13.8) | 5.0 (9.7) | 11.4 (20.0) | 2.4 (4.8) | 1.88 (1.36-2.61) |
| HLA-B*07:02 | 17.6 (32.4) | 12.5 (23.4) | 8.8 (15.8) | 7.3 (14.0) | 8.3 (16.5) | 5.8 (11.2) | 1.4 (2.9) | 2.9 (5.8) | 1.42 (1.19-1.70) |
| HLA-A*02:01 | 28.4 (50.5) | 27.2 (46.9) | 12.9 (24.2) | 12.2 (23.0) | 17.0 (29.4) | 21.0 (37.5) | 7.1 (11.4) | 8.4 (16.2) | 0.99 (0.86-1.15) |
| HLA-B*08:01 | 9.5 (17.8) | 10.6 (20.0) | 3.8 (7.5) | 3.7 (7.2) | 4.6 (9.2) | 4.2 (8.2) | 0 (0) | 1.8 (3.5) | 0.92 (0.72-1.17) |
| HLA allele | Allele frequency, % (phenotype frequency, %) | Odds ratio of allele frequency (95% CI) | |||||||
|---|---|---|---|---|---|---|---|---|---|
| White | African American | Hispanic | Asian | ||||||
| AA n = 275 | Control n = 3 912 440 | AA n = 120 | Control n = 505 250 | AA n = 109 | Control n = 712 764 | AA n = 35 | Control n = 568 597 | ||
| HLA-B*14:02 | 7.3 (12.7) | 2.7 (5.4) | 5.0 (10.0) | 2.2 (4.3) | 7.8 (14.7) | 4.2 (8.3) | 0.0 (0) | 0.2 (0.3) | 2.44 (1.91-3.12) |
| HLA-B*40:02 | 2.4 (4.7) | 1.3 (2.7) | 0.4 (0.8) | 0.3 (0.7) | 7.3 (13.8) | 5.0 (9.7) | 11.4 (20.0) | 2.4 (4.8) | 1.88 (1.36-2.61) |
| HLA-B*07:02 | 17.6 (32.4) | 12.5 (23.4) | 8.8 (15.8) | 7.3 (14.0) | 8.3 (16.5) | 5.8 (11.2) | 1.4 (2.9) | 2.9 (5.8) | 1.42 (1.19-1.70) |
| HLA-A*02:01 | 28.4 (50.5) | 27.2 (46.9) | 12.9 (24.2) | 12.2 (23.0) | 17.0 (29.4) | 21.0 (37.5) | 7.1 (11.4) | 8.4 (16.2) | 0.99 (0.86-1.15) |
| HLA-B*08:01 | 9.5 (17.8) | 10.6 (20.0) | 3.8 (7.5) | 3.7 (7.2) | 4.6 (9.2) | 4.2 (8.2) | 0 (0) | 1.8 (3.5) | 0.92 (0.72-1.17) |
Allele frequency is copy number/2n. The phenotype frequency in the NMDP donors was estimated by the equation of (phenotype frequency) = 1 - (1 - [allele frequency])2.
Patient age did not correlate with HLA loss in general, but did correlate with 2 mutation-associated genotypes HLA-B*07:02 (median age [IQR], 46 [22-64] years vs 27 [18-49] years, P < .0001) (supplemental Table 4) and HLA-B*40:01 (median age [IQR], 46 [27-65] years vs 28 [18-53] years, P = .0022), and therefore, HLA-B*40:01 was exclusively overrepresented among patients 40 years or older compared with healthy individuals (odds ratio 1.56 [95% CI, 1.06-2.52]). Moreover, 7 and 2 patients who lost HLA-B*07:02 and HLA-B*40:01 were also older, with a median age of 65 and 68 years, respectively (supplemental Figure 15; supplemental Table 5). These findings suggested an age-associated immune pathogenesis involving specific HLA-B alleles.
Correlations of HLA loss and HLA genotypes with pretreatment blood values and hematologic responses to IST were weak (supplemental Table 4). However, loss of HLA-B*40:02 and certain minor alleles was related to higher blood counts, and all patients who lost these alleles responded to IST (supplemental Figure 16; supplemental Table 5). These relationships are consistent with the predictive value of reticulocytes for IST response,40,41 and the generally high response rate to IST in Japanese patients with HLA loss, in whom HLA-B*40:02 was most frequently inactivated,18,42 but HLA-B*14:02 and HLA-B*08:01 genotypes are virtually absent.43
High-risk clonal evolution was correlated with the most frequently inactivated allele HLA-B*14:02 (P = .0092) (Figure 3A), as previously suggested by Babushok et al,19 and with HLA loss (P = .00040) (Figure 3B), especially with the loss of HLA-A*02:01 and HLA-B*14:02 (supplemental Figure 17A; supplemental Table 5). In particular, HLA-B*14:02 genotype was strongly correlated among patients without HLA loss (P = .0072), while none of 43 patients who did not carry any of the mutation-associated HLA genotypes exhibited high-risk clonal evolution (supplemental Figure 17B-C); this suggested that the presence of specific HLA class I-mediated immunity, rather than HLA loss itself, predisposed to high-risk clonal evolution. A combined HLA risk, defined as the presence of HLA loss or HLA-B*14:02 genotype (irrespective of loss), yielded a better prediction factor for high-risk clonal evolution than did either HLA loss alone or HLA-B*14:02 alone (supplemental Figure 17D). The HLA associations with high-risk clonal evolution were independent of patient age (Table 3; supplemental Table 6), a known predictor of secondary myeloid malignancies (Figure 3C).44,45 A prediction model for high-risk clonal evolution incorporating the combined HLA risk and age 40 years or older clearly divided patients into 3 risk groups (Figure 3D; supplemental Figure 17E), which was validated in various subgroups of patients (supplemental Figure 18).
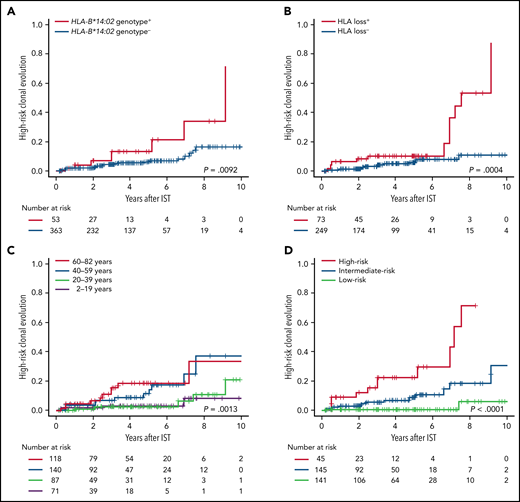
High-risk clonal evolution. Cumulative incidence of high-risk clonal evolution, defined as the acquisition of either chromosome 7 abnormalities, complex cytogenetics, myelodysplastic syndrome, or AML after institution of hATG-based IST, are shown according to the presence or absence of HLA-B*14:02 genotype (A), the presence or absence of HLA loss (B), age groups (C), and 3 risk groups of the prediction model for high-risk clonal evolution incorporating HLA-B*14:02 genotype, HLA loss, and age (D): a high-risk group, any HLA risk (HLA-B*14:02 genotype or HLA loss) present and aged 40 years or older; a low-risk group, no HLA risk present and aged less than 40 years; and an intermediate-risk group, not meeting the criteria for groups of high-risk nor low-risk.
High-risk clonal evolution. Cumulative incidence of high-risk clonal evolution, defined as the acquisition of either chromosome 7 abnormalities, complex cytogenetics, myelodysplastic syndrome, or AML after institution of hATG-based IST, are shown according to the presence or absence of HLA-B*14:02 genotype (A), the presence or absence of HLA loss (B), age groups (C), and 3 risk groups of the prediction model for high-risk clonal evolution incorporating HLA-B*14:02 genotype, HLA loss, and age (D): a high-risk group, any HLA risk (HLA-B*14:02 genotype or HLA loss) present and aged 40 years or older; a low-risk group, no HLA risk present and aged less than 40 years; and an intermediate-risk group, not meeting the criteria for groups of high-risk nor low-risk.
Fine-Gray proportional hazard regression for high-risk clonal evolution
| Univariate model | Multivariate model 1 | Multivariate model 2 | |||||||
|---|---|---|---|---|---|---|---|---|---|
| HR | 95% CI | P | HR | 95% CI | P | HR | 95% CI | P | |
| Age ≥40 y | 4.00 | 1.90-8.40 | .00026 | 4.87 | 2.16-11.0 | .00014 | 4.73 | 2.10-10.7 | .00018 |
| HLA-B*14:02 genotype | 2.83 | 1.32-6.10 | .0077 | 2.47 | 1.01-6.05 | .048 | |||
| HLA loss | 3.68 | 1.77-7.66 | .00050 | 2.70 | 1.20-6.08 | .017 | |||
| Combined HLA risk† | 4.44 | 2.04-9.68 | .00018 | 4.26 | 1.02-9.48 | .00038 | |||
| Univariate model | Multivariate model 1 | Multivariate model 2 | |||||||
|---|---|---|---|---|---|---|---|---|---|
| HR | 95% CI | P | HR | 95% CI | P | HR | 95% CI | P | |
| Age ≥40 y | 4.00 | 1.90-8.40 | .00026 | 4.87 | 2.16-11.0 | .00014 | 4.73 | 2.10-10.7 | .00018 |
| HLA-B*14:02 genotype | 2.83 | 1.32-6.10 | .0077 | 2.47 | 1.01-6.05 | .048 | |||
| HLA loss | 3.68 | 1.77-7.66 | .00050 | 2.70 | 1.20-6.08 | .017 | |||
| Combined HLA risk† | 4.44 | 2.04-9.68 | .00018 | 4.26 | 1.02-9.48 | .00038 | |||
Presence of HLA loss or HLA-B*14:02 genotype.
Origin of monosomy 7
We further studied somatic HLA class I gene mutations in monosomy 7 clones evolved in 11 patients with immune AA, using cryopreserved PBMCs; 4 of the patients had HLA allele lacking-leukocytes present before clonal evolution, and 3 did not, and in the other 4 cases, samples prior to the evolution event were not available. In 2 patients with known HLA allele loss, monosomy 7 clonal evolution was associated with the expansion of clones with a preexisting mutation within intron 1 of HLA-A*02:01 (c.74-19A>G) and HLA-B*40:02 (c.74-9C>A); before clonal evolution, their blood was supported by 15 and 19 clones with HLA gene mutations including the intron 1 mutant clone and a PNH clone (Figure 4; supplemental Tables 7-9). The clone with an HLA-B*40:02 intron 1 mutation could be isolated by flow cytometry, as this mutation partially expressed HLA-B4002 (B4002dim); fluorescent in situ hybridization (FISH) confirmed monosomy 7 was a subclone of this B4002dim intron 1 mutant clone. A monosomy 7 clone accounting for 3% of total monocytes and <1% of total mononuclear cells was detected by FISH from the sorted B4002dim clone (11% in B4002dim cells) at least 2 years before clinical diagnosis of monosomy 7, although bone marrow (BM) cytogenetics at this time and 1 year later were normal. In the other 9 patients, somatic mutations were not detected from HLA-A, HLA-B, and HLA-C genes in the monosomy 7 clones. Similar intron 1 mutations in HLA class I genes were observed in 2 other patients with AA, and 1 of the 2 acquired 7p deletion and t(7;17)(q22;q11.2), implying a possible relationship of this intron 1 mutation with chromosome 7 abnormalities. No PNH clones in 9 patients expanded after monosomy 7 clonal evolution (supplemental Figure 13D), consistent with previous observations.25
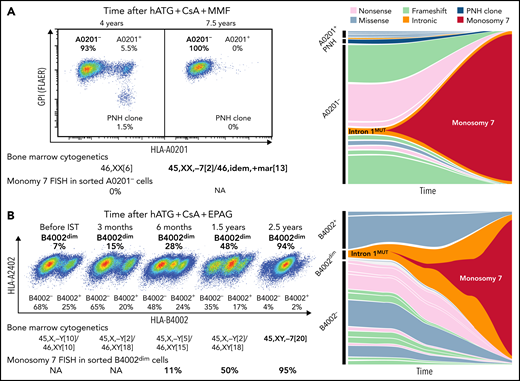
Clonal evolution of monosomy 7 from HLA class I allele-lacking clones. (A) A 56-year-old female at the time of severe AA diagnosis, who received hATG, CsA, and mycophenolate mofetil (MMF), and multiple rounds of salvage IST, including rabbit ATG, daclizumab and danazol, and alemtuzumab, without adequate response, had clonal evolution to monosomy 7 approximately 7.5 years after her initial diagnosis of AA and died 6 months later. Her blood, sampled 4 years after the institution of initial IST, contained a PNH clone and HLA-A0201-lacking cells (A0201-), consistent with the diagnosis of immune AA. Multiple clones with HLA-A*02:01 deactivating mutations constituted sorted A0201- cells (multiple clones with missense mutations also did A0201+ cells), from which chromosome 7 deletion was not detected by fluorescent in situ hybridization (FISH). After 3.5 years, virtually all hematopoiesis was replaced by a preexisting A0201- clone with an intron 1 mutation (intron 1mut; c.74-19A>G) that had acquired monosomy 7. (B) A 72-year-old man at the time of severe AA diagnosis was treated with hATG, CsA, and EPAG with a partial response at 6 months but had declining cytopenia which responded to reinitiation of CsA. About 2.5 years from the initial diagnosis and IST treatment, BM cytogenetics revealed monosomy 7, and myelodysplastic syndrome was diagnosed. The patient received symptomatic treatment (infrequent blood transfusions twice a year) and died of pneumonia 3.5 years later. HLA flow cytometry of cryopreserved cells revealed a gradual expansion of a clone partially lacking HLA-B4002 (B4002dim), which was attributed to an HLA-B*40:02 intron 1 mutation (c.74-9C>A). A small monosomy 7 clone that was not visible by conventional BM cytogenetics by Giemsa-banding was detected by FISH from the sorted B4002dim cells at least 2 years before clinical diagnosis of monosomy 7. NA, not assessed (due to deficient cryopreserved cells for analysis). See also supplemental Tables 8 and 9.
Clonal evolution of monosomy 7 from HLA class I allele-lacking clones. (A) A 56-year-old female at the time of severe AA diagnosis, who received hATG, CsA, and mycophenolate mofetil (MMF), and multiple rounds of salvage IST, including rabbit ATG, daclizumab and danazol, and alemtuzumab, without adequate response, had clonal evolution to monosomy 7 approximately 7.5 years after her initial diagnosis of AA and died 6 months later. Her blood, sampled 4 years after the institution of initial IST, contained a PNH clone and HLA-A0201-lacking cells (A0201-), consistent with the diagnosis of immune AA. Multiple clones with HLA-A*02:01 deactivating mutations constituted sorted A0201- cells (multiple clones with missense mutations also did A0201+ cells), from which chromosome 7 deletion was not detected by fluorescent in situ hybridization (FISH). After 3.5 years, virtually all hematopoiesis was replaced by a preexisting A0201- clone with an intron 1 mutation (intron 1mut; c.74-19A>G) that had acquired monosomy 7. (B) A 72-year-old man at the time of severe AA diagnosis was treated with hATG, CsA, and EPAG with a partial response at 6 months but had declining cytopenia which responded to reinitiation of CsA. About 2.5 years from the initial diagnosis and IST treatment, BM cytogenetics revealed monosomy 7, and myelodysplastic syndrome was diagnosed. The patient received symptomatic treatment (infrequent blood transfusions twice a year) and died of pneumonia 3.5 years later. HLA flow cytometry of cryopreserved cells revealed a gradual expansion of a clone partially lacking HLA-B4002 (B4002dim), which was attributed to an HLA-B*40:02 intron 1 mutation (c.74-9C>A). A small monosomy 7 clone that was not visible by conventional BM cytogenetics by Giemsa-banding was detected by FISH from the sorted B4002dim cells at least 2 years before clinical diagnosis of monosomy 7. NA, not assessed (due to deficient cryopreserved cells for analysis). See also supplemental Tables 8 and 9.
Discussion
Our retrospective study of HLA genes in many hundreds of patients with immune AA demonstrated novel findings regarding HLA allele frequency, loss of HLA class I alleles, and important clinical correlations.
We confirmed the high prevalence of HLA class I alleles recurrently inactivated by somatic mutations across 4 major ethnic groups in the United States, which included previously reported HLA-B*14:02 and HLA-B*40:02,18,19,46 and an unreported allele HLA-B*07:02. Further, correlations of HLA loss with patient age at disease onset, peripheral blood counts, IST-response, and high-risk clonal evolution suggest specific pathogeneses related to certain HLA class I alleles.
In cancer generally, HLA allele loss is frequent and represents an escape from immune surveillance,47,48 which clinically correlates with poor outcomes after treatment with immune checkpoint inhibitors49 and relapse of AML after HCT.50 While HLA loss in immune AA is protective of the stem cell under self-destructive immune attack, it also should allow escape from immune surveillance and therefore of malignant clonal evolution. Indeed, we observed this association and tracked it in 2 patients. Nevertheless, high-risk clonal evolution is more likely consequent to HLA class I–mediated destruction of BM cells than due to immune escape, as HLA-B*14:02, the most frequently mutated and overrepresented class I allele in AA, was associated with high-risk clonal evolution irrespective of HLA mutation status, as previously suggested.19 Most (but not all) monosomy 7 clonal evolution arose from cells with intact HLA genes; high-risk evolution was not observed in any patient without an HLA allele prone to loss. A simple prediction model incorporating HLA loss and HLA-B*14:02, as a convincing indicator of class I-mediated pathogenesis, in addition to age, stratified the incidence of high-risk clonal evolution, even within subgroups. Monitoring for clonal evolution after IST is desirable in patients with HLA allele loss or HLA-B*14:02; early HCT could be considered for the high-risk group patients aged 40 years or older rather than IST as generally recommended for the aged individuals.51
The underlying mechanisms responsible for the predominant development of monosomy 7 in immune AA (and other marrow failure syndromes) remain elusive.52 We detected a small monosomy 7 clone not visible by conventional BM cytogenetics at least 2 years before the clinical diagnosis of clonal evolution in 1 patient; extended time periods may be necessary for aneuploid clones (and somatically mutated clones53) to expand and become clinically evident, either by accumulating additional genetic aberrations or altered environmental selection. Relatively infrequent mutations in non-HLA genes among patients with HLA loss argue against HLA gene mutations arising due to an intrinsically higher somatic mutation rate or from genetic drift in a paucicellular stem cell pool.24 More sensitive methods would benefit both the monitoring and deeper understanding of secondary myeloid malignancies in AA.
PNH clones and HLA allele-lacking clones are both assumed to represent immune escape of HSCs from immune cell attack and destruction, manifested by the recurrent presence of multiple clones with somatic mutations in PIGA54 or HLA class I genes.18 PNH and HLA absent clones were always mutually exclusive, as in the 27 patients who had both types of clones. Loss of surface GPI-anchored proteins may confer no additional survival advantage on HLA allele loss and the reverse. A remarkable difference between the 2 types of escape clones was their relationship to clonal evolution, which occurred regardless of the presence of PNH clones but much more frequently in patients with HLA allele loss. Malignant clones always evolved from non-PNH phenotype cells as previously reported,25 but they did arise from clones with HLA allele loss.
Our deep sequencing method for HLA class I genes allowed detection of somatic mutations at an allele frequency as low as 0.3%. Many coexisting small clones with somatic mutations in HLA-A and HLA-B alleles were identified, most of which functionally confirmed by flow cytometry. Even intronic mutations not involving canonical splice sites and a synonymous mutation were found to reduce cell surface expression of HLA class I alleles; such mutations likely have been underreported in studies using exome or RNA sequencing data and in those lacking functional validation. A limitation of our sequencing method was the lower sensitivity for 6p LOH detection, the result of unbalanced amplification of heterozygous HLA alleles, and the use of whole blood for some patients. Underestimation of 6p LOH would not be predicted to affect the overall detection of HLA loss, as most 6p LOH clones coexist with clones with inactivating mutations in an HLA allele, but detection of such small clones may be of value if they predicted secondary myeloid malignancies.
In summary, our study suggested the presence of distinct pathogenic pathways related to HLA genotype and loss in immune AA. Most important in the clinic was the relationship between high-risk malignant clonal evolution and class I-mediated immunity, as for the most frequently inactivated HLA-B*14:02 genotype and HLA loss in general, which provide a simple prediction tool for this serious complication.
Acknowledgments
This work was supported by the Intramural Research Program of the National Heart, Lung, and Blood Institute (NIH), and the NIH Clinical Center (ZIC CL002128). NCT01623167 is funded by Novartis and previously by GSK through a cooperative research and development agreement (CRADA). The authors acknowledge the support of the National Heart, Lung, and Blood Institute core facilities and the CCR Genomics Core facility: cell sorting was performed at the Flow Cytometry Core Facility, deep sequencing was performed at the DNA Sequencing and Genomics Core Facility, bioinformatics analysis was performed at the Bioinformatics and Computational Biology Core Facility, and droplet digital PCR was performed at the CCR Genomics Core facility.
Authorship
Contribution: Y.Z., S.K., and N.S.Y. designed research; B.A.P., O.J.R., H.E., E.M.G., and N.S.Y. provided clinical care and collected clinical data; Y.Z., S.D.A., A.A.C.L., S.K., X.F., L.A., and D.Q.R. performed research; Y.Z. analyzed and interpreted data; Y.Z., R.S., and C.O.W. performed statistical analysis; and Y.Z., B.A.P., S.K., W.A.F., and N.S.Y. wrote the manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Yoshitaka Zaimoku, Hematology Branch, National Heart, Lung, and Blood Institute, National Institutes of Health, Bethesda, MD, 20892; e-mail: zaimokuyoshitaka@gmail.com.
Presented at the sixty-first annual meeting of the American Society of Hematology, Orlando, FL, 9 December 2019.
There is a Blood Commentary on this article in this issue.
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal