

Key Points
The protein landscape of CLL is governed by trisomy 12 and IGHV mutational status.
Reduced protein abundance buffering implicates the PI3K-AKT-MTOR pathway in the tumorigenic function of trisomy 12.

Abstract
Many functional consequences of mutations on tumor phenotypes in chronic lymphocytic leukemia (CLL) are unknown. This may be in part due to a scarcity of information on the proteome of CLL. We profiled the proteome of 117 CLL patient samples with data-independent acquisition mass spectrometry and integrated the results with genomic, transcriptomic, ex vivo drug response, and clinical outcome data. We found trisomy 12, IGHV mutational status, mutated SF3B1, trisomy 19, del(17)(p13), del(11)(q22.3), mutated DDX3X and MED12 to influence protein expression (false discovery rate [FDR] = 5%). Trisomy 12 and IGHV status were the major determinants of protein expression variation in CLL as shown by principal-component analysis (1055 and 542 differentially expressed proteins, FDR = 5%). Gene set enrichment analyses of CLL with trisomy 12 implicated B-cell receptor (BCR)/phosphatidylinositol 3-kinase (PI3K)/AKT signaling as a tumor driver. These findings were supported by analyses of protein abundance buffering and protein complex formation, which identified limited protein abundance buffering and an upregulated protein complex involved in BCR, AKT, MAPK, and PI3K signaling in trisomy 12 CLL. A survey of proteins associated with trisomy 12/IGHV-independent drug response linked STAT2 protein expression with response to kinase inhibitors, including Bruton tyrosine kinase and mitogen-activated protein kinase kinase (MEK) inhibitors. STAT2 was upregulated in unmutated IGHV CLL and trisomy 12 CLL and required for chemokine/cytokine signaling (interferon response). This study highlights the importance of protein abundance data as a nonredundant layer of information in tumor biology and provides a protein expression reference map for CLL.
Introduction
Chronic lymphocytic leukemia (CLL) is marked by genetic and clinical heterogeneity.1-5 The relationship between genetic and epigenetic changes in CLL and clone expansion and evolution is complex. Recently, integrated omics profiling including genomics (somatic mutations and copy-number variations), epigenomics (DNA methylation), transcriptomics (RNA expression), and ex vivo drug response (viability) phenotypes in large cohorts of patients has provided insight into CLL biology.6-8 Nonetheless, the relations of molecular layers of biology to tumor phenotypes remain partially unknown due to missing data on protein expression. Initial studies of the proteome landscape of CLL using mass spectrometry (MS)–based quantitative proteomics involved smaller patient sets (n = 6-18).9-12 Nevertheless, clear signatures of CLL were found, including overexpression of B-cell receptor (BCR) signaling components,9 dysregulation of spliceosome proteins,9 repression of protein kinase C signaling members,10 and distinct proteomic profiles for IGHV-unmutated CLL (U-CLL) and IGHV-mutated CLL (M-CLL)11,12. Studies in larger cohorts in the context of epigenetic, transcriptomic, drug response, and clinical outcome data, which could provide a multilayered understanding, have been missing. To address this, we analyzed 117 CLL patient samples with data-independent acquisition (DIA) MS using 2 different measurement platforms (Lumos and timsTOF [trapped ion mobility spectrometry coupled with quadrupole time-of-flight MS]).
Methods
CLL patient samples
Blood samples from CLL patients were obtained with informed consent and ethics approval (S-206/2011; S-356/2013).6 Selection criteria for the main cohort included (1) overall balanced sample numbers with respect to major genetic disease drivers, (2) sufficient proportion of lymphocytes in the peripheral blood (white blood cell counts >20 000 cells per mL), and (3) multiomics data availability. Additional patient samples were collected at the Hospital Clinic of Barcelona with informed consent and approval of the Institutional Ethics Committee. The distribution of mutations and patient characteristics are shown in Figure 1 and supplemental Tables-3 (available on the Blood Web site).

Overview. (A) Study outline.66 (B) Characteristics of patient samples included. Numbers shown in parentheses with each genetic aberration indicate the number of mutant samples. CNA, copy number alteration; FISH, fluorescence in situ hybridization; M, mutated; NA, not available; U, unmutated; WES, whole-exome sequencing; WGS, whole-genome sequencing; WT, wild type.
Overview. (A) Study outline.66 (B) Characteristics of patient samples included. Numbers shown in parentheses with each genetic aberration indicate the number of mutant samples. CNA, copy number alteration; FISH, fluorescence in situ hybridization; M, mutated; NA, not available; U, unmutated; WES, whole-exome sequencing; WGS, whole-genome sequencing; WT, wild type.
Cell preparation for MS
Blood was separated by Ficoll gradient centrifugation (GE Healthcare), and mononuclear cells (MNCs) were viably frozen. For patient samples 1 to 49 (batch 1) of the main cohort, MNCs were thawed and washed 3 times with phosphate-buffered saline. For patient samples 50 to 91 (batch 2) of the main cohort, CD19+ CLL cells were selected from MNCs using magnetic activated cell sorting following manufacturer’s instructions (Miltenyi Biotec). In both scenarios, 1 million cells per sample were pelleted and snap frozen in protein low-binding microcentrifuge tubes (Eppendorf). For patient samples 92 to 117 (batch 3) of the additional cohort, malignant B cells were isolated by fluorescence-activated cell sorting. A total of 100 000 CD19+, CD5+, light-chain restricted cells per patient sample were isolated into protein low-binding microcentrifuge tubes (Eppendorf), pelleted, and snap frozen. Further processing was performed as previously described,13 with small adjustments as outlined in supplemental Methods.
MS analysis
MS analysis was performed as described previously,13,14 with minor modifications. Details are given in supplemental Methods.
Raw data processing and quality control
For Lumos MS analysis, DDA and DIA files were imported into Spectronaut (Biognosys) v.13.415 (batch 1 of main cohort) or v.14.5 (batch 2 of main cohort and batch 3 of additional cohort) to create a hybrid library, which was used to search the DIA data against the SwissProt-reviewed subset of the human UniProt database (version 2019-07-09, 20 913 entries) with decoy sequences generated by sequence reversal. For timsTOF MS data analysis, the library was constructed from DIA files only using Spectronaut v14.4 and used to search the DIA data against SwissProt-reviewed data (version 2020-01-01, 20 367 entries) with decoys generated using “mutation”. Details on the Spectronaut settings are provided in supplemental Methods. The Spectronaut reports were exported and further processed in R (version 3.6.0). Normalization, quality control and transformation of raw protein abundance data were done with the R/Bioconductor package DEP.16 Proteins were selected for analysis if they showed <50% missing values across patient samples. The 50% cutoff choice follows similar previous studies.17-19 The protein abundance data were background corrected, scaled, and transformed using the variance stabilizing transformation approach of Huber et al and Karp et al.20,21
Integrative data analysis
To detect associations between protein abundance and categorical variables (gene mutations and copy-number variations), the proDA package was used.22 For the associations between protein abundance and continuous variables such as RNA expression and drug responses, the limma package was used, omitting missing values.23 For principal-component (PC) analysis and hierarchical clustering, missing values were imputed using the quantile regression imputation of left-censored data method.24 Gene set enrichment analysis was performed using CAMERA (correlation adjusted mean rank gene set test)25 from the limma package against gene sets from the Molecular Signature Database (MSigDB).26 Survival analysis was performed using the coxph package.27 Batch information was included as a covariate (“blocking factor”) in all regression models and hypothesis tests to avoid confounding effects. For exploratory analyses and visualizations, the comBat function implemented in the sva R package was used with the aim of removing batch effects.28 Differential RNA expression analysis was performed with DESeq229 and differential exon usage analysis with DEXSeq.30 We included IGHV mutation status and trisomy 12 as blocking factors in regression models for differential RNA and protein expression analyses. All association P values were adjusted for multiple testing using the Benjamini-Hochberg (>5 tests) or Bonferroni procedure (≤5 tests).
DNA-sequencing/RNA-sequencing/drug screen data
Multiomics profiling, including DNA sequencing and RNA sequencing, were previously performed, and these data are available in the R data package BloodCancerMultiOmics2017 from the Bioconductor project (http://bioconductor.org).6 The drug-sensitivity phenotypes, including the sensitivities of 45 patient samples to a panel of 63 small-molecule compounds at 5 concentrations, were characterized and processed as described previously.6
Western blot analysis for STAT2 expression in CLL
Western blots were performed on whole-cell protein lysates of patient MNC pellets according to standard techniques. Antibody details are provided in supplemental Methods. Western blots were developed using Clarity Western ECL Substrate (1705061; Bio-Rad). Chemiluminescence was detected using the imager machine ECL ChemoStar (INTAS). Signal intensities were recorded by densitometry (ImageJ software, version 1.53).
Generation of transgenic cell lines (CRISPR-Cas9)
The human CLL cell lines MEC-1, MEC-2, CII, HG-3, and PGA-1 (DSMZ) were transduced to stably express Cas9 and plasmids containing a nontargeting control or hSTAT2-targeting single guide RNA (34A and 36D) by transfection with lentiCas9-Blast vector (52962; Addgene), single guide RNA expressing vector (U6gRNA5 (BbsI)-PKGpuro2ABFP-W), lentiviral packaging plasmids pCMV-VSVg (8454; Addgene), and psPAX2 (12260; Addgene). Sequences and transfection details are provided in supplemental Methods.
Interferon α stimulation assay
One million cells of STAT2 wild-type and STAT2 knockout MEC-1, MEC-2, CII, HG-3, and PGA-1 cell lines were cultured in RPMI supplemented with 10% fetal calf serum, 50 U/mL penicillin, 50 μg/mL streptomycin, 1 μg/mL puromycin, 2 mM glutamine (all from Gibco) and stimulated with 500 IU/mL human interferon α A (Enzo Life Science) or maintained unstimulated. Cells were harvested after 6 hours for RNA isolation and quantitative polymerase chain reaction.
Quantitative polymerase chain reaction
RNA was isolated according to manufacturer’s instructions using the RNeasy Plus Mini Kit (Qiagen). Isolated messenger RNA (mRNA) was reverse transcribed using the SuperScript IV VILO Master Mix with ezDNase enzyme (SuperScript IV Vilo Master Mix with ezDNase Enzyme; Thermo Fisher Scientific) following the manufacturer’s instructions. We used TaqMan probes and master mix (TaqMan Fast Advanced Master Mix; Thermo Fisher Scientific) for quantitative analysis of complementary DNA. Details are provided in supplemental Methods.
Results
To study the consequences of gene mutations on protein expression in CLL, we measured protein abundances with DIA-MS on a total of 117 CLL samples and integrated it with genomic and transcriptomic data. A total of 91 patient samples constituted the main cohort, and 26 patient samples formed an additional cohort processed differently and enriched for individual genetic drivers (Figure 1). Only proteins identified by ≥2 proteotypic peptides were considered. A total of 3314 proteins were quantified in the main cohort, and 49 samples from the main cohort were also measured with an independent DIA-MS technology (timsTOF).14
To explore relationships between proteome and transcriptome, we computed Pearson correlation coefficients between RNA and protein levels for each gene. These varied widely, with a median of 0.18 (Figure 2A). There was no discernible influence of protein or RNA abundance on the protein-RNA correlation coefficient (supplemental Figure 1). While individual proteins showed high correlation, including ZAP70 (0.71), CD22 (0.74), and CD79a (0.51) (Figure 2B), the overall low correlation between RNA and protein abundance suggests that proteome abundance profiles contain independent information not implied by other omics data.
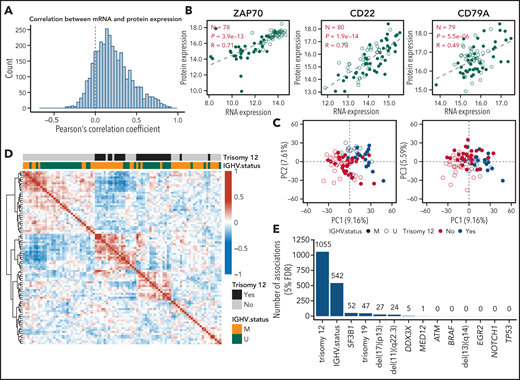
Global protein and RNA expression in CLL. (A) Distribution of Pearson’s correlation coefficient of mRNA and protein expression across 91 patient samples (main cohort). (B) Scatterplots comparing mRNA and protein expression (log2-transformed and median-normalized mRNA and protein intensities) for ZAP70, CD22, and CD79A. Each point represents a patient sample. P values and coefficients are from Pearson’s correlation tests. (C) Scatterplots showing PC analyses based on the expression of all quantified proteins. (D) Heatmap visualization of the Pearson correlation coefficients for all samples, calculated from the expression values of all detected proteins. Matrix rows and columns are ordered according to hierarchical clustering. (E) Number of differentially expressed proteins (FDR = 5%, Benjamini-Hochberg procedure) in the main cohort for individual disease drivers in CLL.
Global protein and RNA expression in CLL. (A) Distribution of Pearson’s correlation coefficient of mRNA and protein expression across 91 patient samples (main cohort). (B) Scatterplots comparing mRNA and protein expression (log2-transformed and median-normalized mRNA and protein intensities) for ZAP70, CD22, and CD79A. Each point represents a patient sample. P values and coefficients are from Pearson’s correlation tests. (C) Scatterplots showing PC analyses based on the expression of all quantified proteins. (D) Heatmap visualization of the Pearson correlation coefficients for all samples, calculated from the expression values of all detected proteins. Matrix rows and columns are ordered according to hierarchical clustering. (E) Number of differentially expressed proteins (FDR = 5%, Benjamini-Hochberg procedure) in the main cohort for individual disease drivers in CLL.
Association of protein expression with disease drivers of CLL
To obtain a global overview of the proteome variation in CLL, we performed principal component (PC) analysis. PC1 and PC2, which explained 9.16% and 7.61% of the total variance of protein expression, showed strong associations with trisomy 12, while PC3 showed association with IGHV status (Figure 2C). Proteins associated with PC1 were enriched, for example, in phosphatidylinositol 3-kinase [PI3K]-AKT-MTOR and MTORC1 signaling (supplemental Figure 2). Unsupervised clustering based on protein expression showed that patient samples were grouped by the presence of trisomy 12 and IGHV mutational status (Figure 2D). Our data suggest that trisomy 12 and IGHV mutational status are main determinants of protein expression variability in CLL.
We next characterized the impact of genetic disease drivers on protein expression in CLL. We considered mutations and copy-number variations with ≥5 occurrences among the 91 patient samples of the main cohort (Figure 1). We found trisomy 12, IGHV mutational status, mutated SF3B1, trisomy 19, del(17)(p13), del(11)(q22.3), mutated DDX3X, and mutated MED12 associated with variable protein expression (method of Benjamini and Hochberg for false discovery rate (FDR) = 5%, P values were assessed by proDA package as described in “Methods”) (Figure 2E). We did not identify significant associations at the 5% FDR cutoff for del(13)(q14), mutated NOTCH1, mutated EGR2, mutated BRAF, ATM and TP53. Previously reported associations for del(13)(q14) (CD22 and CD72)33 did not reach discovery thresholds in the multiple testing setting, but were reproduced with individual P values of 0.02 and 0.04 (supplemental Figure 3). The results can be queried at http://mozi.embl.de/public/proteomExplorer.
For del(11)(q22.3), 24 differentially regulated proteins (FDR = 5%) were detected (Figure 2E). Among encoding genes, 11 were located on chromosome 11, including ATM, a gene commonly deleted in cases with del(11)(q22.3), and CUL5, a protein involved in proteasomal degradation of p53 (supplemental Figure 4).34,35 We detected 52 proteins whose expression was associated with mutated splicing factor SF3B1, among these the splicing factor SUGP1 (supplemental Figures 5A-B), which showed differential splicing (supplemental Figure 5C). Differential protein expression was also observed for CLL with trisomy 19 (Figure 2E). Since trisomy 19 cases were limited to CLL with mutated IGHV and trisomy 12,36 we restricted this analysis to CLL with mutated IGHV and trisomy 12 (n = 15). We found 47 differentially abundant proteins (FDR = 5%), of which 27 were on chromosome 19. Upregulated proteins in trisomy 19 CLL included the metabolic protein GPI, the unfolded protein response chaperone CALR,37 the negative regulator of transforming growth factor beta signaling RANBP3, and the BCR signaling–associated transcription factor POU2F2 (supplemental Figure 6). Independent proteomic measurements on a different instrument platform (timsTOF)14 confirmed the above findings (supplemental Figure 7). The 26 CLL patient samples of the additional cohort were specifically selected for the genetic disease driver of mutated spliceosomal RNA U1 (g.3A>C mutation38 and mutated TP53). For CLL with mutated U1, we observed upregulation of splicing factors, including SF1 and downregulation of the tumor suppressor TES (supplemental Figure 8). For mutated TP53, we observed upregulation of the DNA damage response protein BCAS2 (supplemental Figure 9).39,40 Upregulated BCAS2 in CLL with mutated TP53 was confirmed in the main cohort (supplemental Figure 9).
Trisomy 12 CLL
We identified 1055 proteins differentially abundant in CLL with trisomy 12 (516 upregulated and 539 downregulated; FDR = 5%) (Figure 3A). Of these, 115 (9.5%) were encoded on chromosome 12. We performed pathway enrichment analysis using the cancer hallmark gene sets (FDR = 5%)41 and found upregulation of PI3K-AKT-MTOR, MTORC1, interferon α, and BCR signaling in CLL with trisomy 12 (Figure 3B-C; supplemental Figure 10). Upregulated proteins encoded on chromosome 12 were enriched in PI3K-AKT-MTOR signaling, whereas upregulated proteins encoded elsewhere were enriched in BCR signaling (supplemental Figure 11). Upregulated proteins included signal transducer and activator of transcription STAT2 and protein phosphatase PTPN11 (both on chromosome 12), as well as SMAD2, a mediator of transforming growth factor beta signaling, and the inflammasome protein PYCARD (Figures 3D-3E). CD72 and BCL10 (involved in BCR signaling) also showed higher abundance in trisomy 12 CLL. The RNA expression of STAT2, PTPN11, PYCARD, and CD72 was upregulated in trisomy 12 as well and showed good correlations with protein levels (supplemental Figures 12 and 13). Higher expression of SMAD2 and BCL10 was only evident at the protein level (supplemental Figure 12). Differential protein expression of STAT2, PTPN11, SMAD2, PYCARD, CD72, and BCL10 was validated in independent proteomic measurements (timsTOF)14 (supplemental Figure 14) and with western blot analyses for STAT2 (supplemental Figure 15).

Protein expression in trisomy 12 CLL. (A) Number of differentially expressed proteins encoded by chromosome 12 genes (red) and genes on other chromosomes (blue). (B) Gene set enrichment analysis of differentially expressed genes (FDR = 5%). (C) Heatmaps of expression Z score of differentially expressed proteins annotated as BCR or PI3K-AKT-MTOR signaling pathway components in samples with and without trisomy 12. (D) Volcano plot of differential protein expression. (E) Normalized expression values for selected proteins. (F) A circular heatmap plot showing the protein expression (outer layer) and RNA expression (inner layer) of genes encoded on chromosome 12 in patient samples with and without trisomy 12. The 2 tracks in the middle indicate the differential expression call (summarized across samples) at the protein and RNA levels. Genes are ordered by genomic coordinates on chromosome 12.
Protein expression in trisomy 12 CLL. (A) Number of differentially expressed proteins encoded by chromosome 12 genes (red) and genes on other chromosomes (blue). (B) Gene set enrichment analysis of differentially expressed genes (FDR = 5%). (C) Heatmaps of expression Z score of differentially expressed proteins annotated as BCR or PI3K-AKT-MTOR signaling pathway components in samples with and without trisomy 12. (D) Volcano plot of differential protein expression. (E) Normalized expression values for selected proteins. (F) A circular heatmap plot showing the protein expression (outer layer) and RNA expression (inner layer) of genes encoded on chromosome 12 in patient samples with and without trisomy 12. The 2 tracks in the middle indicate the differential expression call (summarized across samples) at the protein and RNA levels. Genes are ordered by genomic coordinates on chromosome 12.
We visualized RNA and protein expression along chromosomal coordinates (Figure 3F; supplemental Figure 16). This showed chromosomal regions with consistent changes in abundance of the encoded RNAs and proteins, regions with no or little changes, and few regions with changes in RNA, but not in protein expression (Figure 3F; supplemental Figure 16). Since many proteins function in complexes, we examined the proteome data for protein complexes using the CORUM and REACTOME databases of protein interactions.42,43 Gene-dosage effects of trisomy 12 may propagate to proteins encoded on other chromosomes through complex formation and protein stabilization. We connected upregulated chromosome 12 proteins in trisomy 12 CLL to upregulated non-chromosome 12 proteins, if the protein pairs were annotated to form stable complexes in CORUM or REACTOME (Figure 4A). This identified several complexes of potential relevance for trisomy 12 CLL, including STAT2-JAK1-PTPN6-CD79A-PTPN11-PIK3CD, a complex involved in BCR, AKT, MAPK, and PI3K signaling.

Protein complex and protein abundance buffering analysis. (A) Networks connecting proteins that are annotated as components of stable protein complexes by either CORUM or REACTOME databases. The source nodes (red dots) in the networks are proteins that are encoded on chromosome 12 and are upregulated in trisomy 12 CLL at both protein and RNA levels. The target nodes (blue triangles) are proteins that are encoded elsewhere and are upregulated in trisomy 12 CLL. (B-C) The shape of the edges indicates whether the target nodes are upregulated at RNA level (solid lines) or not (dotted lines). Normalized expression levels for chromosome 12 proteins/RNAs in trisomy 12 CLL (B) and chromosome 19 proteins/RNAs in trisomy 19 CLL (C) compared with diploid patient samples. Dosage effects of copy-number variants are seen both for protein and RNA expression and are more pronounced for RNA. (D) Classification of differentially expressed genes (trisomy 12 vs diploidy 12) on chromosome 12 and differentially expressed genes (trisomy 19 vs diploidy 19) on chromosome 19 into buffered (defined in main text), nonbuffered, enhanced, and undetermined groups. (E) Gene set enrichment analysis (Fisher’s exact test) for proteins in the nonbuffered group of trisomy 12 CLL. (F) Comparison of cis-effects of trisomy 12 at RNA and protein levels. The y axis shows the difference between log2(RNA fold change) and log2(protein fold change) for genes on chromosome 12. The comparison is stratified by whether a protein is annotated as a member of a stable complex in the CORUM or REACTOME database. The error bar indicates 1 standard deviation around the mean. (G) Empirical cumulative distribution function curves of copy-number variant mRNA (dashed lines) and copy-number variant protein (solid lines) expression correlations for genes on chromosome 12 (magenta) and chromosome 19 (blue).
Protein complex and protein abundance buffering analysis. (A) Networks connecting proteins that are annotated as components of stable protein complexes by either CORUM or REACTOME databases. The source nodes (red dots) in the networks are proteins that are encoded on chromosome 12 and are upregulated in trisomy 12 CLL at both protein and RNA levels. The target nodes (blue triangles) are proteins that are encoded elsewhere and are upregulated in trisomy 12 CLL. (B-C) The shape of the edges indicates whether the target nodes are upregulated at RNA level (solid lines) or not (dotted lines). Normalized expression levels for chromosome 12 proteins/RNAs in trisomy 12 CLL (B) and chromosome 19 proteins/RNAs in trisomy 19 CLL (C) compared with diploid patient samples. Dosage effects of copy-number variants are seen both for protein and RNA expression and are more pronounced for RNA. (D) Classification of differentially expressed genes (trisomy 12 vs diploidy 12) on chromosome 12 and differentially expressed genes (trisomy 19 vs diploidy 19) on chromosome 19 into buffered (defined in main text), nonbuffered, enhanced, and undetermined groups. (E) Gene set enrichment analysis (Fisher’s exact test) for proteins in the nonbuffered group of trisomy 12 CLL. (F) Comparison of cis-effects of trisomy 12 at RNA and protein levels. The y axis shows the difference between log2(RNA fold change) and log2(protein fold change) for genes on chromosome 12. The comparison is stratified by whether a protein is annotated as a member of a stable complex in the CORUM or REACTOME database. The error bar indicates 1 standard deviation around the mean. (G) Empirical cumulative distribution function curves of copy-number variant mRNA (dashed lines) and copy-number variant protein (solid lines) expression correlations for genes on chromosome 12 (magenta) and chromosome 19 (blue).
Protein abundance buffering
For trisomy 12 and trisomy 19, gene-dosage effects were detected on RNA and protein levels, with greater expression change for RNA than for protein (Figure 4B-C). We divided proteins and transcripts encoded on chromosomes 12 and 19 into buffered (significant upregulation on RNA but not protein level), nonbuffered (significant upregulation both on RNA and protein level), enhanced (significant upregulation on protein but not RNA level), and other genes (no differential abundance on RNA and protein level, or differential abundance in opposite directions) (Figure 4D). For trisomy 12 CLL, the nonbuffered group was the largest (107 out of 171). The signal transducer STAT2 and the phosphatase PTPN11 were among the least buffered proteins for trisomy 12 CLL. Gene set enrichment analysis identified PI3K-AKT-MTOR signaling as enriched among nonbuffered proteins, suggesting that this pathway may be involved in mediating the fitness advantage of cells with trisomy 12 during tumorigenesis (Figure 4E). No significant enrichment was observed in the buffered group. Chromosome 12–encoded proteins known to be part of stable protein complexes had significantly higher levels of buffering compared with others (Figure 4F). This suggests that formation of protein complexes may help maintain the stoichiometric balance of protein levels in CLL cells. In contrast, for trisomy 19 CLL, nonbuffered gene products were rare (12 out of 185) (Figure 4D). Significantly more buffering was observed for trisomy 19 compared with trisomy 12 CLL (P = .0002, Kolmogorov-Smirnov test) (Figure 4G). The different degrees of protein abundance buffering in trisomy 12 and trisomy 19 CLL highlight the nonredundant information in protein expression measurements as compared with gene dosage or RNA expression.
IGHV mutational status
IGHV mutational status was associated with 542 differentially regulated proteins (273 upregulated and 269 downregulated in M-CLL; FDR = 5%) (Figure 5A). As expected, the expression of the ZAP70 protein was higher in CLL patient samples with U-CLL (Figure 5A).44 Differentially regulated proteins included BANK1 (which functions in BCR-induced calcium mobilization), CASP3, and the transcription factor STAT2 (Figure 5C; supplemental Figure 17). CASP3 was previously linked to enhanced CLL cell viability by stimulation through myosin-exposed apoptotic cells.45 STAT2 demonstrated altered expression only on the protein level and not on the RNA level (Figure 5C; supplemental Figure 17). We validated these findings by independent proteomic measurements using timsTOF14 (supplemental Figure 18).

Protein expression in M-CLL vs U-CLL. (A) Volcano plot. (B) Normalized expression for specific proteins in M-CLL and U-CLL.
Protein expression in M-CLL vs U-CLL. (A) Volcano plot. (B) Normalized expression for specific proteins in M-CLL and U-CLL.
Protein biomarkers for clinical outcome and drug responses
To identify protein markers that might add predictive power to current risk factors, we used multivariate Cox regression models using known risk factors (age, sex, IGHV status, trisomy 12, and TP53/del(17)(p13); supplemental Figure 19) and the expression level of one of the quantified proteins in turn, to predict time-to-next-treatment (TTT) and overall survival. PRMT5 (protein arginine methyltransferase 5), an enzyme responsible for histone methylation,46 the telomerase component PES1,47 and glycogen phosphorylase B (PYGB) were found to be significant predictors for TTT (FDR = 5%, method of Benjamini-Hochberg) (Figure 6A).

Protein expression in CLL explains drug response and clinical outcome. (A) Hazard ratios and P values of known risks plus protein expressions (PRMT5, PYGB, and PES1) using multivariate Cox regression models for predicting TTT. (B) Number of significant associations (FDR = 5%) of protein expression with responses to individual drugs as assessed in an ex vivo drug screen. (C) Exemplary ex vivo drug response and protein expression correlations. (D) Association between ex vivo response to the PI3K inhibitor duvelisib and trisomy 12. (E) Explanatory power of STAT2 protein expression, genetics, and combined STAT2 protein expression and genetics for drug response to cobimetinib. (F) Exemplary ex vivo drug response and protein expression correlation.
Protein expression in CLL explains drug response and clinical outcome. (A) Hazard ratios and P values of known risks plus protein expressions (PRMT5, PYGB, and PES1) using multivariate Cox regression models for predicting TTT. (B) Number of significant associations (FDR = 5%) of protein expression with responses to individual drugs as assessed in an ex vivo drug screen. (C) Exemplary ex vivo drug response and protein expression correlations. (D) Association between ex vivo response to the PI3K inhibitor duvelisib and trisomy 12. (E) Explanatory power of STAT2 protein expression, genetics, and combined STAT2 protein expression and genetics for drug response to cobimetinib. (F) Exemplary ex vivo drug response and protein expression correlation.
We next examined the role of protein expression as a predictor of the response to anticancer drugs, as measured by drug response profiling ex vivo.6 We blocked for trisomy 12 and IGHV mutational status to identify independent proteins. For 28 drugs (of 63, 44%), we found significant (FDR = 5%) relationships between protein expression and drug response. The highest number of protein associations was observed for MEK/ERK inhibitors, such as cobimetinib (n = 150, FDR = 5%) and trametinib (n = 111, FDR = 5%) (Figure 6B). For cobimetinib, the most significant associations were with STAT2, ANP32E, DOCK10, KLHL14, and GRB2 (adjusted value of P < .001). Other examples included the response to the MEK inhibitor trametinib and expression of PTPN11 (Figure 3), response to the Bruton tyrosine kinase inhibitor ibrutinib and expression of the lymphoma survival factor ANXA2,48 as well as response to p53 destabilizing Nutlin-3a and expression of p53-suppressing USP5 (Figure 6C).49 We observed strong responses of trisomy 12 CLL patient samples to PI3K inhibitors such as duvelisib (Figure 6D). We tested the ability of protein expression to add to the explanatory power of gene mutations for drug response using multivariate linear regression models. STAT2 expression added to the explanatory power of genomics (Figure 6E) for responses toward MEK/ERK inhibitors, including cobimetinib, trametinib, and SCH772984, based on multivariate tests (FDR = 5%). STAT2 regulates several signaling pathways, including interferon signaling, JAK-STAT, and Ras/MEK,50,51 and our data indicate a role of STAT2 in the response to MEK inhibitors.
Factors affecting STAT2 expression in CLL
To understand factors associated with STAT2 expression further, we used multivariate linear regression with L1 regularization. STAT2 protein expression was determined by trisomy 12 and IGHV mutational status with additional determinants from protein expression and RNA expression (Figure 7A-C; supplemental Figure 20). STAT2 protein expression was associated with interferon pathway activation, irrespective of IGHV and trisomy 12 status (Figure 7D; supplemental Figures 21 and 22). Higher STAT2 protein levels were associated with increased expression of interferon target genes, including OAS2 and IFI44 (Figure 7E; supplemental Figure 23). Similar to STAT2, the RNA expression of OAS2 and IFI44 was highest in U-CLL patient samples with trisomy 12 (Figure 7F). We measured interferon target gene expression (STAT2, OAS1, and OAS2) in wild-type and STAT2 knockout cells of 5 different CLL/non-Hodgkin lymphoma B-cell lines. In all of these, STAT2 knockout led to reduced expression of interferon target genes in unstimulated state and impaired induction after interferon α stimulation (supplemental Figure 24).

IGHV and trisomy 12 jointly affect interferon signaling through STAT2. (A) Visualization of fitted adaptive L1 (lasso) regularization multivariate models using IGHV status, mutations, and copy-number variations. The z scores of STAT2 protein expression are shown in the scatterplot at the bottom. The heatmap in the middle shows the predictor values, with black indicating the presence of mutation or copy-number variation. The model coefficients (averaged over 50 bootstrap samples) are shown by horizontal bars on the left. Only the features that were selected in all bootstrap samples are shown. (B) Analogous to A, for multivariate models using RNA and protein expression values, which are shown in the heatmap. (C) STAT2 protein and RNA expression stratified by IGHV and trisomy 12 status. (D) Cancer hallmark pathways enriched for RNAs and proteins associated with STAT2 protein expression after blocking for trisomy 12 and IGHV mutational status. (E) Associations between STAT2 protein expression and 2 interferon-induced genes, OAS2 and IFI44. (F) OAS2 and IFI44 RNA expression stratified by IGHV and trisomy 12 status.
IGHV and trisomy 12 jointly affect interferon signaling through STAT2. (A) Visualization of fitted adaptive L1 (lasso) regularization multivariate models using IGHV status, mutations, and copy-number variations. The z scores of STAT2 protein expression are shown in the scatterplot at the bottom. The heatmap in the middle shows the predictor values, with black indicating the presence of mutation or copy-number variation. The model coefficients (averaged over 50 bootstrap samples) are shown by horizontal bars on the left. Only the features that were selected in all bootstrap samples are shown. (B) Analogous to A, for multivariate models using RNA and protein expression values, which are shown in the heatmap. (C) STAT2 protein and RNA expression stratified by IGHV and trisomy 12 status. (D) Cancer hallmark pathways enriched for RNAs and proteins associated with STAT2 protein expression after blocking for trisomy 12 and IGHV mutational status. (E) Associations between STAT2 protein expression and 2 interferon-induced genes, OAS2 and IFI44. (F) OAS2 and IFI44 RNA expression stratified by IGHV and trisomy 12 status.
In summary, STAT2 was upregulated in U-CLL and trisomy 12 CLL, associated with drug response independently of these disease drivers, and required for chemokine/cytokine signaling (interferon response).
Discussion
We used mass spectrometry (MS) to quantify protein abundance in 117 patient samples covering the range of genetic and clinical heterogeneity of the disease. The study provides a protein expression landscape of CLL. We identified trisomy 12 and IGHV status as the strongest determinants of protein expression in CLL and found evidence that the tumor driver role of trisomy 12 is linked to BCR/PI3K/AKT signaling.
Previous proteomic studies in CLL patients were restricted to relatively small cohorts (n = 6-18).9-12,52-55 Our study confirms 11 (trisomy 12) and 8 (U-CLL) out of the 20 strongly differentially expressed subtype-specific proteins highlighted by Johnston et al.9 Diez et al10 reported 4 differentially regulated proteins for IGHV mutational status, 2 of which are confirmed by our study (PTK2 and PRKCG).10 Eagle et al12 reported downregulation of the lymphocyte chemotaxis pathway in U-CLL.12 Our study confirms 3 (GNAI2, RASGRP2, and ARF6) out of 9 differentially regulated proteins of this pathway reported by Eagle et al.12 A more recent publication by Eagle and colleagues11 in U-CLL and M-CLL addressed more technical aspects.11 Six out of the 20 most significantly changed proteins (adjusted P value) reported for IGHV mutational status are confirmed in our study (TFRC, LGALS9, PFKP, SERPINH1, AHCY, and MSI2).11 None of the proteins reported as differentially expressed between U-CLL and M-CLL in small prior studies (n = 6-12) were confirmed by our data.52-55 This highlights the importance of high resolution technologies and large cohorts for discovery of new protein markers.
Protein abundance is regulated by multiple mechanisms and cannot be inferred merely from DNA copy number or RNA abundance.56,57 Whereas most copy-number variants in HeLa cells and colon cancer patient samples drove mRNA abundance changes, few translated into consistent changes in protein abundance. This phenomenon, termed protein abundance buffering,56-58 relies on posttranslational mechanisms such as protein degradation counteracting effects of gene dosage.59-61 Here we studied the phenomenon of protein abundance buffering in CLL. The amount of buffering we observed for trisomy 12 was less than may have been expected from prior studies in colon cancer.57 The relative absence or reduction of protein abundance buffering seen in trisomy 12 CLL highlights the functional relevance. STAT2 and PTPN11 were upregulated in trisomy 12 CLL, and the PI3K-AKT-MTOR pathway was enriched in nonbuffered proteins. The same proteins correlated with drug response in an ex vivo system, including response to inhibitors of the PI3K-AKT-MTOR pathway (Figure 6). Lack of buffering combined with functional relevance for chromosome 12 proteins in CLL is consistent with the specific occurrence of trisomy 12 in B-cell cancers.
STAT2 has not been linked to the pathogenesis of CLL. Our findings suggest that STAT2 levels are dependent on IGHV mutation status and trisomy 12 and mediate CLL response to chemokines and cytokines such as interferon α. Interferon α has been shown to be produced by monocytes (cells known to protect CLL survival) in peripheral blood.62,63 Once activated, interferon α signaling prevents apoptosis of CLL cells in vitro and in vivo.64,65 Our data support a model in which high STAT2 levels (in case of trisomy 12 due to gene dosage) render CLL cells more receptive to chemokines and cytokines. Future studies are required to understand whether chemokines and cytokines other than interferon α are implicated. We further identified STAT2 protein expression as a determinant of drug response to Bruton tyrosine kinase and MEK inhibitors.
Our study provides a high-resolution map of the protein landscape of CLL integrated with genetics, RNA expression, methylation, and drug response data. This study represents a comprehensive basal analysis of CLL and will serve as a valuable resource for the research community.
Acknowledgments
The authors thank Paolo Nanni and Witold Wolski from the Functional Genomics Center Zurich for help with proteomic data acquisition and processing, and Sabine Amon for help with sample preparation.
This research was funded by grants from Swiss Cancer Research foundation (KFS-4439-02-2018), KFSP Next Generation Drug Response Profiling for Personalized Cancer Care, Monique Dornonville-de-la-Cour Foundation, Personalized Health and Related Technologies (PHRT) at ETHZ (Swiss Federal Institute of Technology in Zürich), and the Hairy Cell Leukemia and Promedica foundations. J.L. and W.H. were supported by the German Federal Ministry of Education and Research (CompLS Project MOFA under grant agreement 031L0171A, and SMART-CARE under grant agreement 031L0212E). This study was also supported by “la Caixa” Foundation (CLLEvolution-LCF/PR/HR17/52150017, Health Research 2017 Program “HR17-0022” [E. Campo]). E. Campo is an Academia Researcher of Catalan Institution for Research and Advanced Studies.
Authorship
Contribution: T.Z. and F.M.-A. conceived of the study; F.M.-A., L.K., S.P., E. Cannizzaro, M.F.P., S.K., B.C.C., K.S.L., P.X., M.G., J.H., S.S., and S.D. performed experiments; J.L., F.M.-A., M.R., B.C.C., K.S.L., and T.Z. analyzed data; F.N. and E. Campo contributed patient samples; and F.M.-A., T.Z., J.L., R.A., and W.H. wrote the manuscript.
Conflict-of-interest disclosure: R.A. holds shares of Biognosys AG, which operates in the proteomics field. The remaining authors declare no competing financial interests.
Correspondence: Thorsten Zenz, Department of Medical Oncology and Hematology, University Hospital Zurich, Raemistrasse 100, 8091 Zurich, Switzerland; e-mail: thorsten.zenz@usz.ch; Ruedi Aebersold, Department of Biology, Institute of Molecular Systems Biology, ETH Zurich, Otto-Stern-Weg 3, 8093 Zurich, Switzerland; e-mail: aebersold@imsb.biol.ethz.ch; and Wolfgang Huber, European Molecular Biology Laboratory, 69117 Heidelberg, Germany; e-mail: wolfgang.huber@embl.org.
All scripts and data sets for the bioinformatic analyses are available at: https://github.com/Huber-group-EMBL/CLLproteomics and https://www.huber.embl.de/users/jlu/CLL_Proteomics_public/index.html in the form of Rmarkdown documents organized by workflowr package.31 An R Shiny app (http://mozi.embl.de/public/proteomExplorer) is provided for interactive exploration of the data set and analysis results. The MS proteomics data were deposited to the ProteomeXchange Consortium via the PRIDE32 partner repository with the data set identifiers PXD022198 (Lumos) and PXD022216 (timsTOF).
The online version of this article contains a data supplement.
There is a Blood Commentary on this article in this issue.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal