

In medicine, the unraveling of etiologies, risk factors, disease mechanisms, and genotypes/phenotypes is essential for effective therapy, particularly for complex disorders. In this issue of Blood, 1 report a complex systems analysis of chronic lymphocytic leukemia (CLL) using an integrative OMICS approach together with ex vivo drug response and clinical outcome data. Using this multidimensional approach, they obtain both confirmatory and novel data that offer many opportunities for further elucidation and/or investigation to improve the therapeutic outcome of CLL.
All elements (eg, DNAs, RNAs, proteins, lipids, metabolites) of a cell play an important role in maintaining homeostasis, whereas imbalances or defects in these elements may cause diseases. Large-scale analysis of single elements (ie, genomics, transcriptomics, proteomics, lipidomics, and metabolomics) is useful for addressing disease mechanisms and defining novel biomarkers.2 However, it cannot capture the interplay of these factors to present a complete picture of a complex disease such as CLL.3,4 This underscores the need for more robust analyses using an integrative approach (ie, integrative omics).2,5
Meier-Abt et al used unbiased data-independent acquisition mass spectrometry (in which ions of all peptides within a mass range, not only the predefined or preselected ones, are fragmented and further analyzed)6 using 2 different mass spectrometric modalities (Orbitrap Fusion Lumos and timsTOF) for proteomics analysis of 117 CLL patients’ mononuclear cells and CD19+, CD5+, and light chain restricted malignant B cells. Integrating the obtained proteomics data with the available transcriptomics, genomics, ex vivo drug response, and clinical outcome data, analyses were then performed using bioinformatic tools and statistical packages to determine associations between/among parameters or elements.
This complex systems analysis has revealed that CLL with trisomy 12, immunoglobulin heavy-chain variable region gene mutational status, mutated SF3B1, trisomy 19, del(17)(p13), del(11)(q22.3), mutated DDX3X, and/or mutated MED12 is associated with differentially expressed proteins as compared with CLL without each of these factors. Correlation analysis of protein-RNA expression of the affected genes on chromosome 12 in patients with or without trisomy 12 has demonstrated coordinated, uncoordinated, and contradictory changes of protein and RNA expression (see figure). It is not surprising that the correlation between alterations in protein abundance and changes of RNA expression is relatively low (median coefficient of correlation is only 0.18), suggesting that a variety of protein forms/fragments and posttranslational modifications, which commonly occur in cancers, should not be overlooked.7,8 If these are considered during analyses, there may be more clues of a protein-RNA relationship. Multidimensional association study has shown that trisomy 12 and immunoglobulin heavy-chain variable region gene mutational status are the most prominent disease drivers via phosphoinositide 3-kinase (PI3K), a serine/threonine protein kinase (AKT), mammalian target of rapamycin (MTOR) signaling and STAT2-JAK1-PTPN6-CD79A-PTPN11-PIK3CD interacting complex (or interactome).
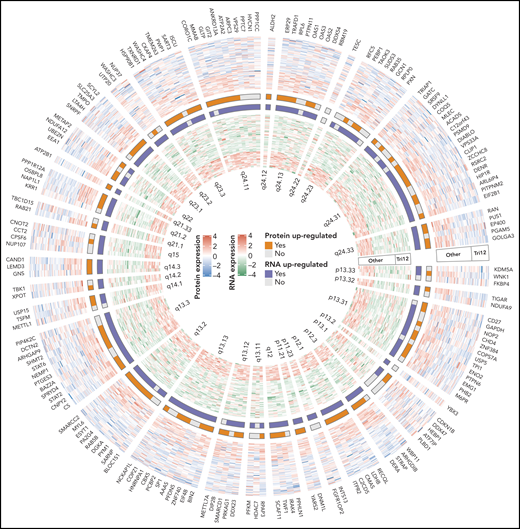
A part of complex systems analysis done by Meier-Abt et al. The figure illustrates circular heatmap plot of the data derived from integrative proteomics and transcriptomics analyses of proteins and RNAs encoded by genes on chromosome 12 of CLL patients with or without trisomy 12. Coordinated, uncoordinated, and contradictory changes of these two elements are clearly demonstrated. See figure 3F in the article by Meier-Abt et al that appears on page 2514.
A part of complex systems analysis done by Meier-Abt et al. The figure illustrates circular heatmap plot of the data derived from integrative proteomics and transcriptomics analyses of proteins and RNAs encoded by genes on chromosome 12 of CLL patients with or without trisomy 12. Coordinated, uncoordinated, and contradictory changes of these two elements are clearly demonstrated. See figure 3F in the article by Meier-Abt et al that appears on page 2514.
In addition, protein arginine methyltransferase 5, pescadillo ribosomal biogenesis factor 1, and glycogen phosphorylase B can predict the clinical outcome as shown by time to next treatment. In ex vivo drug response profiles, cobimetinib is associated with the greatest number of differentially expressed proteins, especially STAT2. Interestingly, high STAT2 expression is associated with activation of the interferon pathway and upregulation of the interferon target genes OAS2 and IFI44, which are downregulated in the STAT2-knockout CLL cells. From the many differentially expressed proteins identified, only STAT2 has been validated for its functional relevance in disease pathway. Therefore, more target proteins should be further explored for their impact on the pathogenic mechanisms of CLL.
Multiple element analysis (ie, combined genomics, epigenomics, and transcriptomics) has previously been used for investigation of their relationship with ex vivo drug response and clinical outcome data in CLL by the same network of investigators.9,10 The study by Meier-Abt et al, however, has expanded the dimensions of the complex systems analysis by integrating proteomics with more extensive analyses of the associations with genomics, transcriptomics, ex vivo drug response and clinical outcome data. The authors are to be commended for their efforts, especially in the robustness of the OMICS data, the cross-validation capability of the methods used, and the data sharing.
Conflict-of-interest disclosure: The author declares no competing financial interests.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal