

Key Points
WGS describes IgM-MM as predominantly a pre–germinal center malignancy and provides molecular differences to WM.
High BCL2/BCL2L1 ratio and high expression of CD20 and Bruton tyrosine kinase in IgM-MM provide potential for targeted therapeutic options.

Abstract
Immunoglobulin M (IgM) multiple myeloma (MM) is a rare disease subgroup. Its differentiation from other IgM-producing gammopathies such as Waldenström macroglobulinemia (WM) has not been well characterized but is essential for proper risk assessment and treatment. In this study, we investigated genomic and transcriptomic characteristics of IgM-MM samples using whole-genome and transcriptome sequencing to identify differentiating characteristics from non–IgM-MM and WM. Our results suggest that IgM-MM shares most of its defining structural variants and gene-expression profiling with MM, but has some key characteristics, including t(11;14) translocation, chromosome 6 and 13 deletion as well as distinct molecular and transcription-factor signatures. Furthermore, IgM-MM translocations were predominantly characterized by VHDHJH recombination-induced breakpoints, as opposed to the usual class-switching region breakpoints; coupled with its lack of class switching, these data favor a pre–germinal center origin. Finally, we found elevated expression of clinically relevant targets, including CD20 and Bruton tyrosine kinase, as well as high BCL2/BCL2L1 ratio in IgM-MM, providing potential for targeted therapeutics.
Medscape Continuing Medical Education online
In support of improving patient care, this activity has been planned and implemented by Medscape, LLC and the American Society of Hematology. Medscape, LLC is jointly accredited by the Accreditation Council for Continuing Medical Education (ACCME), the Accreditation Council for Pharmacy Education (ACPE), and the American Nurses Credentialing Center (ANCC), to provide continuing education for the healthcare team.
Medscape, LLC designates this Journal-based CME activity for a maximum of 1.00 AMA PRA Category 1 Credit(s)™. Physicians should claim only the credit commensurate with the extent of their participation in the activity.
Successful completion of this CME activity, which includes participation in the evaluation component, enables the participant to earn up to 1.0 MOC points in the American Board of Internal Medicine's (ABIM) Maintenance of Certification (MOC) program. Participants will earn MOC points equivalent to the amount of CME credits claimed for the activity. It is the CME activity provider's responsibility to submit participant completion information to ACCME for the purpose of granting ABIM MOC credit.
All other clinicians completing this activity will be issued a certificate of participation. To participate in this journal CME activity: (1) review the learning objectives and author disclosures; (2) study the education content; (3) take the post-test with a 75% minimum passing score and complete the evaluation at http://www.medscape.org/journal/blood; and (4) view/print certificate. For CME questions, see page 2007.
Disclosures
Editor Andrew Roberts's organization, the Walter and Eliza Hall Institute, received grants for clinical research from AbbVie Inc, and Janssen Pharmaceuticals, Inc. The Walter and Eliza Hall Institute also received royalties related to venetoclax and will control any distribution based on their institutional policies about scientific contribution to commercial income. Author Kenneth C. Anderson serves as advisor or consultant for Bristol-Myers Squibb Company; Celgene Corporation; Gilead Sciences, Inc; Janssen Pharmaceuticals, Inc; Precision BioSciences; Sanofi; Takeda Pharmaceuticals North America, Inc; and Tolero Pharmaceuticals; he is on the board of directors of and holds stock options in OncoPep. Author Nikhil C. Munshi serves as advisor or consultant for AbbVie Inc; Amgen Inc; Bristol-Myers Squibb Company; GlaxoSmithKline; Janssen Pharmaceuticals, Inc; Karyopharm Therapeutics Inc; Legend Biotech; OncoPep; and Takeda Pharmaceuticals North America, Inc; he is on the board of directors of and holds stock options in Oncopep. CME questions author Laurie Barclay, freelance writer and reviewer, Medscape, LLC, and the remaining authors declare no competing financial interests.
Learning objectives
Upon completion of this activity, participants will:
- 1.
Describe genomic and transcriptomic characteristics of immunoglobulin M (IgM) -multiple myeloma (MM) through whole-genome sequencing and transcriptome sequencing of IgM-MM cases compared with non-IgM-MM, Waldenström macroglobulinemia (WM), and healthy donor PCs
- 2.
Determine driver mutations in IgM-MM compared with non-IgM-MM, WM, and healthy donor plasma cells
- 3.
Identify which IgM monoclonal gammopathy of undetermined significance has the potential to progress to IgM MM or WM
- 4.
Identify therapeutic targets in IgM-MM, according to this genomic and transcriptomic study
Release date: November 18, 2021; Expiration date: November 18, 2022
Introduction
Immunoglobulin M (IgM) multiple myeloma (MM) is a rare condition with an estimated incidence of <0.5%.1 Its differentiation from more common IgM-producing plasma cell (PC) disorders, chiefly Waldenström macroglobulinemia (WM), is challenging but essential because of distinct treatment modalities and prognoses.2 Recent advancements in molecular techniques have shed light on the genomic characteristics and unique genetic alterations of MM and WM3,4; however, comprehensive profiling has not yet been performed for IgM-MM. This study investigates genomic and transcriptomic characteristics of IgM-MM through whole-genome (WGS) and transcriptome sequencing of IgM-MM cases, comparing them to non–IgM-MM, WM, and healthy donor PC.
Study design
All MM patient samples and deidentified clinical data were collected after written informed consent. We performed and compared WGS on CD138-sorted myeloma cells from 15 IgM-MM, 211 non–IgM-MM, and 55 WM patient samples, and transcriptomic data from 15 IgM-MM, 30 non–IgM-MM, 35 WM, and 3 healthy donor PCs. We analyzed WGS data with Mutect2, Facets, and Manta, and RNAseq analysis with Salmon, and R (supplemental Methods, available on the Blood Web site).
Results and discussion
VDJ-t(11;14)
We first evaluated the WGS data for structural variants and observed that all patients with IgM-MM harbored a t(11;14)(q13;q32) translocation that fused immunoglobulin superenhancers with CCND1 (Figure 1A). Sixty percent of t(11;14) translocations in IgM-MM involved VH-DH-JH regions on IGH (Figure 1B), compared with only 25% of non–IgM-t(11;14)-MM that predominantly involved immunoglobulin switch regions (Figure 1C-D). None of the WM samples featured any translocation (Figure 1E).
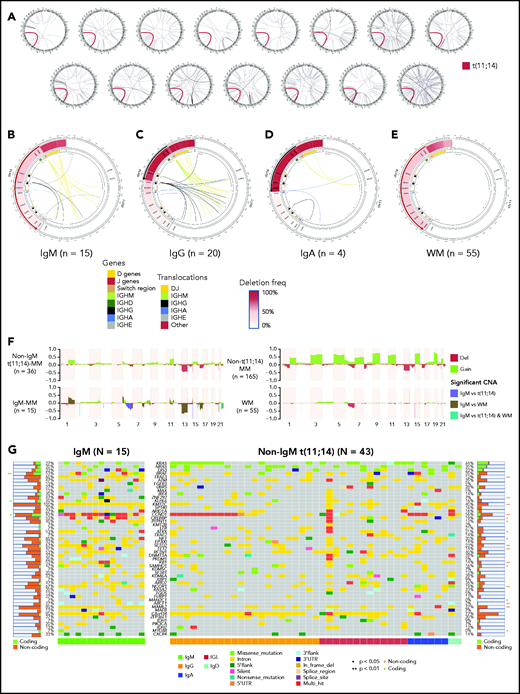
Cytogenetic abnormalities in IgM-MM. (A) Translocations in 15 patients with IgM MM. All 15 samples (each circos plot represents 1 patient) from newly diagnosed patients with IgM-MM had t(11;14) translocation (shown in red). Other translocations are shown with black links. Chromosomes from chr1 to chrY are ordered clockwise. (B) Patients with IgM (n = 15) displaying t(11;14) translocations involving predominantly DJ genes and deletions suggestive of VDJ recombination. Only 4 patients had deletions suggestive of class-switch recombination, and 6 had translocations involving class-switch regions. The IgH region is shown on the left side of the panel, and the CCND1 region is shown on the right side. IgH region genes and switch regions are color coded. Translocations (inner links) are also color coded by their classes. (C) Patients with IgG (n = 20) with t(11;14) translocations involving DJ genes and class-switch regions and deletions suggestive of VDJ recombination and class switch to IgG subtype. (D) Patients with IgA (n = 4) displaying t(11;14) translocations involving DJ genes and class-switch regions and deletions suggestive of VDJ recombination and class switch to IgA subtype. (E) WM samples (n = 55) displaying deletions suggestive of VDJ recombination, and 4 patients displaying class switching to IgG and IgA. None of the WM samples had translocation involving IgH region. (F) Copy-number alterations (CNA) in non–t(11;14)-MM samples (n = 165), non−IgM-t(11;14) samples (n = 36), IgM-MM samples (n = 15), and WM samples (n = 55). For each group gain (green) and deletion (red), frequencies are shown (y-axis). Regions that are statistically different between IgM-MM vs non−IgM-t(11;14)-MM or IgM-MM vs WM or both are color coded, and significant alterations are highlighted (P < .05). (G) Oncoplot of coding and noncoding mutations in MM driver genes for IgM (n = 15) and non−IgM-t(11;14) (n = 43) patients. Significant differences for coding mutations in each gene are marked with an asterisk on the left side and for noncoding mutations on the right side. Immunoglobulin type of each patient is shown at the bottom with color codes (green for IgM, orange for IgG, red for light chain only, blue for IgA, and turquoise for IgD). Mutation types are also shown in each cell with color codes explained at the bottom. IGL, immunoglobulin light chain; 3′UTR, 3′ untranslated region.
Cytogenetic abnormalities in IgM-MM. (A) Translocations in 15 patients with IgM MM. All 15 samples (each circos plot represents 1 patient) from newly diagnosed patients with IgM-MM had t(11;14) translocation (shown in red). Other translocations are shown with black links. Chromosomes from chr1 to chrY are ordered clockwise. (B) Patients with IgM (n = 15) displaying t(11;14) translocations involving predominantly DJ genes and deletions suggestive of VDJ recombination. Only 4 patients had deletions suggestive of class-switch recombination, and 6 had translocations involving class-switch regions. The IgH region is shown on the left side of the panel, and the CCND1 region is shown on the right side. IgH region genes and switch regions are color coded. Translocations (inner links) are also color coded by their classes. (C) Patients with IgG (n = 20) with t(11;14) translocations involving DJ genes and class-switch regions and deletions suggestive of VDJ recombination and class switch to IgG subtype. (D) Patients with IgA (n = 4) displaying t(11;14) translocations involving DJ genes and class-switch regions and deletions suggestive of VDJ recombination and class switch to IgA subtype. (E) WM samples (n = 55) displaying deletions suggestive of VDJ recombination, and 4 patients displaying class switching to IgG and IgA. None of the WM samples had translocation involving IgH region. (F) Copy-number alterations (CNA) in non–t(11;14)-MM samples (n = 165), non−IgM-t(11;14) samples (n = 36), IgM-MM samples (n = 15), and WM samples (n = 55). For each group gain (green) and deletion (red), frequencies are shown (y-axis). Regions that are statistically different between IgM-MM vs non−IgM-t(11;14)-MM or IgM-MM vs WM or both are color coded, and significant alterations are highlighted (P < .05). (G) Oncoplot of coding and noncoding mutations in MM driver genes for IgM (n = 15) and non−IgM-t(11;14) (n = 43) patients. Significant differences for coding mutations in each gene are marked with an asterisk on the left side and for noncoding mutations on the right side. Immunoglobulin type of each patient is shown at the bottom with color codes (green for IgM, orange for IgG, red for light chain only, blue for IgA, and turquoise for IgD). Mutation types are also shown in each cell with color codes explained at the bottom. IGL, immunoglobulin light chain; 3′UTR, 3′ untranslated region.
Chromosomal copy-number variation
IgM-MM displayed copy-number variation patterns similar to those reported in non–IgM-MM (Figure 1F; supplemental Figure 1). We observed clonal changes involving del13q and del6q in 67% and 40% of patients with IgM-MM, respectively, with 1q21 and 6p gains in 40% of 15 patients. Subclonal events involving del16q were observed in 47%, with del1p and del17p in 1 patient each. Del6q and del13 were the only events that were more frequently observed in IgM-MM vs non–IgM-MM and non–IgM-t(11;14)-MM (P < .05).5 It is interesting to note that del6q occurred in >31% of WM and is associated with progression from IgM monoclonal gammopathy of undetermined significance (MGUS) to WM.6
Driver mutations
Previously described MM driver mutations7 involving KRAS, NRAS, BRAF, CCND1, and DIS3 were also encountered in IgM-MM (Figure 1G). We have observed a high frequency of clock-like signature in IgM-MM and WM compared with non–IgM-MM and high APOBEC in IgM-MM compared with WM, suggesting an alternate mechanism in these groups (supplemental Figure 2).8 Matched RNA-seq variant allele frequency (VAF) was linearly correlated with genome-called VAF for other mutations, suggesting a lack of impact on transcription, except for CCND1, which displayed a disproportionate expression of the mutant allele (supplemental Table 1). Interestingly, MYD88 mutation, a hallmark of WM observed in 95% to 97% of patients with WM,4 was detected in 1 IgM-MM sample; however, it was a subclonal S219C mutation and not the usual L265P variant observed in WM.9
RNA-seq data from IgM-MM displayed elevated expression of transcripts relevant to MM-associated proteins, including CD38, CD138, BCMA, and SLAMF7 (Figure 2A), as well as t(11;14)-specific proteins, including CCND1, CD20, CD23, and CD79A (Figure 2B). Furthermore, CD56 and CD117 are highly expressed in MM but low or absent in IgM-MM and WM (Figure 2C). Gene set enrichment analysis showed upregulation of MYC target genes in IgM-MM compared with both non–IgM-MM and WM (Figure 2D; supplemental Figure 3; supplemental Tables 2 and 3).
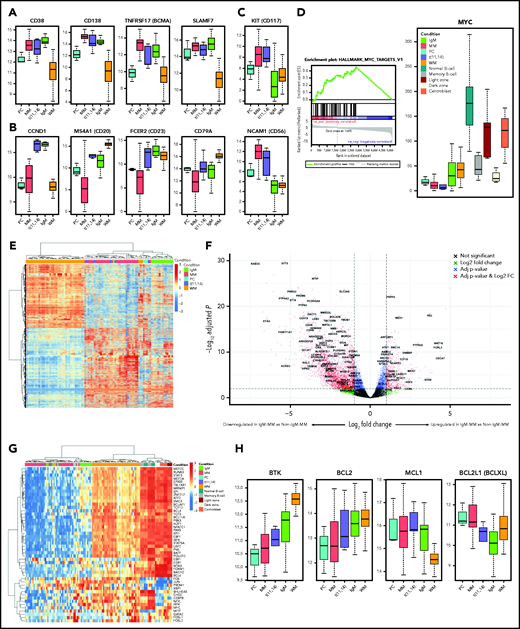
Gene expression profiles. (A) IgM-MM shows similar transcript expression of key MM proteins. (B) IgM-MM shows similar transcript expression of key t(11;14) MM proteins. (C) IgM-MM–specific transcript expression shows low levels of CD56 and CD117. (D) MYC gene set enrichment analysis and transcript levels showing upregulation in IgM-MM compared with non–IgM-MM. (E). Clustering of protein coding genes differentially expressed between non–IgM-MM and WM, showing preferential clustering of IgM-MM subtype with other MM samples and suggesting a unique transcriptional signature in this subgroup. (F) Volcano plot of differentially expressed genes between IgM-MM and non–IgM-MM with similarly differentially expressed genes between WM and non–IgM-MM labeled. (G) Heat map of B-cell–specific transcription factors expression profiles showing preferential clustering of IgM-MM subtype with other MM samples and separation in IgM-specific signature, and closer homology between WM and immature B cells. (H) Expression levels of clinically relevant genes. FC, fold change; NS, not significant.
Gene expression profiles. (A) IgM-MM shows similar transcript expression of key MM proteins. (B) IgM-MM shows similar transcript expression of key t(11;14) MM proteins. (C) IgM-MM–specific transcript expression shows low levels of CD56 and CD117. (D) MYC gene set enrichment analysis and transcript levels showing upregulation in IgM-MM compared with non–IgM-MM. (E). Clustering of protein coding genes differentially expressed between non–IgM-MM and WM, showing preferential clustering of IgM-MM subtype with other MM samples and suggesting a unique transcriptional signature in this subgroup. (F) Volcano plot of differentially expressed genes between IgM-MM and non–IgM-MM with similarly differentially expressed genes between WM and non–IgM-MM labeled. (G) Heat map of B-cell–specific transcription factors expression profiles showing preferential clustering of IgM-MM subtype with other MM samples and separation in IgM-specific signature, and closer homology between WM and immature B cells. (H) Expression levels of clinically relevant genes. FC, fold change; NS, not significant.
Unsupervised hierarchical clustering using differentially expressed genes between MM and WM revealed a closer molecular homology of IgM-MM to non–IgM-MM compared with WM (Figure 2E). We identified 230 genes unique to IgM-MM and WM and differentially expressed from MM (Figure 2F; supplemental Figure 4). A similar analysis using only B-cell–specific transcription factors10-12 showed separation of WM and MM and preferential clustering of IgM-MM with non–IgM-MM (Figure 2G; supplemental Figure 5). WM was noted to have closer homology to peripheral memory and germinal center (GC) B cells compared with MM, including IgM-MM, which appeared to have a more mature phenotype. It appears that IgM-MM has an intermediate phenotype compared with non–IgM-MM, contrasted with WM, which retains a fully immature signature with upregulated PAX5, MYC, and BCL-6 and downregulated PRDM1, XBP1, and IRF4. WM is theorized to originate from peripheral memory B cells, marginal zone cells, or GC B cells,13-15 which was observed in the unsupervised clustering (Figure 2G).
IGH translocations have been observed in >40% of patients with MM, most frequently involving CCND1 on chromosome 11 (15% to 20%).16 It has been suggested that different mechanisms in B-cell development are responsible for breakpoints generated on the IGH locus. In fact, the majority (>60%) occur within the switch-region upstream of IGH constant genes, generated through class-switch recombination in mature B cells in GCs.17 A fifth of t(11;14) translocations, however, are thought to be generated through VH-DH-JH recombination, occurring earlier at the pro–B-cell stage in the bone marrow (BM).17 All of the IgM-MM samples showed CCND1 translocation, the most probable MM initiating event in this subgroup, and 60% of these appear to originate from VH-DH-JH recombination, suggesting their occurrence in the BM at pre-GC progenitor level. Also, although IgG/IgA-MM displayed evidence of class switch recombination (CSR) with deletions between the IGHM switch region and IGHG/IGHA switch regions, most IgM-MM samples (73%) had no such alterations. It is interesting to note that 2 IgM samples appear to have acquired the t(11;14) translocation during CSR but failed to class switch, which could explain their persistence in the IgM state, and perhaps even more intriguing, 4 samples appear to have fully classed switched and acquired the t(11:14) translocation during CSR, yet the translocated allele is inactivated through an unclear mechanism and the cells exclusively produce IgM. Although both IgM-MM and WM share the same precursor condition, namely IgM-MGUS, the presence of t(11;14) and del13 coupled with the lack of MYD88 mutation at a precursor level may serve as predictors for progression of MGUS to IgM-MM as opposed to WM.18
Clinically relevant targets
Three clinically relevant upregulated targets, BCL-2, CD20, and Bruton tyrosine kinase (BTK), were identified in IgM-MM (Figure 2H). We have confirmed that all 15 patients with IgM-MM are t(11;14), and all express CD20 at the RNA level. This is in line with reported CD20 expression in t(11;14)-MM.19 Moreover, using immunoperoxidase and in situ hybridizations as well as flow cytometric methods, we have confirmed CD20 and cyclin D1 expression in CD138+ PCs with monotypic cytoplasmic reactivity for λ light chain and IgM heavy chain in BM from a patient with IgM-MM. This provides the rationale for use of rituxan in patients with IgM-MM. BTK inhibitors, such as ibrutinib, are very effective in treating WM,20 but have only shown modest activity in MM.21 BTK expression was significantly higher in IgM-MM and WM compared with non–IgM-MM, indicating a possible role for BTK inhibitors in IgM-MM. Finally, both IgM and non–IgM-t(11;14)-MM had elevated levels of BCL-2, which is a known predictor of venetoclax efficacy.22 Interestingly, the higher BCL2/BCL-XL and BCL2/MCL1 ratios observed in IgM-MM compared with non–IgM-t(11;14)-MM may suggest even better sensitivity to venetoclax in this group of patients.23 Although certainly not unique to IgM-MM, CD38, BCMA, and SLAMF7 are well-established targets for treatment in MM, and given their high expressions in IgM-MM, similar responses are expected in this subgroup.24,25 To date, clinical data are lacking, and further investigations are needed to fully understand the potential role of these drugs in treating IgM-MM.
In summary, we here describe a molecular and transcriptomic landscape of IgM-MM, which both identifies its origin at an early stage of PC differentiation and provides clues for a selective therapeutic targeting.
Acknowledgments
This study was supported by National Institutes of Health, National Cancer Institute grants P01 CA155258 and P50 CA100707, VA Healthcare System grant 5I01BX001584, and a Paula and Roger Riney Foundation grant.
Authorship
Contribution: A.H.B. designed the research, analyzed the data, and wrote the manuscript; H.A.-L., R.S., A.A.S., Z.H., M.S., J.C., M.F., K.C.A., G.P., S.P.T., and M.M. reviewed the manuscript and discussed the results; A.H.B., H.A.-L., R.S., Z.H., S.P.T., M.K.S., N.C.M. collected the samples and generated the data; M.K.S. and N.C.M. designed the research and wrote the manuscript.
Conflict-of-interest disclosure: K.C.A. has received consulting fees from Bristol Myers Squibb, Celgene, Gilead, Janssen, Precision Biosciences, Sanofi-Aventis, Takeda, and Tolero and serves on the board of directors of and has stock options in Oncopep. N.C.M. is a consultant for BMS, Janssen, Oncopep, Amgen, Karyopharm, Legend, Abbvie, Takeda, and GSK and serves on the board of directors of and has stock options in Oncopep. The remaining authors declare no competing financial interests.
Correspondence: Mehmet Kemal Samur, Dana-Farber Cancer Institute, Harvard T.H. Chan School of Public Health, 450 Brookline Ave, Boston, MA 02215; e-mail: mehmet_samur@dfci.harvard.edu; and Nikhil C. Munshi, Dana-Farber Cancer Institute, Harvard Medical School, 450 Brookline Ave, Boston, MA 02215; e-mail: nikhil_munshi@dfci.harvard.edu.
For original data, please contact Nikhil_Munshi@dfci.harvard.edu.
The online version of this article contains a data supplement.
There is a Blood Commentary on this article in this issue.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal