

Key Points
KIN-8194 is a highly potent dual HCK and BTK inhibitor with superior antitumor activity over ibrutinib in MYD88-mutated B-cell lymphomas.
KIN-8194 overcomes ibrutinib resistance with a survival benefit in TMD-8 ABC DLBCL xenografted mice and synergizes with venetoclax.

Abstract
Activating mutations in MYD88 promote malignant cell growth and survival through hematopoietic cell kinase (HCK)–mediated activation of Bruton tyrosine kinase (BTK). Ibrutinib binds to BTKCys481 and is active in B-cell malignancies driven by mutated MYD88. Mutations in BTKCys481, particularly BTKCys481Ser, are common in patients with acquired ibrutinib resistance. We therefore performed an extensive medicinal chemistry campaign and identified KIN-8194 as a novel dual inhibitor of HCK and BTK. KIN-8194 showed potent and selective in vitro killing of MYD88-mutated lymphoma cells, including ibrutinib-resistant BTKCys481Ser-expressing cells. KIN-8194 demonstrated excellent bioavailability and pharmacokinetic parameters, with good tolerance in rodent models at pharmacologically achievable and active doses. Pharmacodynamic studies showed sustained inhibition of HCK and BTK for 24 hours after single oral administration of KIN-8194 in an MYD88-mutated TMD-8 activated B-cell diffuse large B-cell lymphoma (ABC DLBCL) and BCWM.1 Waldenström macroglobulinemia (WM) xenografted mice with wild-type BTK (BTKWT)– or BTKCys481Ser-expressing tumors. KIN-8194 showed superior survival benefit over ibrutinib in both BTKWT- and BTKCys481Ser-expressing TMD-8 DLBCL xenografted mice, including sustained complete responses of >12 weeks off treatment in mice with BTKWT-expressing TMD-8 tumors. The BCL_2 inhibitor venetoclax enhanced the antitumor activity of KIN-8194 in BTKWT- and BTKCys481Ser-expressing MYD88-mutated lymphoma cells and markedly reduced tumor growth and prolonged survival in mice with BTKCys481Ser-expressing TMD-8 tumors treated with both drugs. The findings highlight the feasibility of targeting HCK, a key driver of mutated MYD88 pro-survival signaling, and provide a framework for the advancement of KIN-8194 for human studies in B-cell malignancies driven by HCK and BTK.
Introduction
Activating mutations in MYD88 are prevalent in Waldenström macroglobulinemia (WM; 95% to 97%), primary central nervous system lymphoma (PCNSL; 60% to 80%), activated B-cell diffuse large B-cell lymphoma (ABC DLBCL; 30% to 40%), chronic lymphocytic leukemia (CLL; 5% to 10%), and marginal zone lymphoma (MZL; 5% to 10%).1-6 Mutated MYD88 triggers auto-assembly of a myddosome complex that includes Bruton tyrosine kinase (BTK), which triggers downstream NF-kB pro-survival signaling.7 Moreover, mutated MYD88 transcriptionally upregulates the SRC family member hematopoietic cell kinase (HCK) through regulation that involves PAX5 and activates HCK through interleukin-6 (IL-6).8,9 HCK in turn activates BTK permitting it to join the myddosome signaling complex and trigger other pro-survival pathways, including PI3K/AKT, MAPK/ERK1/2, and SYK/STAT3/AKT.7,9,10 Ibrutinib blocks BTK activity by covalently binding to BTKCys481 and is active in MYD88-mutated WM, PCNSL, ABC DLBCL, CLL, and MZL.11-14 Other covalent and noncovalent inhibitors of BTK are also in clinical use or are under development for MYD88-mutated B-cell malignancies.
Acquired resistance to ibrutinib as a result of BTKCys481 mutations is common in WM, CLL, and MZL.15-18 Our previous studies showed that mutations in BTKCys481 restore downstream pro-survival signaling that includes NF-kB and ERK1/2.19 The latter facilitates release of inflammatory mediators, including IL-6 and IL-10, that trigger resistance in neighboring wild-type BTK (BTKWT) cells. Because BTK is activated by HCK, we sought to develop HCK inhibitors to overcome ibrutinib resistance in mutated BTKCys481-expressing WM and ABC DLBCL lymphomas driven by mutated MYD88. In a previous study, we showed that a toolbox HCK inhibitor, A419259, overcame in vitro ibrutinib resistance mediated by mutated BTKCys481.8 We therefore screened >220 clinical and preclinical kinase inhibitors and performed lead optimization with iterative rounds of synthesis of >400 analogs. These efforts resulted in the identification of 2 lead compounds, KIN-8193 and KIN-8194. KIN-8193, a potent HCK inhibitor derived from a preclinical toolbox compound, TLR-2-59, blocked BTK activity and overcame ibrutinib resistance related to mutated BTKCys481 but demonstrated a tight therapeutic index.20 KIN-8194, a dual BTK/HCK inhibitor described herein, has a chemical structure similar to but distinct from that of A419259 (RK-20449).21,22 In this report, we present our findings characterizing the in vitro and in vivo activity of KIN-8194 in MYD88-mutated lymphomas, including in ibrutinib-resistant tumor models.
Materials and methods
Cell lines and reagents
MYD88-mutated (MYD88L265P) WM (BCWM.1 and MWCL-1), ABC DLBCL (TMD-8, HBL1), and wild-type (MYD88WT) germinal center B-cell like (GCB) DLBCL (OCI-Ly7, OCI-Ly19), Burkitt’s lymphoma (Ramos), and myeloma (RPMI-8226) cells were used. Their identities were confirmed by short tandem repeat profiles with GenePrint 10 System (Promega, Fitchburg, WI). Ibrutinib and venetoclax were obtained from MedChem Express (Monmouth Junction, NJ).
Patient samples
Mononuclear cells from bone marrow (BM) aspirates were isolated using Ficoll-Paque PLUS Media (GE Healthcare) and 2 × 106 BM mononuclear cells treated overnight (for apoptosis) or for 1 to 2 hours (for Phosflow) with ibrutinib or KIN-8194. Apoptosis or Phosflow analyses were performed on CD19+ (for apoptosis) or CD20+ (for Phosflow) lymphoplasmacytic cells (LPCs). MYD88 genotyping was performed as before.1,2 Peripheral blood mononuclear cells from healthy donors were used as controls. Sample use was approved by the Dana-Farber/Harvard Cancer Center Institutional Review Board after patients provided written consent.
Kinase profiling and enzymatic assays
KINOMEscan assays were performed to quantitatively measure interactions between KIN-8194 and 468 kinases in vitro at Eurofins DiscoverX Corporation (San Diego, CA).23 Scores for primary screens were reported as a percent of dimethyl sulfoxide (DMSO) control. Selectivity score was defined as the ratio of kinases inhibited to a specified percentage vs total number of kinases, excluding mutant variants. Enzymatic activities against HCK and BTK were tested in Z-Lyte assays with adenosine triphosphate (ATP) concentrations of at Km [app] adenosine triphosphate (ATP) concentrations (Thermo Fisher Scientific, Waltham, MA).
Live-cell target engagement studies
Live-cell kinase target engagement profiling with KiNativ was performed by ActivX Biosciences (La Jolla, CA).24 Briefly, live (TMD-8) cells were pretreated with DMSO or 1.0 μM KIN-8194 for 90 minutes, then washed twice with ice-cold phosphate-buffered saline. Cell pellets were snap-frozen in liquid nitrogen and sent for KiNativ profiling. HCK- and BTK-specific targeting was also assessed using Pierce Kinase Enrichment Kit with ActivX Desthiobiotin-ATP Probe (Thermo Fisher Scientific) after treatment of BCWM.1 and TMD-8 cells with serially diluted KIN-8194 or ibrutinib for 90 minutes with ATP-binding to HCK or BTK resolved by western blotting.
Computational docking models
Molecular models were constructed by ligand alignment with the corresponding co-crystal structure small molecules of BTK (Protein Data Bank identification number [pdb: 5P9J]) and HCK (pdb: 5ZJ6) and energy minimized by refining the protein-ligand complex protocol with default parameters using Schrodinger Glide Docking (version 2020-04). Water molecules observed in the pdb structures around the ligand were conserved and optimized. Binding energies were estimated with docking scores by using place scoring method (Glide version 2020-04).
In vitro cellular efficacy studies
In vitro cellular efficacies were assessed by using a CellTiter-Glo Luminescent Cell Viability Assay (Promega).7,10,19 Dose-response curves were calculated with GraphPad Prism software. Drug interactions were assessed by using CalcuSyn 2.0 software (Biosoft, Cambridge, United Kingdom). Apoptosis was assessed by using an Apoptosis Detection Kit (BD Pharmingen, San Diego, CA). Cells (1 × 106 cells per well) were treated with inhibitors overnight in 24-well plates. A minimum of 10 000 events was acquired by using a BD FACSCanto II flow cytometer, and the results were analyzed with FlowJo software. WM patient LPCs (2 × 106 cells per well) were treated with inhibitors, and CD19-APC-Cy7 antibody (BD Biosciences, San Jose, CA) was used with an annexin V antibody to evaluate apoptosis.
Signaling studies
Cells were treated for 1 to 2 hours before Phosflow, and immunoblotting studies were performed using antibodies to phycoerythrin-conjugated pBTK (Y223), APC-Cy7–conjugated CD20 (BD Biosciences); pBTK(Y223), pHCK(Y410), pIRAK1 (T209), DyLight 650–conjugated goat F(ab′)2 anti-rabbit immunoglobulin G (IgG) secondary antibody (Abcam, Cambridge, MA); pSYK(Y525/526) (R&D Systems); and Alexa Fluor 647–conjugated pSYK(Y525/526), SYK, pAKT(S473), AKT, pERK1/2(T202/Y204), ERK1/2, BTK, HCK (Cell Signaling Technologies, MA).7,8 Glyceraldehyde-3-phosphate dehydrogenase antibody (Santa Cruz Biotechnology, Dallas, TX) was used as loading control for immunoblotting.
Generation of HCKThr333Met- or BTKCys481Ser-expressing cell lines
Cell lines expressing HCKWT or a mutated gatekeeper residue (HCKThr333Met) along with BTKWT or BTKCys481Ser were generated by lentiviral transduction in a pLVX-EF1α-IRES-Puro vector (Clontech Laboratories, Palo Alto, CA).8,19 After lentiviral transduction, stable cell lines were selected with 0.5 to 1.0 μg/mL puromycin. Expression of transduced coding sequences was confirmed by Sanger sequencing after gene-specific reverse transcription polymerase chain reaction, and protein expression was confirmed by immunoblotting. For xenografted murine models, TMD-8 ABC DLBCL cells were co-transduced to express luciferase and green fluorescent protein (GFP) using a pLenti-II-CMV-Luc-IRES-GFP lentiviral vector (Applied Biological Materials, Vancouver, BC, Canada).
Pharmacokinetic studies
Pharmacokinetic (PK) studies were performed at Scripps Florida under an Institutional Animal Care and Use Committee (IACUC)–approved protocol. C57BL/6 mice were administered A419259 or KIN-8194 in normal saline by tail vein intravenous injection at 2 mg/kg or orally at 10 or 25 mg/kg. Plasma concentrations were determined by liquid chromatography-tandem mass spectrometry after mass transition 49600→340 AMU. PK parameters were calculated by using Phoenix WinNonlin.
Pharmacodynamic and in vivo efficacy studies
BTKWT- or BTKCys481Ser-expressing TMD-8 ABC DLBCL cells were xenografted into mice for pharmacodynamic (PD) and in vivo efficacy studies. Female NOD-SCID mice (Strain Code 394) age 6 to 8 weeks weighing 16 to 20 g were obtained from Charles River Laboratories (Shrewsbury, MA). Animal studies were performed under IACUC-approved protocols at the Experimental Therapeutics Core at Dana-Farber Cancer Institute. KIN-8194 tolerability was determined in a 14-day daily dosing study which showed that doses up to 75 mg/kg per day were tolerable. Then, 1 × 107 GFP+ BTKWT- or BTKCys481Ser-expressing TMD-8 cells were subcutaneously inoculated in a Matrigel suspension (Corning Life Sciences, Tewksbury, MA). In PD studies, when tumor size approximated 300 mm3, mice were treated with KIN-8194 at the indicated dose by oral gavage. At 6 or 24 hours after administration of KIN-8194, mice were euthanized in a CO2 chamber, and tumors that were harvested mechanically were dissociated to form single-cell suspensions. Phosflow studies were performed for pHCKY410 and pBTKY223 in GFP+ tumor cells. PD studies showing changes in HCK and BTK activity after oral administration of KIN-8194 were also performed in NOD-SCID mice xenografted with BTKWT BCWM.1 WM cells by intravenous injection. Phosflow for pHCKY410 and pBTKY223 in GFP+ BCWM.1 WM cells was performed on mononuclear cells obtained from mouse femurs at 6 and 24 hours after oral administration of vehicle control or KIN-8194 at 25 or 50 mg/kg. For in vivo efficacy studies, mice were randomly assigned to each cohort when tumors reached ∼200 mm3 and were treated once per day with vehicle, ibrutinib, A419259, KIN-8194, or any of those combined with venetoclax at the indicated dose by oral gavage (n = 8 mice per cohort). Tumor volumes were measured twice per week and calculated as tumor volume ([length × width2] × 0.5).
Statistical analysis
Statistical significance of differences was analyzed using one-way analysis of variance with Tukey’s multiple comparisons test by Prism software. Differences were considered significant at P < .05. Error bars where shown denote standard deviation. The Kaplan-Meier method was used to estimate survival curves, and the curves were compared using the log-rank test.
Results
KIN-8194 selectively targets HCK and BTK in MYD88-mutated WM and ABC DLBCL cells
The chemical structure of KIN-8194 is depicted in Figure 1A. KIN-8194 has a chemical structure similar to but distinct from that of A419259 (RK-20449), a toolbox compound we previously characterized in MYD88 lymphomas, including ibrutinib-resistant BTKCys481 mutant-expressing cells (supplemental Figure 1).8 KIN-8194 demonstrated greater selectivity compared with A419259 with no KIT, RET, PDGFRA, EPHB6, or CDPK1 inhibition (at 90% kinase inhibition), an important consideration because several of these targets have well-known toxicity concerns (supplemental Table 1). Biochemical kinase assays demonstrated that KIN-8194 strongly inhibited HCK (50% inhibitory dose [IC50] <0.495 nM) and BTK (IC50, 0.915 nM). By comparison in a fixed time-point assay, ibrutinib exhibited similar BTK inhibition (IC50, 0.614 nM), and 100-fold less potent HCK inhibition (IC50, 49 nM). To assess the kinase target selectivity of KIN-8194, we performed a KINOMEscan assay against a panel of 468 kinases. KIN-8194 at 0.1 and 1.0 μM showed S10 selectivity scores of 0.07 and 0.12, with targets mainly limited to Src (ie, HCK, BLK, LYN, and FRK) and Tec (ie, BTK) family kinases (Figure 1B; supplemental Table 2). To further evaluate the kinome selectivity of KIN-8194 and verify its target engagement in living cells, we performed KiNativ profiling in TMD-8 ABC DLBCL cells at 1.0 μM which confirmed that KIN-8194 strongly engaged both HCK and BTK (Figure 1C; supplemental Table 3). An ATP-competitive assay was also performed using Pierce Kinase Enrichment Kit with an ActivX desthiobiotin-ATP probe after live cell were pretreated with KIN-8194 in MYD88-mutated BCWM.1 WM cells and TMD-8 ABC DLBCL cells. The enriched kinases were resolved by western blots and showed robust engagement of HCK by KIN-8194 and a level of BTK binding for KIN-8194 similar to that of ibrutinib (Figure 1D).
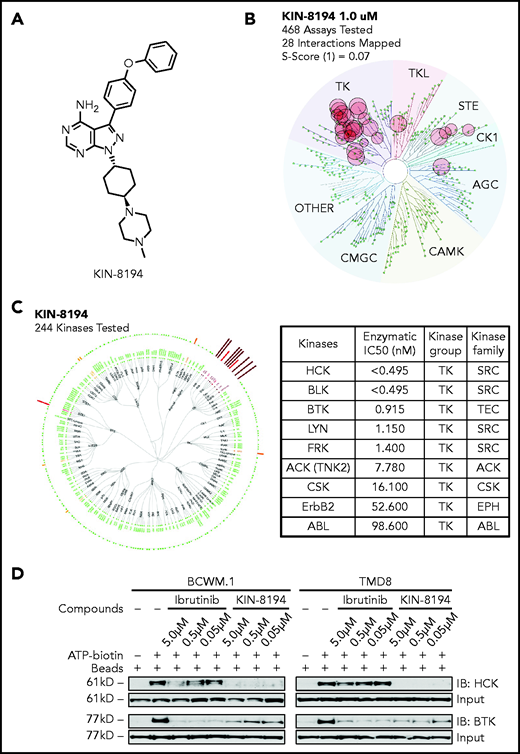
KIN-8194 selectively targets HCK and BTK in vitro in MYD88-mutated WM and ABC DLBCL cells. (A) Chemical structure of KIN-8194. (B) KINOMEscan kinase selectivity profile for KIN-8194. KIN-8194 was profiled at a concentration of 1.0 μM against a diverse panel of 464 kinases by using DiscoverX. Kinases that exhibited a score of 10% or below are marked in red circles. (C) KiNativ cellular target engagement profile for KIN-8194. Kinases engaged >50% with 1.0 µM KIN-8194 treatment in TMD-8 cells are labeled. Kinases demonstrating >90% inhibition in KiNativ profiling (as shown in supplemental Table 2) were subjected to enzymatic IC50 determination as shown here. (D) Activated kinases enriched by pulldown assay using Desthiobiotin-ATP Probe after BCWM.1 and TMD-8 live cells pretreated with KIN-8194 or ibrutinib at indicated concentrations for 90 minutes and the ATP-binding HCK and BTK were resolved by western blotting. IB, ibrutinib.
KIN-8194 selectively targets HCK and BTK in vitro in MYD88-mutated WM and ABC DLBCL cells. (A) Chemical structure of KIN-8194. (B) KINOMEscan kinase selectivity profile for KIN-8194. KIN-8194 was profiled at a concentration of 1.0 μM against a diverse panel of 464 kinases by using DiscoverX. Kinases that exhibited a score of 10% or below are marked in red circles. (C) KiNativ cellular target engagement profile for KIN-8194. Kinases engaged >50% with 1.0 µM KIN-8194 treatment in TMD-8 cells are labeled. Kinases demonstrating >90% inhibition in KiNativ profiling (as shown in supplemental Table 2) were subjected to enzymatic IC50 determination as shown here. (D) Activated kinases enriched by pulldown assay using Desthiobiotin-ATP Probe after BCWM.1 and TMD-8 live cells pretreated with KIN-8194 or ibrutinib at indicated concentrations for 90 minutes and the ATP-binding HCK and BTK were resolved by western blotting. IB, ibrutinib.
KIN-8194 binds to gatekeeper residue Thr333 of HCK but not to Cys481 of BTK
To clarify the mode of recognition of the ATP pocket by KIN-8194, we performed a docking study of KIN-8194 into the co-crystal structure of HCK with A419259 (pdb:5zj6) and BTK with ibrutinib (pdb:5p9i). The docking study predicted that KIN-8194 would recognize the ATP-binding pocket of HCK with an estimated binding energy (ΔG) of 13.4 kcal/mol. The 4-amino group of KIN-8194 is predicted to form a hydrogen bond with the carbonyl groups of the gatekeeper residue Thr333 and also with Glu334 of HCK; its 3-nitrogen atom as an H-bond acceptor interacted with the backbone amino group of Met336 of HCK (supplemental Figure 2A). KIN-8194 also exhibited similar interactions involving Thr474, Glu475, and Met477, but not Cys481 of BTK with an estimated binding energy of −13.0 kcal/mol (supplemental Figure 2B).
KIN-8194 shows robust inhibition of both HCK and BTK activity in MYD88-mutated cells
We next assessed the ability of KIN-8194 and ibrutinib to inhibit HCK and BTK in MYD88-mutated cells by evaluating changes in phosphorylation of HCKTyr410 and BTKTyr223. Ibrutinib modestly inhibited HCK phosphorylation, whereas KIN-8194 showed robust inhibition of HCKTyr410 phosphorylation (Figure 2A; supplemental Figure 3). A similar level of inhibition for BTKTyr223 phosphorylation was also observed for ibrutinib and KIN-8194 (Figure 2B). KIN-8194 more potently suppressed both HCK and BTK activity compared with ibrutinib in primary patient BM LPCs in the presence of cells that support the microenvironment (Figure 2C).
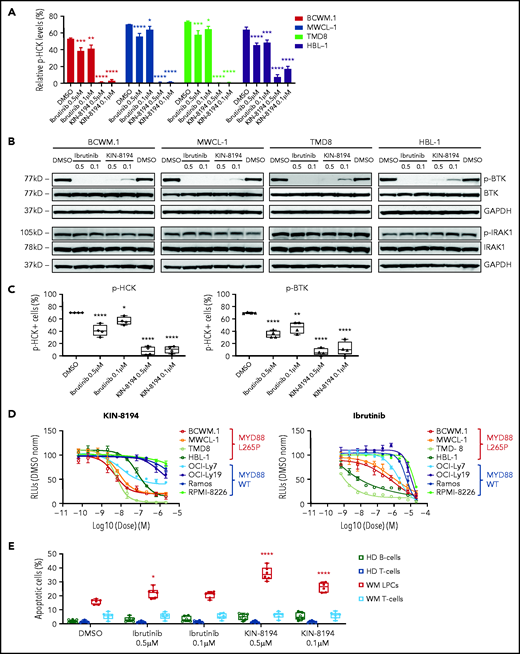
KIN-8194 blocks HCK and BTK and inhibits the growth and survival of MYD88-mutated WM and ABC DLBCL cells. (A) Representative experiments showing changes in phosphorylated HCK (pHCKTyr410) or (B) phosphorylated BTK (pBTKTyr223) levels after 1.0-hour treatment with KIN-8194 or ibrutinib at the indicated concentrations in MYD88-mutated WM (BCWM.1, MWCL-1) and ABC DLBCL (TMD-8, HBL-1) cell lines (n = 3 mice per treatment group). (C) Phosflow results for pHCKTyr410 and pBTKTyr223 in BM LPCs for MYD88-mutated WM patients after treatment with KIN-8194 or ibrutinib at indicated concentrations for 2.0 hours in the presence of whole BM mononuclear cells (n = 4). (D) Dose-response curves for MYD88WT and MYD88L265P cell lines after treatment with KIN-8194 or ibrutinib for 72 hours at indicated concentrations. (E) Apoptotic studies using annexin V/propidium iodide (PI) staining for primary BM LPCs and T cells from MYD88-mutated patients (n = 6) and peripheral blood B cells and T cells from healthy donors (HDs) (n = 6) after treatment with ibrutinib or KIN-8194 at indicated concentrations for 16 hours. *P < .05; **P < .01; ****P < .0001. DMSO, dimethyl sulfoxide; GAPDH, glyceraldehyde-3-phosphate dehydrogenase; norm, normalized; RLU, relative light unit.
KIN-8194 blocks HCK and BTK and inhibits the growth and survival of MYD88-mutated WM and ABC DLBCL cells. (A) Representative experiments showing changes in phosphorylated HCK (pHCKTyr410) or (B) phosphorylated BTK (pBTKTyr223) levels after 1.0-hour treatment with KIN-8194 or ibrutinib at the indicated concentrations in MYD88-mutated WM (BCWM.1, MWCL-1) and ABC DLBCL (TMD-8, HBL-1) cell lines (n = 3 mice per treatment group). (C) Phosflow results for pHCKTyr410 and pBTKTyr223 in BM LPCs for MYD88-mutated WM patients after treatment with KIN-8194 or ibrutinib at indicated concentrations for 2.0 hours in the presence of whole BM mononuclear cells (n = 4). (D) Dose-response curves for MYD88WT and MYD88L265P cell lines after treatment with KIN-8194 or ibrutinib for 72 hours at indicated concentrations. (E) Apoptotic studies using annexin V/propidium iodide (PI) staining for primary BM LPCs and T cells from MYD88-mutated patients (n = 6) and peripheral blood B cells and T cells from healthy donors (HDs) (n = 6) after treatment with ibrutinib or KIN-8194 at indicated concentrations for 16 hours. *P < .05; **P < .01; ****P < .0001. DMSO, dimethyl sulfoxide; GAPDH, glyceraldehyde-3-phosphate dehydrogenase; norm, normalized; RLU, relative light unit.
KIN-8194 blocks HCK and BTK and inhibits the growth and survival of MYD88-mutated WM and ABC DLBCL cells
We next evaluated the antiproliferative effects of KIN-8194 in MYD88-mutated and WT cell lines. KIN-8194 showed marked and targeted antitumor activity against MYD88-mutated cell lines with cellular efficacies more potent than those of ibrutinib (Figure 2D; supplemental Table 4). Conversely, MYD88-mutated OCI-Ly3 cells bearing the CARD11 mutation showed no antitumor effects from either ibrutinib or KIN-8194 (supplemental Figure 4). We also assessed the antitumor activity of KIN-8194 by using primary MYD88-mutated BM LPCs from 6 WM patients and compared its effect in autologous T cells from these patients, and in peripheral blood B and T cells from 6 healthy donors. KIN-8194 produced more robust apoptosis compared with ibrutinib against primary WM LPCs. No notable apoptosis was observed for KIN-8194 against T cells from patients who had received an autologous stem cell transplantation or B and T cells from healthy donors (Figure 2E).
HCK and BTK are key targets of KIN-8194 in MYD88-mutated lymphoma cells
To confirm the key functional targets of KIN-8194 in MYD88-mutated B-cell lymphomas, we generated HCK gatekeeper mutant HCKT333M transduced cell models and performed rescue experiments in MYD88-mutated BCWM.1 and MWCL-1 cells. Expression of HCKT333M produced an approximately log2 fold-change increase in resistance to KIN-8194 compared with vector or HCKWT-transduced BCWM.1 and MWCL-1 cells (Figure 3A; supplemental Table 4). In addition to these findings, we observed that expression of HCKT333M, but not HCKWT, led to persistent activation of HCK in the presence of KIN-8194 (Figure 3B).
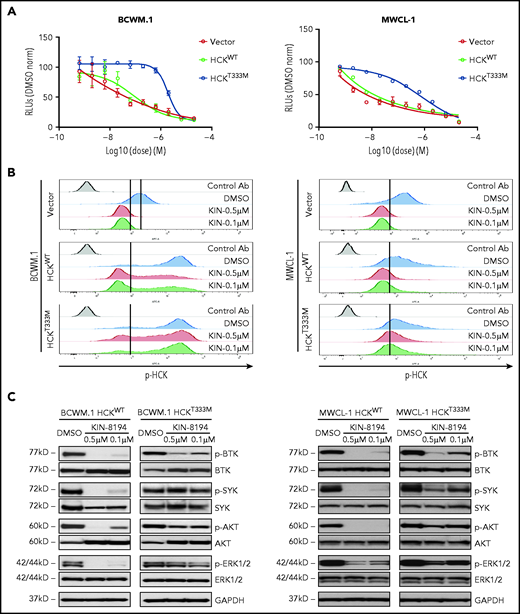
HCK and BTK are key targets of KIN-8194 activity in MYD88-mutated WM cells. (A) Dose-response curves for vector only, HCKWT, or HCKT333M transduced BCWM.1 and MWCL-1 WM cells after treatment with KIN-8194 for 72 hours. (B) Relative pHCKTyr410 levels by Phosflow analysis treatment of KIN-8194 at indicated concentrations for 1.0 hours in vector only, HCKWT, or HCKT333M transduced MYD88-mutated BCWM.1 and MWCL-1 WM cells. (C) pBTKY223, pSYKY525/526, pAKTS473, pERK1/2T202/Y204 expression by western blot analysis in HCKWT or HCKT333M transduced BCWM.1 and MWCL-1 WM cells after treatment with KIN-8194 at indicated concentrations for 1.0 hours. The expression levels of total BTK, SYK, AKT, and ERK1/2 in these cells and protein loading control GAPDH are also shown. Experiments were performed at least twice, with representative blots shown. Ab, antibody.
HCK and BTK are key targets of KIN-8194 activity in MYD88-mutated WM cells. (A) Dose-response curves for vector only, HCKWT, or HCKT333M transduced BCWM.1 and MWCL-1 WM cells after treatment with KIN-8194 for 72 hours. (B) Relative pHCKTyr410 levels by Phosflow analysis treatment of KIN-8194 at indicated concentrations for 1.0 hours in vector only, HCKWT, or HCKT333M transduced MYD88-mutated BCWM.1 and MWCL-1 WM cells. (C) pBTKY223, pSYKY525/526, pAKTS473, pERK1/2T202/Y204 expression by western blot analysis in HCKWT or HCKT333M transduced BCWM.1 and MWCL-1 WM cells after treatment with KIN-8194 at indicated concentrations for 1.0 hours. The expression levels of total BTK, SYK, AKT, and ERK1/2 in these cells and protein loading control GAPDH are also shown. Experiments were performed at least twice, with representative blots shown. Ab, antibody.
Because HCK triggers multiple pro-survival pathways in MYD88-mutated lymphoma cells, we next treated HCKWT- or HCKT333M-expressing BCWM.1 and MWCL-1 cells with KIN-8194 and assessed the activation state of BTK, AKT, ERK1/2, and SYK.7-10 KIN-8194 blocked pBTK, pAKT, pERK1/2, and pSYK in a dose-dependent manner in HCKWT-expressing cells (Figure 3C). Conversely, attenuation of pAKT, pERK1/2, and pSYK was abrogated in HCKT333M-expressing BCWM.1 and MWCL-1 cells. Importantly, pBTK continued to be inhibited by KIN-8194 in HCKT333M-expressing BCWM.1 and MWCL-1 cells, which is consistent with our earlier binding studies showing BTK as an additional direct target of KIN-8194 (Figure 3C). Taken together, these data support that HCK and BTK are key targets of KIN-8194 activity in MYD88-mutated lymphoma cells, and their inhibition by KIN-8194 suppresses known downstream pro-survival signaling pathways.
KIN-8194 overcomes ibrutinib resistance related to BTKCys481Ser-expressing MYD88-mutated B-cell lymphoma cells
We next examined whether KIN-8194 could overcome ibrutinib resistance caused by the mutation of the nucleophilic cysteine residue BTKCys481. The expression of BTKCys481Ser resulted in a log1 fold-change to log2 fold-change in ibrutinib resistance in MYD88-mutated BCWM.1 and TMD-8 cells when compared with their BTKWT- or vector only–expressing counterparts (Figure 4A). KIN-8194 overcame BTKCys481Ser-related resistance in these cells with similar antitumor activity observed in vector only–, BTKWT-, and BTKCys481Ser-expressing BCWM.1 or TMD-8 cells (Figure 4A; supplemental Table 4). Moreover, KIN-8194 showed more robust induction of apoptosis relative to ibrutinib in BTKCys481Ser-expressing BCWM.1 and TMD-8 cells and at levels similar to those observed in BTKWT cells (Figure 4B). KIN-8194 robustly blocked HCK as well as BTK and downstream ERK1/2 activation in vector-, BTKWT-, and BTKCys481Ser-expressing BCWM.1 and TMD-8 cells (Figure 4C-D). These findings demonstrate that KIN-8194 blocks activation of HCK, BTK, and downstream ERK1/2 signaling in BTKCys481-mutated cells and overcomes ibrutinib resistance related to mutated BTKCys481 expression in MYD88-mutated cells.
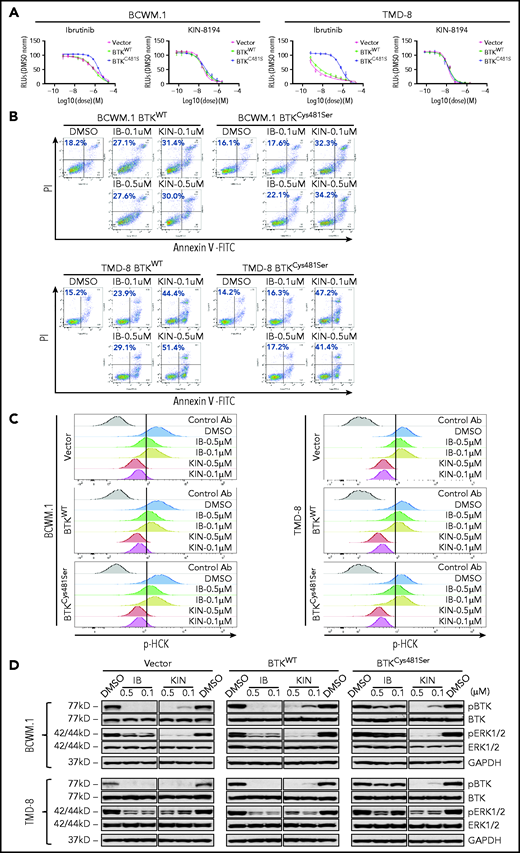
In vitro studies of KIN-8194 in ibrutinib-resistant BTKCys481Ser-expressing WM and ABC DLBCL cells. (A) Dose-response curves for vector only, BTKWT, or BTKCys481Ser transduced BCWM.1 and TMD-8 cells after treatment with ibrutinib or KIN-8194 for 72 hours. (B) Apoptosis analyses with annexin V-fluorescein isothiocyanate (FITC) and PI staining for native, BTKWT-, and BTKCys481Ser-expressing BCWM.1 WM and TMD-8 ABC DLBCL cells after treatment with KIN-8194 or ibrutinib at the indicated concentrations for 16 hours. (C) Changes in pHCKTyr410 expression after treatment with ibrutinib or KIN-8194 at indicated concentrations for 1.0 hours in vector only, BTKWT, or BTKCys481Ser transduced BCWM.1 and TMD-8 cells. (D) Changes in pBTKY223 and pERK1/2T202/Y204 levels after treatment with ibrutinib or KIN-8194 at indicated concentrations for 1.0 hours in vector only, BTKWT, or BTKCys481Ser transduced BCWM.1 and TMD-8 cells. The expression levels of total BTK and ERK1/2 in these cells and protein loading control GAPDH are also shown. KIN, KIN-8194.
In vitro studies of KIN-8194 in ibrutinib-resistant BTKCys481Ser-expressing WM and ABC DLBCL cells. (A) Dose-response curves for vector only, BTKWT, or BTKCys481Ser transduced BCWM.1 and TMD-8 cells after treatment with ibrutinib or KIN-8194 for 72 hours. (B) Apoptosis analyses with annexin V-fluorescein isothiocyanate (FITC) and PI staining for native, BTKWT-, and BTKCys481Ser-expressing BCWM.1 WM and TMD-8 ABC DLBCL cells after treatment with KIN-8194 or ibrutinib at the indicated concentrations for 16 hours. (C) Changes in pHCKTyr410 expression after treatment with ibrutinib or KIN-8194 at indicated concentrations for 1.0 hours in vector only, BTKWT, or BTKCys481Ser transduced BCWM.1 and TMD-8 cells. (D) Changes in pBTKY223 and pERK1/2T202/Y204 levels after treatment with ibrutinib or KIN-8194 at indicated concentrations for 1.0 hours in vector only, BTKWT, or BTKCys481Ser transduced BCWM.1 and TMD-8 cells. The expression levels of total BTK and ERK1/2 in these cells and protein loading control GAPDH are also shown. KIN, KIN-8194.
PK and tolerability of KIN-8194 in rodents
KIN-8194 is metabolized slowly after incubation with purified liver microsomes across multiple species: terminal half-life (t1/2) for mice is 106.4 minutes, t1/2 for rats is >120 minutes, t1/2 for canines is 60 minutes, and t1/2 for humans is 49.5 minutes. By comparison, microsomal stability was shorter for A419259 across species, including humans with a t1/2 of 33.6 minutes (supplemental Figure 5). Cross-species studies showed comparable protein binding for KIN-8194: mice, 97.7%; rats, 95.5%; canines, 95.0%; and humans, 95.7%. By comparison, tighter protein binding was observed for A419259 across species, including 99.8% for humans (supplemental Figure 5). Consistent with the long microsomal stability of KIN-8194, mouse PK studies showed bioavailability of 55% and 49% after oral administration of 10 and 25 mg/kg, respectively (supplemental Figure 5). The serum half-life after intravenous administration was 11.5 hours, after oral administration of 10 mg/kg, it was 15.1 hours, and after oral administration of 25 mg/kg, it was 16.9 hours. By comparison, the serum half-life for A419259 was unpredictable and greatly prolonged at 176.0 hours (supplemental Figure 5).
KIN-8194 was well tolerated at oral doses up to 75 mg/kg once per day with no adverse events observed in NOD-SCID mice after 6 weeks of treatment. In vitro CYP inhibitions (percent at 10 μM) for KIN-8194 were as follows: CYP1A, –27.9%; CYP2B6, +64.5%; CYP2CB, –3.8%; CYP2C9, –10.9%; CYP2C19, +0.8%; CYP2D6, +24.9%; CYP3A, –22.7% and −4.9%). Ames genotoxicity testing was negative for 4 Salmonella typhimurium tester strains (TA98, TA100, TA1535, and TA1537) up to 50 μM with and without metabolic activation by rat liver S9 fraction. hERG testing (patch clamp) for cardiotoxicity was negative at 16 μM.
KIN-8194 shows superior activity over ibrutinib in MYD88-mutated TMD-8 ABC DLBCL xenograft mouse model
We performed PD studies using ibrutinib-sensitive BTKWT TMD-8 ABC DLBCL cells xenografted into mice. Xenografted mice were administered KIN-8194 by oral gavage, and tumor cells were harvested at 6 and 24 hours after single administration of KIN-8194 at 12.5, 25, or 50 mg/kg. Fluorescence-activated cell sorting studies on excised tumors using phospho-specific antibodies demonstrated that KIN-8194 blocked pHCK and pBTK in a dose-dependent manner and maintained suppression for 24 hours for both HCK and BTK, particularly at higher dose levels (Figure 5A-B). Findings were similar after oral administration of KIN-8194 in mice xenografted with BTKWT BCWM.1 WM cells by intravenous injection. Phosflow studies showed sustained reduction in both pHCK and pBTK in GFP+ BCWM.1 WM cells obtained from mouse femurs at 6 and 24 hours after oral administration of KIN-8194 at 25 or 50 mg/kg (supplemental Figure 6).
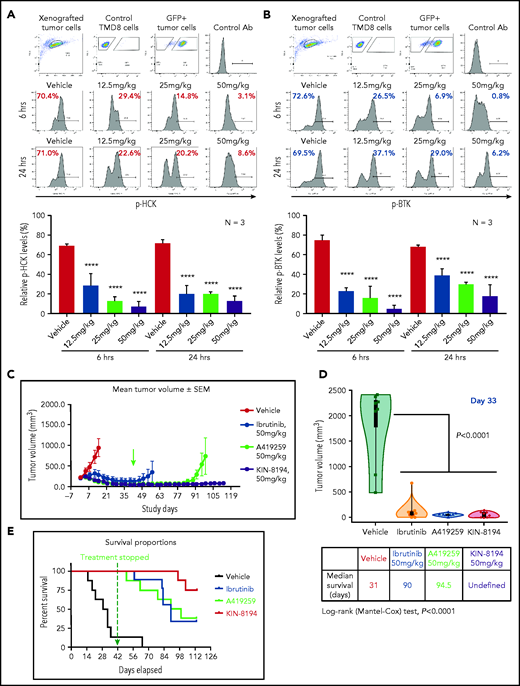
PD and efficacy studies of KIN-8194 in ibrutinib-sensitive BTKWT-expressing TMD-8 ABC DLBCL xenografted mice. PD studies showing the activity of HCK and BTK after oral administration of KIN-8194 in NOD-SCID mice subcutaneously xenografted with ibrutinib-sensitive BTKWT TMD-8 ABC DLBCL cells. Phosflow plots for (A) pHCKY410 and (B) pBTKY223 in GFP+ TMD-8 tumor cells excised at 6 and 24 hours after oral administration of KIN-8194 at the indicated doses (n = 3 per group). (C) Efficacy studies in NOD-SCID mice (n = 8 per cohort) bearing ibrutinib-sensitive BTKWT TMD-8 cells after daily oral administration of vehicle control, ibrutinib, A419259, or KIN-8194 at 50 mg/kg. Tumor volume (mm3) was measured twice per week and reported as the mean volume ± standard error of the mean (SEM). Treatment was stopped (indicated by green arrow) at day 42 and tumor volumes were monitored until day 113. (D) Tumor volume comparisons at day 33. P values for comparisons against mice treated with vehicle control are shown. (E) Survival curve estimations using the Kaplan-Meier method. The median survival (days) for cohorts were determined by using Prism software. P < .0001 for log-rank comparisons between cohorts. ****P < .0001.
PD and efficacy studies of KIN-8194 in ibrutinib-sensitive BTKWT-expressing TMD-8 ABC DLBCL xenografted mice. PD studies showing the activity of HCK and BTK after oral administration of KIN-8194 in NOD-SCID mice subcutaneously xenografted with ibrutinib-sensitive BTKWT TMD-8 ABC DLBCL cells. Phosflow plots for (A) pHCKY410 and (B) pBTKY223 in GFP+ TMD-8 tumor cells excised at 6 and 24 hours after oral administration of KIN-8194 at the indicated doses (n = 3 per group). (C) Efficacy studies in NOD-SCID mice (n = 8 per cohort) bearing ibrutinib-sensitive BTKWT TMD-8 cells after daily oral administration of vehicle control, ibrutinib, A419259, or KIN-8194 at 50 mg/kg. Tumor volume (mm3) was measured twice per week and reported as the mean volume ± standard error of the mean (SEM). Treatment was stopped (indicated by green arrow) at day 42 and tumor volumes were monitored until day 113. (D) Tumor volume comparisons at day 33. P values for comparisons against mice treated with vehicle control are shown. (E) Survival curve estimations using the Kaplan-Meier method. The median survival (days) for cohorts were determined by using Prism software. P < .0001 for log-rank comparisons between cohorts. ****P < .0001.
Given these findings, we next performed in vivo efficacy studies evaluating once-per-day oral administration of vehicle, ibrutinib (50 mg/kg), A419259 (50 mg/kg), and KIN-8194 (50 mg/kg) for 6 weeks (day 42), at which point treatment was stopped because 7 of 8 mice in the vehicle control cohort were moribund or had succumbed. Animals receiving KIN-8194 or ibrutinib showed good tolerance toward treatment during the 6-week treatment period, whereas labored breathing, ruffled fur indicative of lack of grooming, and toxicity were observed in mice receiving A419259. After 6 weeks, all surviving mice were observed off treatment for adverse events, tumor volume evolution, and survival. Significant volumetric differences in tumor size were observed among mice treated with vehicle control, ibrutinib, A419259, or KIN-8194 (Figure 5C-D). Mice that received vehicle control showed continued tumor growth, whereas those treated with ibrutinib, A419259, or KIN-8194 had significantly smaller tumors on day 33 (Figure 5D), which was near the median survival time for mice in the vehicle control group. After cessation of treatment at week 6, tumors in mice treated with ibrutinib and A419259 began to increase, whereas those in mice treated with KIN-8194 were undetectable or remained stable. By study end (day 113), 4 of 8 mice treated with KIN-8194 had no observable tumors, including by necropsy. For the other 4 mice, small palpable but not measurable subcutaneous tumors were detected. Consistent with these observations, mice treated with KIN-8194 showed superior survival with 6 of 8 mice alive at study end (Figure 5E). Conversely, the median survival was shorter at 31, 90, and 94.5 days for mice treated with vehicle control, ibrutinib, and A419259, respectively (Figure 5E).
KIN-8194 is active against ibrutinib resistance in BTKCys481Ser-expressing ABC DLBCL xenograft mouse model
We performed PD studies in mice engrafted with BTKCys481Ser-expressing TMD-8 cells. Xenografted mice received a single administration of KIN-8194 (25, 50, or 75 mg/kg) by oral gavage, and tumor cells were collected 6 and 24 hours later. KIN-8194 effectively blocked pHCK and pBTK in a dose-dependent manner with inhibition of pHCK and pBTK maintained through 24 hours (Figure 6A-B). We next evaluated KIN-8194 activity in BTKCys481Ser-expressing TMD-8 xenografted mice. Xenografted mice were treated with vehicle, ibrutinib (50 mg/kg), A419259 (50 mg/kg), or KIN-8194 (50 mg/kg) by oral administration once per day. Treatment was stopped for all animals at day 49 because of labored breathing and ruffled fur in the A419259 cohort, and tumor volumes were monitored to day 59. Mice treated with KIN-8194 showed superior suppression of tumor growth and survival compared with mice receiving vehicle, ibrutinib, or A419259 (supplemental Figure 7). However, in contrast to our earlier findings, tumors in BTKWT TMD-8 xenografted mice treated with KIN-8194 continued to show time-dependent progression.
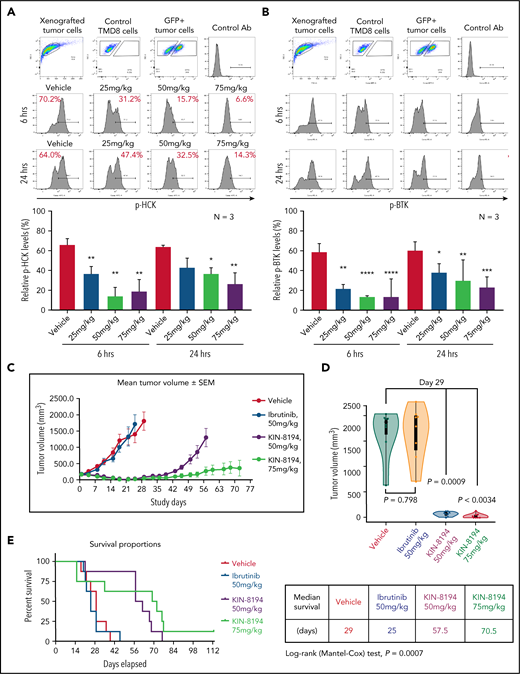
PD and efficacy studies of KIN-8194 in ibrutinib-resistant BTKCys481Ser-expressing TMD-8 ABC DLBCL xenografted mice. PD studies showing the activity of HCK and BTK after oral administration of KIN-8194 in NOD-SCID mice subcutaneously xenografted with ibrutinib-resistant BTKCys481Ser TMD-8 ABC DLBCL cells. Phosflow plots for (A) pHCKY410 and (B) pBTKY223 in GFP+ TMD-8 tumor cells excised at 6 and 24 hours after oral administration of KIN-8194 at the indicated doses (n = 3 per group). (C) Efficacy studies in NOD-SCID mice (n = 8 per cohort) bearing ibrutinib-resistant BTKCys481Ser expressing TMD-8 cells after daily oral administration of vehicle control, ibrutinib (50 mg/kg), or KIN-8194 (50 or 75 mg/kg). Tumor volume (mm3) was measured twice per week and reported as the mean volume ± SEM. (D) Tumor volume comparisons at day 29. P values are for cohort comparisons. (E) Survival curve estimations using the Kaplan-Meier method. Prism software was used to determine the median survival in days for cohorts. Mantel-Cox long-rank P value of .0007 is for comparisons between cohorts. *P < .05; **P < .01; ***P < .005; ****P < .0001.
PD and efficacy studies of KIN-8194 in ibrutinib-resistant BTKCys481Ser-expressing TMD-8 ABC DLBCL xenografted mice. PD studies showing the activity of HCK and BTK after oral administration of KIN-8194 in NOD-SCID mice subcutaneously xenografted with ibrutinib-resistant BTKCys481Ser TMD-8 ABC DLBCL cells. Phosflow plots for (A) pHCKY410 and (B) pBTKY223 in GFP+ TMD-8 tumor cells excised at 6 and 24 hours after oral administration of KIN-8194 at the indicated doses (n = 3 per group). (C) Efficacy studies in NOD-SCID mice (n = 8 per cohort) bearing ibrutinib-resistant BTKCys481Ser expressing TMD-8 cells after daily oral administration of vehicle control, ibrutinib (50 mg/kg), or KIN-8194 (50 or 75 mg/kg). Tumor volume (mm3) was measured twice per week and reported as the mean volume ± SEM. (D) Tumor volume comparisons at day 29. P values are for cohort comparisons. (E) Survival curve estimations using the Kaplan-Meier method. Prism software was used to determine the median survival in days for cohorts. Mantel-Cox long-rank P value of .0007 is for comparisons between cohorts. *P < .05; **P < .01; ***P < .005; ****P < .0001.
Given these findings, we subsequently treated xenografted mice bearing BTKCys481Ser-expressing TMD-8 tumors with vehicle, ibrutinib (50 mg/kg), or KIN-8194 at 50 or 75 mg/kg once per day. Mice received treatment continuously for 16 weeks (day 113). Mice treated with KIN-8194 showed good tolerance to treatment and superior suppression of tumor growth, particularly at the highest dose (75 mg/kg), compared with animals treated with vehicle or ibrutinib (Figure 6C). Comparison of tumor volumes on day 29, near the median survival time for mice in the vehicle control group, showed significantly smaller tumor volumes for mice treated with KIN-8194 compared with mice treated with vehicle or ibrutinib (Figure 6D). Importantly, mice treated with KIN-8194 showed significantly longer survival, with a median survival of 57.5 days for those treated at 50 mg/kg and 70.5 days for those treated at 75 mg/kg. Conversely, median survival was 25 days for xenografted mice treated with vehicle and 29 days for xenografted mice treated with ibrutinib (Figure 6E).
KIN-8194 synergizes with the BCL_2 inhibitor venetoclax in MYD88-mutated B-cell lymphoma cells
Because inhibition of BCL_2 enhances ibrutinib apoptotic activity in MYD88-mutated WM and ABC DLBCL cells,25,26 we next examined whether the combination of KIN-8194 and venetoclax had a similar effect. By combination index and by normalized isobologram analyses, KIN-8194 showed synergistic interactions with venetoclax in both BTKWT- and BTKCys481Ser-expressing BCWM.1 and TMD-8 cells at most dose combinations (Figure 7A). To evaluate the feasibility of using lower doses of KIN-8194 with venetoclax, mice xenografted with ibrutinib-resistant BTKCys481Ser TMD-8 cells were treated with this combination. A dose of 30 mg/kg per day representing ∼50% effective dose for KIN-8194 (Figure 6C) and an intermediate effective dose of venetoclax (50 mg/kg per day) were used in these studies. Mice received treatment once per day with vehicle, KIN-8194, venetoclax, or KIN-8194 and venetoclax. Mice receiving KIN-8194 and venetoclax showed good tolerance to therapy as well as superior suppression of tumor growth compared with mice treated with vehicle, KIN-8194, or venetoclax alone (Figure 7). Tumor volume comparisons at the median survival time for mice in the vehicle control group (ie, day 22) showed significantly smaller tumors in xenografted mice that received KIN-8194 and venetoclax compared with those treated with vehicle, KIN-8194, or venetoclax alone (Figure 7C). Importantly, mice that received combination therapy showed significantly longer median survival (63 days) compared with mice that received vehicle control (22 days), venetoclax (27.5 days), or KIN-8194 (36 days) alone (Figure 7D).
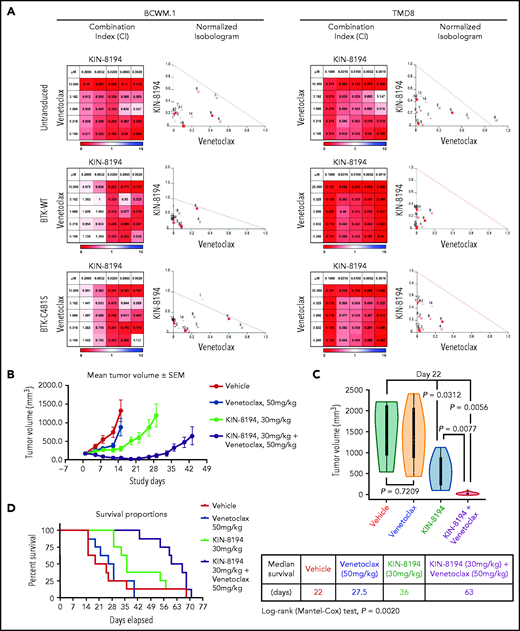
Studies assessing synergistic interactions of KIN-8194 and the BCL_2 inhibitor venetoclax. (A) In vitro studies assessed for synergistic interactions of KIN-8194 and venetoclax in native (untransduced), BTKWT-, and BTKCys481Ser-expressing MYD88-mutated BCWM.1 WM and TMD-8 ABC DLBCL cells. The combination index (CI) and normalized isobologram analyses are depicted. CI <1 (indicated in shades of red) or the dots under the oblique line in the isobologram plots indicate a synergistic effect for the combination. (B) Efficacy studies in NOD-SCID mice (n = 8 per cohort) bearing ibrutinib-resistant BTKCys481Ser-expressing TMD-8 cells after daily oral administration of vehicle control, venetoclax (50 mg/kg), KIN-8194 (30 mg/kg), or the combination of venetoclax (50 mg/kg) and KIN-8194 (30 mg/kg). Tumor volume (mm3) was measured twice per week and reported as the mean volume ± SEM. (D) Tumor volume comparisons at day 22. P values are for cohort comparisons. (E) Survival curve estimations using the Kaplan-Meier method. Prism software was used to determine the median survival in days for cohorts. P = .002 for log-rank comparisons between cohorts.
Studies assessing synergistic interactions of KIN-8194 and the BCL_2 inhibitor venetoclax. (A) In vitro studies assessed for synergistic interactions of KIN-8194 and venetoclax in native (untransduced), BTKWT-, and BTKCys481Ser-expressing MYD88-mutated BCWM.1 WM and TMD-8 ABC DLBCL cells. The combination index (CI) and normalized isobologram analyses are depicted. CI <1 (indicated in shades of red) or the dots under the oblique line in the isobologram plots indicate a synergistic effect for the combination. (B) Efficacy studies in NOD-SCID mice (n = 8 per cohort) bearing ibrutinib-resistant BTKCys481Ser-expressing TMD-8 cells after daily oral administration of vehicle control, venetoclax (50 mg/kg), KIN-8194 (30 mg/kg), or the combination of venetoclax (50 mg/kg) and KIN-8194 (30 mg/kg). Tumor volume (mm3) was measured twice per week and reported as the mean volume ± SEM. (D) Tumor volume comparisons at day 22. P values are for cohort comparisons. (E) Survival curve estimations using the Kaplan-Meier method. Prism software was used to determine the median survival in days for cohorts. P = .002 for log-rank comparisons between cohorts.
Discussion
Given the importance of HCK and BTK in pro-survival signaling in MYD88-mutated lymphomas, we undertook an extensive medicinal chemistry effort to target these kinases. We describe KIN-8194, a pyrazolopyrimidine-based inhibitor that suppressed both HCK and BTK at sub-nanomolar levels with a high degree of selectivity. Target engagement studies confirmed both HCK and BTK binding at doses below pharmacologically achievable levels in rodents. KIN-8194 strongly abrogated HCK and BTK activity in MYD88-mutated cell lines as well as primary MYD88-mutated WM cells. KIN-8194 also abrogated downstream pro-survival signaling related to HCK and BTK, including AKT, ERK, and SYK. Although treatment of HCKWT tumor cells with KIN-8194 abolished both pHCK and pBTK activities, treatment of HCKThr333Met-expressing cells that harbor a mutation at the gatekeeper ATP binding site resulted in loss of activity against HCK. Despite this, KIN-8194 still attenuated pBTK activity consistent with our molecular modeling and target engagement studies that showed direct binding of KIN-8194 to BTK. These studies support on-target activity of KIN-8194 against both HCK and its downstream mediator BTK.
KIN-8194 showed selective killing of MYD88-mutated lymphoma cells and primary WM cells which rely on both HCK and BTK for pro-survival signaling and had more potent anti-tumor activity compared with ibrutinib. Notably, neither ibrutinib nor KIN-8194 showed any activity in OCI-Ly-3 DLBCL cells that carry both mutated MYD88 and the CBM complex member CARD11. Mutations in CARD11 are downstream of BTK and are associated with ibrutinib resistance in ABC DLBCL patients, and therefore may be of consequence to the activity of KIN-8194 in certain ibrutinib-resistant patients.12 In mice xenografted with ibrutinib-sensitive BTKWT TMD-8 ABC DLBCL cells, KIN-8194 showed robust and sustained antitumor activity even after 6 weeks of treatment. Importantly, 4 (50%) of 8 mice showed no evidence of tumor during the 12 weeks of follow-up off treatment. Necropsies revealed no observable tumors in these animals suggestive of a curative potential for KIN-8194 after a limited duration of treatment. Compared with ibrutinib, KIN-8194 showed significantly longer survival at equivalent dosing in this ibrutinib-sensitive xenograft model.
Importantly, our in vitro studies also demonstrated that KIN-8194 abrogated both BTK and downstream ERK activation in ibrutinib-resistant BTKCys481Ser-expressing MYD88-mutated WM and ABC DLBCL cells. Our findings suggest that both BTK inhibition by blockade of upstream HCK activation and binding to non-Cys481 sites within BTK likely account for the activity of KIN-8194 in ibrutinib-resistant BTKCys481Ser-expressing tumor cells. Such a mechanistic approach may represent an advance in MYD88-mutated tumors over noncovalent inhibitors currently being developed for ibrutinib-resistant disease that target BTK alone,27-29 because the addition of HCK activity would block other pro-survival pathways such as ERK, AKT, and SYK in these tumors.8-10,19 Consistent with our in vitro findings, PD studies in mice engrafted with ibrutinib-resistant BTKCys481-mutated TMD-8 ABC DLBCL tumor cells showed that oral administration of KIN-8194 suppressed both BTK and HCK in a dose-dependent manner for 24 hours, which is consistent with a sustainable pharmacodynamic effect with daily dosing. At both 50 and 75 mg/kg per day continuous dosing, KIN-8194 produced superior suppression of tumor growth and a survival advantage in ibrutinib-resistant xenografts compared with mice treated with vehicle control or ibrutinib. The sustained antitumor effect and survival were longest in xenografted mice receiving KIN-8194 at 75 mg/kg per day.
Although KIN-8194 greatly prolonged survival in ibrutinib BTKCys481-mutated xenografts, the curative potential observed in BTKWT xenografts was not recognized. Altered pro-survival signaling and inflammatory cytokine release introduced by mutated BTKCys481 may underpin these findings.19 Indeed, comparison of PD data in BTKWT and mutated BTKCys481 obtained from excised tumors in mice treated with 50 mg/kg per day showed greater levels of HCK and BTK suppression in BTKWT tumors. Given these findings, we sought to examine the potential synergistic benefit of BCL_2 inhibition with KIN-8194. Our previous studies and those of others demonstrated the importance of BCL_2 inhibition in enhancing ibrutinib activity.24,25 Herein, we observed that the addition of BCL_2 inhibitor venetoclax to KIN-8194 led to synergistic interactions across most doses in both BTKWT and BTKCys481Ser-expressing WM and ABC DLBCL cells driven by mutated MYD88. Consistent with the in vitro pharmacologic data, treatment of mice xenografted with ibrutinib-resistant BTKCys481Ser-expressing TMD-8 ABC DLBCL tumors with combined therapy showed superior inhibition of tumor growth and overall survival compared with xenografted mice receiving venetoclax or KIN-8194 alone.
Importantly, KIN-8194 showed distinct biological differences from A419259, a toolbox compound we previously characterized in MYD88 lymphomas, including ibrutinib-resistant BTKCys481 mutant expressing cells.8 A419259 has a chemical structure that is similar to but different from that of KIN-8194. KIN-8194 demonstrated greater selectivity compared with A419259 with no KIT, RET, PDGFRA, EPHB6, or CDPK1 inhibition (at 90% kinase inhibition), an important consideration because several of these targets have well-known toxicity concerns. Moreover, more favorable human microsomal stability, human plasma protein binding, PK, efficacy, and tolerability in animal models, including xenografts with both WT and mutated BTKCys481-expressing tumors were observed with KIN-8194. The more favorable PK characteristics for KIN-8194 over A419259 may have contributed to the enhanced efficacy and tolerability because A419259 exhibited an unpredictable and prolonged serum half-life that could have resulted in drug accumulation.
In addition to MYD88-mutated WM and ABC DLBCL models evaluated herein, other B-cell malignancies driven by MYD88 such as PCNSL, MZL, and CLL may benefit from KIN-8194. Our preliminary rodent PK data showed that high drug concentrations of KIN-8194 in the central nervous system can be achieved, which provides a potentially targetable approach for treating PCNSL. More recently, the importance of HCK as a key driver for the growth and survival of mantle cell lymphoma has been reported.30 Additional modeling studies are needed to examine the activity of KIN-8194 in these entities.
In summary, we described a novel dual inhibitor of HCK and BTK that showed potent and selective in vitro and in vivo activity against MYD88-mutated lymphomas, including ibrutinib-resistant BTKCys481-expressing tumors. These findings highlight the rational translation and targeting of HCK, a key driver of mutated MYD88 pro-survival signaling, and provide a framework for advancing KIN-8194 for B-cell malignancies driven by HCK and BTK.
Acknowledgments
The authors gratefully acknowledge Prafulla Gokhale and Michael J. Poitras at the Experimental Therapeutics Core, Dana-Farber Cancer Institute, for their excellent support for the animal studies and thank the WM patients who provided samples for these studies.
This work was supported by Peter Bing (the International Waldenstrom’s Macroglobulinemia Foundation), by grants from the Leukemia and Lymphoma Society (R6507-18), the National Cancer Institute, National Institutes of Health, SPORE in Multiple Myeloma (2P50CA100707-16A1), Kinogen, the Edward and Linda Nelson Fund for Waldenström Macroglobulinemia (WM) Research, the Kerry Robertson Fund for WM Research, the Bauman Family Trust, and the Siegel Family Fund for WM.
Authorship
Contribution: G.Y., J.W., S.J.B., N.S.G., and S.P.T. conceived and designed the studies; G.Y. and S.P.T. wrote the manuscript; J.W., L.T., S.J.B., N.S.G., G.Y., and S.P.T. supported medicinal chemistry efforts; J.W. performed the data analysis; M.M. performed cellular ex vivo assays using patient biopsies and xenografted mouse tumors; X.L. maintained cell lines and performed lentiviral expression and knockdown studies; M.M. and A.K. performed signaling studies; G.Y. performed target engagement and synergy studies; G.Y. and J.G.C. performed compound screening; N.T., M.G.D., M.L.G., and L.X. processed patients’ samples; Z.R.H. performed the statistical analysis; S.P.T., J.J.C., C.J.P., and K.M. provided patient care and collected patient samples; M.C. performed rodent tolerance studies and PK analyses; J.C. performed docking studies; and N.C.M. and K.C.A. provided data review and input into the writing of this manuscript.
Conflict-of-interest disclosure: S.P.T. received research funding, and/or consulting fees from AbbVie/Pharmacyclics, Janssen Oncology, Beigene, Eli Lilly Pharmaceuticals, Bristol Myers Squibb, and Kinogen. N.S.G. is a founder, scientific advisory board member, and equity holder in Gatekeeper, Syros, Petra, C4, B2S, and Soltego (board member). G.Y., S.J.B., J.W., N.S.G., and S.P.T. are named inventors on a patent application covering KIN-8194 and targeting of HCK in MYD88-mutated diseases. K.C.A. received consulting fees from Bristol Myers Squibb, Celgene, Gilead, Janssen, Precision Biosciences, Sanofi-Aventis, Takeda, and Tolero and is on the board of directors of and stock options in Oncopep. N.C.M. is a consultant for Bristol Myers Squibb, Janssen, Oncopep, Amgen, Karyopharm, Legend, AbbVie, Takeda, and GlaxoSmithKline and is on the board of directors of and stock options in Oncopep. The Gray laboratory receives or has received research funding from Novartis, Takeda, Astellas, Taiho, Jansen, Kinogen, Her2llc, Deerfield and Sanofi. The remaining authors declare no competing financial interests.
Correspondence: Steven P. Treon, Dana-Farber Cancer Institute, M548, 450 Brookline Ave, Boston, MA 02115; e-mail: steven_treon@dfci.harvard.edu.
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal