

Key Points
Leukemia predisposition variants in ETV6 lead to dramatic loss of transcription repressor activity, mainly by disrupting DNA binding.
Germline ETV6 variants influence ALL transcriptional profile with a striking resemblance of ETV6-RUNX1 ALL but unique somatic mutations.

Abstract
There is growing evidence supporting an inherited basis for susceptibility to acute lymphoblastic leukemia (ALL) in children. In particular, we and others reported recurrent germline ETV6 variants linked to ALL risk, which collectively represent a novel leukemia predisposition syndrome. To understand the influence of ETV6 variation on ALL pathogenesis, we comprehensively characterized a cohort of 32 childhood leukemia cases arising from this rare syndrome. Of 34 nonsynonymous germline ETV6 variants in ALL, we identified 22 variants with impaired transcription repressor activity, loss of DNA binding, and altered nuclear localization. Missense variants retained dimerization with wild-type ETV6 with potentially dominant-negative effects. Whole-transcriptome and whole-genome sequencing of this cohort of leukemia cases revealed a profound influence of germline ETV6 variants on leukemia transcriptional landscape, with distinct ALL subsets invoking unique patterns of somatic cooperating mutations. 70% of ALL cases with damaging germline ETV6 variants exhibited hyperdiploid karyotype with characteristic recurrent mutations in NRAS, KRAS, and PTPN11. In contrast, the remaining 30% cases had a diploid leukemia genome and an exceedingly high frequency of somatic copy-number loss of PAX5 and ETV6, with a gene expression pattern that strikingly mirrored that of ALL with somatic ETV6-RUNX1 fusion. Two ETV6 germline variants gave rise to both acute myeloid leukemia and ALL, with lineage-specific genetic lesions in the leukemia genomes. ETV6 variants compromise its tumor suppressor activity in vitro with specific molecular targets identified by assay for transposase-accessible chromatin sequencing profiling. ETV6-mediated ALL predisposition exemplifies the intricate interactions between inherited and acquired genomic variations in leukemia pathogenesis.
Introduction
Acute lymphoblastic leukemia (ALL) is the most common pediatric cancer and a significant cause of childhood morbidity and mortality.1-3 Historically, hereditary predisposition has not been considered a major risk factor contributing to ALL development; however, there is a growing body of evidence indicating that genetic susceptibility plays a much larger role than previously thought. For example, genome-wide association studies have identified common polymorphisms associated with ALL risk affecting, including IKZF1, ARID5B, CEBPE, CDKN2A, BMI1-PIP4K2A, and TP63, and studies of leukemia-prone kindreds or children with the low hypodiploid ALL subtype have revealed rare damaging germline variants in PAX5, IKZF1, and TP53.4-12 More recently, we and others described germline variants in ETV6, the gene encoding the essential hematopoietic transcriptional repressor erythroblast transformation specific (ETS) variant 6, among families exhibiting autosomal-dominant thrombocytopenia and preponderance of ALL.13-15 These ETV6 variants were absent from the general population and cosegregated with thrombocytopenia in all cases. Initial functional evaluation of the small number of index variants revealed that they decreased ETV6 transcriptional repressive activity, and the majority impaired DNA binding and altered subcellular localization.13-15 Additional studies have recently identified familial leukemia cases with inherited structural alterations in the ETV6 gene, further highlighting the role of this gene in ALL predisposition.16,17
To explore the contribution of germline variation in ETV6 more broadly in childhood ALL, we subsequently sequenced 4405 childhood B-ALL cases and identified potential leukemia-related variants in ∼1% of patients.18 Of these, nearly half were located in the essential DNA-binding domain, including 9 variants predicted to be most deleterious by the combined annotation–dependent depletion scores. Nevertheless, the pathogenicity of these purported ALL risk variants remained unclear without experimental characterization of their functional consequences. More importantly, the mechanisms of germline ETV6-mediated leukemogenesis remained undefined.
Here, we systematically characterize how these germline ETV6 variants affect the transcriptional repressor function of the encoded ETV6 protein and elucidate their impacts on downstream gene expression in vitro and in human primary ALL samples. We also comprehensively describe the somatic genomic landscape of a large cohort of childhood leukemia cases with ETV6 risk variants and explore the functional interactions between specific somatic lesions and germline ETV6 variants during oncogene-mediated transformation.
Methods
Thirty-one B-ALL and 2 acute myeloid leukemia (AML) samples with germline ETV6 variants from 32 unique patients at St. Jude Children’s Research Hospital, Dana-Farber Cancer Institute, or the Children’s Oncology Group were included in this study. Genomic DNA was extracted from diagnostic bone marrow or peripheral blood samples using QIAamp DNA Midi or Maxi kit (Qiagen) and total RNA extracted using TRIzol (Thermo Scientific). Bone marrow or peripheral blood collected during clinical remission was used as the source for germline DNA. Whole-genome sequencing of matched germline and tumor DNA was performed on 30 of 32 cases, while in 2 cases, only tumor DNA was sequenced. Whole-exome sequencing was applied on 1 paired sample. Whole-transcriptome sequencing was performed on 22 of these 33 samples (supplemental Table 1, available on the Blood Web site). In addition, 231 children with ALL from the Ma-Spore ALL clinical trial19,20 and 30 ETV6-RUNX1 ALL cases from St. Jude Children’s Research Hospital were included for whom whole-transcriptome and whole-genome sequencing data were used to compare with those of ETV6 ALL, respectively. Somatic and germline variant data as well as transcriptome sequencing data for 13 ALL cases with somatic ETV6 variants (6 nonsense, 4 frameshift, 1 splice site, and 2 missense variants) were obtained and analyzed from St. Jude Children’s Research Hospital and TARGET21,22 (supplemental Table 2). This study was approved by the respective institutional review boards, and informed consent was obtained from the parents, guardians, and/or patients, as appropriate.
Additional details for the genomic profiling, functional assays, and statistical analyses are included in supplemental Methods.
Results
As a transcription factor, ETV6 primarily functions to repress the expression of a wide spectrum of target genes, many of which are highly regulated during hematopoiesis.23-25 Using in vitro biochemical assays, we first sought to systematically characterize the impacts of 34 nonsynonymous germline ETV6 variants that have been identified as ALL associated thus far.18 By directly measuring transcription repressor activity with a luciferase reporter assay in HEK293T cells, we observed that wild-type (WT) ETV6 robustly repressed PF4 promoter-driven transcription, whereas 22 of the 34 variants tested (65%) exhibited significant impairment of transcription repression; in all cases, expression of mutant ETV6 was detectable and comparable to the level of WT ETV6 (not shown) (Figure 1A; these variants are hereafter referred to as “damaging”). In contrast, the other 12 variants functioned similarly to or better than WT ETV6 (these are referred to as “WT-like”).

Functional characterization of germline ETV6 variants identified in pediatric ALL. (A) ETV6 transcription repressor activity was determined using a dual-luciferase reporter assay by cotransfecting an ETV6 expression construct, PF4 luciferase reporter construct, and Renilla luciferase reporter construct in HEK293T cells. Bars show the mean of triplicate experiments and repeated at least 3 times; error bars represent standard deviation (SD). (B) Electrophoretic mobility shift assays were performed using nuclear extracts from HEK293T cells ectopically expressing WT or variant ETV6. Bars show the mean of triplicate experiments; error bars represent SD. (C) Western blotting was performed after subcellular fractionation of HEK293T cells ectopically expressing WT or variant ETV6. Bars show the mean of nuclear to cytoplasmic ratio from 3 individual experiments; error bars represent SD. In panels A-C, a 1-way analysis of variance (ANOVA) with multiple comparison was performed to compare each variant with WT (*P < .01). (D) Germline ETV6 variants were classified as damaging or WT-like on the basis of functional characterization as shown in panels A-C.
Functional characterization of germline ETV6 variants identified in pediatric ALL. (A) ETV6 transcription repressor activity was determined using a dual-luciferase reporter assay by cotransfecting an ETV6 expression construct, PF4 luciferase reporter construct, and Renilla luciferase reporter construct in HEK293T cells. Bars show the mean of triplicate experiments and repeated at least 3 times; error bars represent standard deviation (SD). (B) Electrophoretic mobility shift assays were performed using nuclear extracts from HEK293T cells ectopically expressing WT or variant ETV6. Bars show the mean of triplicate experiments; error bars represent SD. (C) Western blotting was performed after subcellular fractionation of HEK293T cells ectopically expressing WT or variant ETV6. Bars show the mean of nuclear to cytoplasmic ratio from 3 individual experiments; error bars represent SD. In panels A-C, a 1-way analysis of variance (ANOVA) with multiple comparison was performed to compare each variant with WT (*P < .01). (D) Germline ETV6 variants were classified as damaging or WT-like on the basis of functional characterization as shown in panels A-C.
We also tested 12 common ETV6 variants as defined previously18 and found that these exhibited WT levels of transcription repression (supplemental Figure 1). Transcription repressor activity of ETV6 variants tracked closely with bioinformatic prediction score derived from the REVEL algorithm,26 with 11 of 13 damaging missense variants exhibiting a REVEL score >0.5 (supplemental Figure 2A). We then compared the prevalence of ETV6 variants (nonsense/frameshift alterations or missense variants with REVEL score ≥ 0.5) in ALL cases vs non-ALL controls in the Genome Aggregation Database data set (N = 134 187)27 and observed a striking 24.0-fold enrichment of damaging variants in ALL (P = 2.2 × 10−16), whereas WT-like variants were equally distributed between ALL and controls (supplemental Figure 2B).
The ETS domain is critical for ETV6 to suppress transcription.28 Consistent with this notion, 5 of 6 frameshift variants and all 4 nonsense variants predicted to truncate ETV6 upstream or within the DNA-binding domain resulted in nearly complete loss of repressor activity. In fact, while examining bone marrow cells from patients carrying nonsense variant R359X, we could not detect the truncated protein (supplemental Figure 3), suggesting that these variants result in loss of function, plausibly through nonsense-mediated decay. Furthermore, of 13 damaging missense variants, all but one (R433H) resided within the ETS domain. For subsequent functional assays, we tested only the 22 damaging variants and excluded any WT-like variants. In keeping with their loss-of-transcription-repressor activity, the 22 damaging variants exhibited a significant decrease in ability to bind to an ETS DNA consensus sequence (Figure 1B; supplemental Figure 4), as assessed using an electrophoretic mobility shift assay. We also examined the subcellular localization of WT and variant ETV6 proteins. Compared with WT ETV6, which was sequestered within the nucleus, 21 of 22 damaging variants tested (95%) exhibited a significant loss of nuclear localization, albeit to varying degrees (Figure 1C; supplemental Figure 5). A summary of the functional classification for each variant as either damaging or WT-like is shown in Figure 1D.
Because carriers of ETV6 variants always have a heterozygous genotype in the germline, we postulated that variant proteins may function in a dominant-negative fashion. To test this hypothesis, we assessed luciferase activity following cotransfection of constructs encoding WT and increasing amounts of variant ETV6. Across all 22 damaging variants, we consistently observed a dose-dependent inhibition of WT ETV6 activity (Figure 2A), pointing to a potential dominant-negative mechanism of action. In contrast, none of the WT-like variants exhibited such effects (supplemental Figure 6). ETV6 functions as a homodimer or polymer to efficiently bind ETS consensus sequences.29,30 Therefore, we next evaluated the ability of deleterious variants to dimerize with WT protein using a coimmunoprecipitation assay, focusing on 7 variants representative of truncating and missense changes. In this assay, all selected variant ETV6 proteins exhibited robust dimerization with WT ETV6, whereas the double mutant A93D/V112E (known to impair polymerization)31 remained completely uncoupled (Figure 2B). We further asked whether the subcellular localization of WT ETV6 protein would be affected by the presence of mutant ETV6 when both are simultaneously expressed in the same cells. Upon cotransfection, the presence of each of these 7 representative ETV6 variants caused a general reduction in the ratio of nuclear to cytoplasmic WT ETV6 (Figure 2C; supplemental Figure 7). Taken together, these results indicate that damaging ETV6 variants lose their transcriptional repressor function due to impairment of DNA binding and nuclear localization, with minimal effects on dimerization.
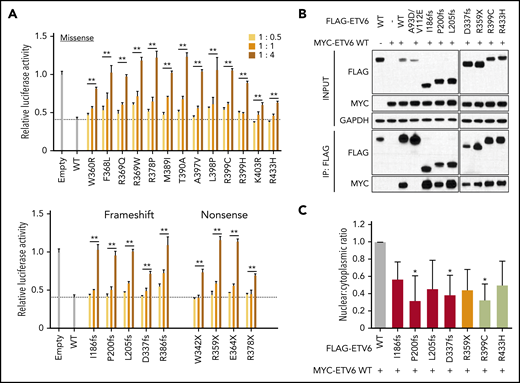
Dominant-negative effects of variant ETV6 on WT ETV6. (A) ETV6 transcription repressor activity was measured by cotransfecting HEK293T with constructs encoding WT ETV6, increasing amounts of variant ETV6, and a luciferase reporter. The top panel denotes missense variants, while the bottom panel indicates nonsense or frameshift variants. Bars show the mean of triplicate experiments and repeated at least 3 times; error bars represent SD. **P < .01 in ≥1 condition. (B) HEK293T cells were cotransfected with MYC-tagged WT and FLAG-tagged variant ETV6 constructs followed by pull-down with anti-FLAG beads for western blotting to examine dimerization. Seven representative variants were chosen for this assay. IP, immunoprecipitation. (C) HEK293T cells were cotransfected with MYC-tagged WT and the same 7 FLAG-tagged variant ETV6 constructs as in panel B followed by subcellular fractionation and western blotting of nuclear and cytoplasmic fractions using an anti-MYC antibody. Red bars indicate frameshift variants, orange bar indicates a nonsense variant, and lime green bars indicate missense variants. Bars represent the nuclear-to-cytoplasmic ratio from triplicate experiments; error bars represent SD. *P < .05. In panels A and C, a 1-way ANOVA with multiple comparison was performed to compare each variant with WT.
Dominant-negative effects of variant ETV6 on WT ETV6. (A) ETV6 transcription repressor activity was measured by cotransfecting HEK293T with constructs encoding WT ETV6, increasing amounts of variant ETV6, and a luciferase reporter. The top panel denotes missense variants, while the bottom panel indicates nonsense or frameshift variants. Bars show the mean of triplicate experiments and repeated at least 3 times; error bars represent SD. **P < .01 in ≥1 condition. (B) HEK293T cells were cotransfected with MYC-tagged WT and FLAG-tagged variant ETV6 constructs followed by pull-down with anti-FLAG beads for western blotting to examine dimerization. Seven representative variants were chosen for this assay. IP, immunoprecipitation. (C) HEK293T cells were cotransfected with MYC-tagged WT and the same 7 FLAG-tagged variant ETV6 constructs as in panel B followed by subcellular fractionation and western blotting of nuclear and cytoplasmic fractions using an anti-MYC antibody. Red bars indicate frameshift variants, orange bar indicates a nonsense variant, and lime green bars indicate missense variants. Bars represent the nuclear-to-cytoplasmic ratio from triplicate experiments; error bars represent SD. *P < .05. In panels A and C, a 1-way ANOVA with multiple comparison was performed to compare each variant with WT.
Because germline ETV6 variants do not always result in malignancy, with only 25% to 30% of carriers developing leukemia,32 we hypothesized that additional genomic abnormalities must be acquired somatically to induce leukemia. To identify these cooperating events and exhaustively define the genomic landscape of ALL with germline ETV6 variants, we performed comprehensive genomic profiling of the leukemia samples from 32 available index cases in whom we identified damaging (n = 24) or WT-like (N = 8) germline ETV6 variants (supplemental Table 1; 1 case [R386fs] had both an ALL and an AML sample available). Using whole-genome sequencing, we examined genome-wide structural alterations (eg, focal copy-number changes and large chromosomal aberrations) and sequence mutations (single-nucleotide variants and small indels); in parallel, we also identified gene fusions and global expression patterns using whole-transcriptome sequencing (supplemental Tables 3-6). Of 23 cases with damaging ETV6 variants, 16 (70%) showed gross chromosomal gains, consistent with our previous report describing an association of germline ETV6 variants with hyperdiploid ALL18 (Figure 3A). Along with DNA index, the other clinical features of damaging ETV6 variant carriers can be found in supplemental Table 7. The hyperdiploid cases exhibited recurrent somatic mutations affecting RAS pathway genes, including NRAS, KRAS, and PTPN11, as is commonly seen in this ALL subtype.33 In contrast, the remaining 7 ETV6 ALL cases had a diploid leukemia genome with a strikingly high prevalence of PAX5 and ETV6 copy-number loss (86% and 57%, respectively). These PAX5 and ETV6 deletions were largely absent in hyperdiploid ALL samples but were frequently observed in ALL harboring the somatic ETV6-RUNX1 fusion (Figure 3A). Chromosomal rearrangement (gene fusion) was relatively rare, detected in only 3 of these 23 cases. We did not observe any recurrent genomic features in the 8 cases with WT-like germline ETV6 variants other than hyperdiploidy in 5 samples. We further performed somatic mutation signature analysis34,35 of the ALL cases with germline ETV6 variants. Mutational signatures corresponding to activity of the AID/APOBEC family of cytidine deaminases (SBS2 and SBS13) and defective DNA repair with BRCA1/2 mutations (SBS3) were identified in 7 and 14 cases, respectively. However, there was no significant difference in mutational signatures between diploid and hyperdiploid subgroups within germline ETV6 variant–positive ALL cases (supplemental Figure 8).

Comprehensive genomic profiling of ALL harboring germline ETV6 variants. (A) Heatmap showing major somatic coding mutations and structural variations identified in cases with a germline ETV6 variant or ETV6-RUNX1 fusion. (B-C) Unsupervised hierarchical clustering of global gene expression profile from cases with germline ETV6 variants (B) or with addition of the 231 pediatric ALL cases of diverse subtypes (C). (D) Somatic genomic features in AML and ALL arising from the same ETV6 germline variant.
Comprehensive genomic profiling of ALL harboring germline ETV6 variants. (A) Heatmap showing major somatic coding mutations and structural variations identified in cases with a germline ETV6 variant or ETV6-RUNX1 fusion. (B-C) Unsupervised hierarchical clustering of global gene expression profile from cases with germline ETV6 variants (B) or with addition of the 231 pediatric ALL cases of diverse subtypes (C). (D) Somatic genomic features in AML and ALL arising from the same ETV6 germline variant.
Similarly, ETV6 ALL cases also exhibited distinct transcriptomic patterns depending upon the functional effects of germline ETV6 variants and somatic genomic alterations. Unsupervised hierarchical clustering analysis first separated cases with damaging vs WT-like germline ETV6 variants, pointing to a global effect of ETV6 on leukemia gene expression (Figure 3B). However, within cases with damaging ETV6 variants, the transcriptional profile was largely driven by leukemia ploidy (diploid vs hyperdiploid) or plausibly by other genomic features tracking with ploidy (eg, ETV6 and/or PAX5 deletion vs RAS pathway mutations in diploid vs hyperdiploid cases, respectively).33,36 When we repeated the clustering analysis using whole-transcriptome sequencing data from 231 pediatric ALL cases representative of diverse subtypes, damaging ETV6 variant ALL samples with diploid karyotype grouped tightly with ETV6-RUNX1 cases, whereas ETV6 ALL with hyperdiploidy shared the expression signature seen in other hyperdiploid cases (Figure 3C). In contrast, 4 of 13 (31%) cases with somatic ETV6 mutations (3 cases with ETV6-RUNX1 fusion and 1 case with somatic copy-number loss of ETV6) clustered with ETV6-RUNX1 ALL based on gene expression, while the 9 remaining somatic ETV6-mutated cases did not exhibit any characteristic expression profile or distinct pattern of genomic alterations (supplemental Table 2 and supplemental Figure 9). Therefore, unlike germline ETV6 variants, acquired ETV6 mutations do not appear to dictate an obvious transcriptomic program or direct the acquisition of specific cooperating alterations in preleukemic cells during the process of malignant transformation.
Notably, in 2 unrelated families, ETV6 germline variants gave rise to both ALL and/or AML (Figure 3D; supplemental Figure 10), raising the possibility that lineage-specific leukemogenesis is driven by secondary somatic lesions. For example, the patient harboring an ETV6 R386fs variant first developed ALL and subsequently therapy-related AML. Genomic profiling of the ALL and AML samples from this individual revealed completely different somatic second events, characteristic of lymphoid (PAX5 deletion) and myeloid malignancies (CBL mutation), respectively.36,37 In the second example, 2 individuals from the same family carrying the ETV6 R399H variant independently developed ALL and AML, respectively. While the ALL genome had a CRLF2 rearrangement with ETV6, PAX5, and IKZF1 deletions, the AML blasts harbored a KMT2A-MLLT3 fusion with an ASXL1 frameshift mutation. These somatic genomic features are uniquely associated with ALL and AML, respectively, and likely contributed to leukemogenesis in distinct lineages arising from the shared germline ETV6 variant.
To explore the effects of ETV6 variants on leukemogenic signaling, we directly compared variant vs WT ETV6 for their impacts on oncogene-driven transformation using a cytokine-dependent growth assay in the murine hematopoietic progenitor cell line Ba/F3. Given the relatively high frequency of RAS mutations in ALL with germline ETV6 variants, we elected to use the NRASG12D mutation as the oncogenic driver in these experiments. We found that coexpression of WT ETV6 strongly inhibited RAS-mediated transformation in this model (ie, interleukin-3 [IL-3]-independent growth), with the WT-like ETV6 R353Q variant showing similar effects (Figure 4A). However, expression of the damaging variants R359X or R399C consistently failed to suppress in vitro transformation in the presence of NRASG12D, suggesting that these variants directly affected a key function of ETV6 as a tumor suppressor. As a control, we expressed ETV6 variants alone and observed that these variants have no transformation capacity in the absence of NRAS G12D (supplemental Figure 11). In parallel, we performed whole-transcriptome sequencing (RNA-seq) and assay for transposase-accessible chromatin sequencing (ATAC-seq) on these Ba/F3 cells to profile the transcriptional and epigenomic changes induced by WT and variant ETV6. Within 48 hours following transduction, there were already significant alterations in the gene expression pattern between cells expressing WT vs variant ETV6, whereas the latter were largely indistinguishable from Ba/F3 cells transduced with empty vector (supplemental Figure 12A). These effects were even more pronounced upon cytokine removal (Figure 4B). Interestingly, of 1189 genes differentially expressed, 597 (50.2%) were upregulated in cells expressing variant ETV6 (supplemental Table 8), consistent with the loss of transcription repressor function. Using ATAC-seq to broadly profile chromatin accessibility as a proxy for epigenomic modifications (eg, chromatin occupancy by transcription factors such as ETV6), we observed an overall increase in ATAC-seq peaks in NRASG12D-transduced Ba/F3 cells expressing variant compared with WT ETV6 (5184 upregulated peaks compared with WT, Figure 4B and supplemental Figure 12A). Footprint analysis revealed that ETV6 and CEBP motifs were significantly enriched in the upregulated ATAC-seq peaks (supplemental Figure 12B). Among these 5184 peaks, 3190 (61.5%) loci encompassed a canonical ETV6-binding motif (supplemental Table 9) (significantly more than what would be expected by chance, P = 7.44 × 10−137). Of the 668 genes with potential loss of ETV6 binding (as evidenced by presence of ATAC-seq peaks and ETV6 binding motifs), 107 showed concordant upregulation in expression in ETV6 variant cells (significantly more than by chance, P = 5.40 × 10−50), suggesting a loss or reduction of ETV6 binding at these targets and, consequently, relief of ETV6-mediated transcriptional repression (Figure 4C).

Effects of ETV6 variants on oncogenic transformation in vitro. Ba/F3 cells were transduced with constructs encoding WT ETV6, damaging ETV6 (R359X or R399C), WT-like ETV6 (R353Q), and NRASG12D, followed by sorting for cells expressing mCherry and ZsGreen double-positive cells and IL-3 withdrawal 48 hours after transduction. (A) Cytokine-independent cell growth was monitored daily as an indicator of transformation in vitro. Data represent the mean from 3 individual experiments; error bars represent SD. Two-way ANOVA with multiple comparison was performed to compare each variant with WT (*P < .05). (B) RNA-seq and (C) ATAC-seq were performed using Ba/F3 cells at 48 hours after IL-3 withdrawal. Cells expressing a damaging ETV6 variant (R359X) exhibited similar RNA-seq and ATAC-seq patterns as cells transduced with empty vector (B). Differential ATAC-seq peaks in Ba/F3 cells expressing a damaging ETV6 variant (R359X) versus WT ETV6 correspond to upregulated genes identified by RNA-seq (C). (D) t-Distributed stochastic neighbor embedding clustering analysis was performed on the 253 ALL cases as shown in Figure 3C using 94 possible target genes identified by RNA-seq and ATAC-seq of Ba/F3 cells.
Effects of ETV6 variants on oncogenic transformation in vitro. Ba/F3 cells were transduced with constructs encoding WT ETV6, damaging ETV6 (R359X or R399C), WT-like ETV6 (R353Q), and NRASG12D, followed by sorting for cells expressing mCherry and ZsGreen double-positive cells and IL-3 withdrawal 48 hours after transduction. (A) Cytokine-independent cell growth was monitored daily as an indicator of transformation in vitro. Data represent the mean from 3 individual experiments; error bars represent SD. Two-way ANOVA with multiple comparison was performed to compare each variant with WT (*P < .05). (B) RNA-seq and (C) ATAC-seq were performed using Ba/F3 cells at 48 hours after IL-3 withdrawal. Cells expressing a damaging ETV6 variant (R359X) exhibited similar RNA-seq and ATAC-seq patterns as cells transduced with empty vector (B). Differential ATAC-seq peaks in Ba/F3 cells expressing a damaging ETV6 variant (R359X) versus WT ETV6 correspond to upregulated genes identified by RNA-seq (C). (D) t-Distributed stochastic neighbor embedding clustering analysis was performed on the 253 ALL cases as shown in Figure 3C using 94 possible target genes identified by RNA-seq and ATAC-seq of Ba/F3 cells.
Comparing ATAC-seq and whole-transcriptome sequencing data and mapping to homologous human genes, we identified 94 putative ETV6 target genes (based on upregulation of expression, increase in chromatin openness, and the presence of an ETV6 binding motif) (supplemental Table 10). These genes are most likely to be transcriptionally regulated by ETV6, with expression altered in cells harboring variant ETV6. Gene Ontology analysis revealed significant enrichment of genes involved in innate immune response and leukocyte migration in inflammatory response (supplemental Table 11). Interestingly, we found that these 94 genes showed significant overlap with ETV6 targets identified using chromatin immunoprecipitation sequencing in ALL cell lines (12 out of 94 genes, P = .038 by Fisher’s exact test) (supplemental Table 10).38 This supports the notion that these genes may be direct transcriptional targets of ETV6. Strikingly, t-distributed stochastic neighbor embedding clustering analysis39 of the transcriptome of 253 primary ALL tumors on the basis of these 94 genes recapitulated the transcriptional relationship of germline ETV6 variant-associated ALL with other subtypes as determined by transcriptome seq (Figure 4D). This finding strongly suggests that these 94 genes are essential ETV6 targets, potentially linking their deregulation to the pathogenesis of germline ETV6 variant ALL. Finally, a comprehensive summary of the functional consequences of each germline ETV6 variant identified in this study can be found in Figure 5.
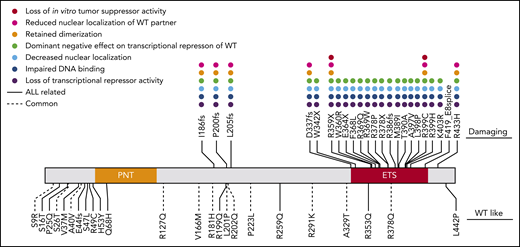
Summary of in vitro functional characterization of ETV6 variants. A variety of in vitro assays were performed to comprehensively characterize the impacts of germline ETV6 variants. Dot colors represent different assays; blanks denote that the variant was not tested in the assay. The top panel includes “damaging” variants as determined by impairment of transcriptional repression in luciferase reporter assays. The bottom panel includes WT-like variants, which showed no impairment of transcriptional repression.
Summary of in vitro functional characterization of ETV6 variants. A variety of in vitro assays were performed to comprehensively characterize the impacts of germline ETV6 variants. Dot colors represent different assays; blanks denote that the variant was not tested in the assay. The top panel includes “damaging” variants as determined by impairment of transcriptional repression in luciferase reporter assays. The bottom panel includes WT-like variants, which showed no impairment of transcriptional repression.
Discussion
In this study, we comprehensively characterized the functional impacts of a large panel of germline ALL risk variants in ETV6, profiled the somatic genomic landscape of ALL arising from this unique genetic predisposition, and explored how these germline and somatic genetic variations interact to enable transformation and alter the epigenomic and transcriptional landscape of leukemia cells. Through these studies, we classified 22 of 34 ETV6 variants as damaging based on the degree of reduction in transcription repressor activity, all of which also lost the ability to bind ETV6 target DNA sequences and properly localize to the nucleus. Nearly all of the 22 damaging variants resided within, or caused truncation of, the ETS DNA-binding domain (Figure 1), and they maintained their ability to dimerize with WT ETV6, leading to cytoplasmic sequestration and impaired repressor activity of the WT protein.
Compared with the damaging germline variants, most WT-like variants were missense and located outside the ETS DNA-binding domain. Despite their normal transcription repressor activity, it remains possible that at least some of these WT-like variants have functional consequences. For example, the PNT domain facilitates homo-oligomerization and formation of corepressor complexes, and the central domain has been implicated in binding of corepressors.29,40-43 Therefore, ETV6 variants in or near these domains could disrupt protein-protein interactions and thus alter the binding of corepressor complexes to specific targets. Such effects might not be observed in a heterologous ectopic expression system using a single target promoter.
Intriguingly, 2 of the 12 WT-like variants demonstrated enhanced repressor activity compared with WT (E44fs and L442P). Close examination of E44fs sequence revealed 2 possible protein products: (1) it encodes an early termination at amino acid residue 44 of the canonical reading frame, which results in a truncated ETV6 missing most of the functional domains; or (2) it uses the +1 reading frame and an alternative start codon, which gives rise to a shortened ETV6 protein that loses the first 45 amino acids at the N-terminal portion but retains the PNT, linker, and ETS domains (supplemental Figure 13). The latter is similar to a previously reported ETV6 isoform (TEL-M43), which lacks the first 42 amino acids, but exerted enhanced repressor activity compared with WT ETV6 protein due to altered nuclear export.44 To test this directly, we transfected 293T cells with constructs encoding WT ETV6 or the TEL-M43 or E44fs variants and then performed western blots to evaluate protein expression. A shortened ETV6 protein was indeed detected in cells expressing the E44fs variant, with the same molecular weight as TEL-M43 (supplemental Figure 13). While evaluation of endogenous protein expression driven by the E44fs variant in patient samples or knockin cells would be ideal to confirm this finding and rule out the possibility of nonsense-mediated decay, our data nevertheless suggest that the E44fs variant encodes a shortened E44fs protein that is highly analogous to the TEL-M43 isoform and exerts a similar mechanism of enhanced repression. By contrast, the L442P variant is located in close proximity to the C-terminal helix that directly interacts with the ETS domain, which potentially blocks ETV6 DNA binding with inhibitory effects.30,45 We postulate that the L442P variant introduces changes to this highly conserved leucine residue and interferes with this autoinhibitory function and thus enhances repressor activity.
Given the consistent damaging effects of ETV6 variants on the activity of this critical hematopoietic transcription factor, we hypothesized that ALL arising from this predisposition syndrome would have a unique global transcriptional landscape. This notion is partly supported by our whole-transcriptome sequencing–based clustering analysis in which ALL cases with damaging ETV6 variants were clearly separated from those with WT-like variants. The impact of these germline variants is profound at a genome-wide level, and the preservation of such effects in leukemia cells suggest their importance in leukemogenesis. On the other hand, within ETV6 ALL, the transcriptional program was also strongly influenced by secondary somatic abnormalities, with an especially striking separation of hyperdiploid vs diploid ALL cases. In contrast to hyperdiploid cases with RAS pathway mutations, diploid ALL cases with germline ETV6 variants exhibited highly recurrent focal deletion of ETV6 and PAX5 (and to a lesser degree IKZF1). This suggests that even with the same underlying genetic predisposition factor, preleukemic cells can invoke a few restricted but distinctive pathways as major cooperating events during leukemogenesis. Because hyperdiploidy can arise in utero, this early and large-scale genomic abnormality could efficiently promote leukemogenesis, coupled with germline ETV6 defects, with a small number of additional somatic hits (eg, RAS mutations). In contrast, diploid ETV6 ALL may take a narrower path to leukemia, requiring loss of several hematopoietic transcription factors (eg, ETV6, PAX5, and IKZF1) that would result in disruption of normal B-cell differentiation. This is further demonstrated by our discovery of distinct lymphoid- and myeloid-related somatic genetic events in the same individual (ETV6 R386fs) with both ALL and AML, and in 2 members of the same family (ETV6 R399H), one with ALL and the other with AML, respectively. While germline ETV6 variants might set the stage for leukemogenesis, the development of leukemia subtypes of differing cellular origin likely results from the acquisition of specific second somatic hits (eg, PAX5 deletion in ALL or AXSL1 frameshift in AML) and the hematopoietic compartment in which they arise. Functional validation of these somatic genomic features in appropriate leukemia model systems is warranted in future studies to determine their cooperativity with germline ETV6 variants.
The shared transcriptional landscape between ALL with germline ETV6 variants and those with the somatic ETV6-RUNX1 fusion is highly reminiscent of the recently described “ETV6-RUNX1-like” ALL subtype.46,47 These notable similarities beg the question as to whether all or a proportion of ETV6-RUNX1-like cases arise in individuals harboring damaging germline (or somatic) ETV6 variants. Also of note is a high frequency of somatic copy-number loss of ETV6 in ETV6-RUNX1 ALL and germline ETV6 variant ALL, arguing that the deletion of the WT ETV6 allele is required for development of leukemia in some, but not all, cases. This raises the possibility of complex mechanisms by which ETV6 is involved in leukemogenesis, similar to the multifaceted roles of the major tumor suppressor p53, which can promote tumorigenesis by loss of tumor suppressor function or conversely by acquisition of dominant-negative or even “gain-of-function” effects.48 Knockin of variant ETV6 in murine or other models is needed to more definitively dissect the leukemogenic properties associated with specific germline ETV6 variants.
Testing NRASG12D-mediated transformation in vitro, we demonstrated that damaging ETV6 variants lack the tumor suppressor activity of WT protein and significantly alter the transcriptional and epigenomic programs. Our sequencing analyses identified 94 potential ETV6 target genes that were upregulated in cells expressing the damaging ETV6 R359X variant. Interestingly, expression profile analysis using this gene set recapitulated the clustering of germline ETV6 variant ALL cases with other ALL subtypes, as observed by whole-transcriptome analysis, pointing to an essential role for these putative ETV6 targets in the global transcriptional landscape. Comparing our gene set with ETV6 and/or ETV6-RUNX1 targets reported previously,25,49,50 we observed a number of overlapping genes, including CLIC5, LRP8, SLC7A11, CHST12, and IL21R (supplemental Figure 14). One of the most consistently upregulated genes in ETV6 mutant ALL is CLIC5, the gene encoding chloride intracellular channel 5. CLIC5 was recently reported as a direct target of ETV6 and is overexpressed in ETV6-RUNX1 pediatric ALL cases.25 Overexpression of CLIC5 in that study revealed its ability to suppress lysosomal-mediated apoptosis induced by hydrogen peroxide. This represents a potential mechanism by which variants in ETV6 could lead to CLIC5-mediated resistance to oxidative stress and accumulation of DNA damage. Given these intriguing findings, there is a pressing need to elucidate the molecular mechanisms by which ETV6 and its downstream target genes contribute to normal and malignant hematopoiesis.
In summary, we provide a comprehensive functional characterization of ALL risk variants in ETV6 that result in profound alterations in biochemical, transcriptional, and epigenomic functions of ETV6. The functional data gathered in this study will be helpful to improve upon clinical classification of germline ETV6 variants. Genomic profiling of ALL arising from this rare genetic syndrome highlights remarkable similarities in the effects of damaging germline and somatic ETV6 aberrations on the transcriptional landscape of leukemic blasts, implying intricate interactions between the host and leukemia genomes in ALL pathogenesis.
For original data, please contact Jun J. Yang (jun.yang@stjude.org) or Kim E. Nichols (kim.nichols@stjude.org).
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Acknowledgments
The authors thank the patients and parents who participated in the St. Jude Children’s Research Hospital and Children’s Oncology Group protocols included in this study, as well as the clinicians and research staff at St. Jude Children’s Research Hospital and Children’s Oncology Group institutions.
This work was supported by the US National Institutes of Health National Cancer Institute (grants CA21765, CA98543, CA114766, CA98413, CA180886, CA180899, and CA241452) and National Institute of General Medical Science (grant GM115279) and the American Lebanese Syrian Associated Charities.
The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Authorship
Contribution: R.N., R.B.-D., Wentao Yang, M. Bloom, G.W., K.E.N., and J.J.Y. wrote the manuscript; R.N., R.B.-D., Wentao Yang, N.O., X.Z., Wenjian Yang, K.H., M. Bloom, K.V., M. Burns, Z.L., T.-N.L., M.Q., T.M., J.M.G.-F., K.R.R., E.R., C.M., C.-H.P., A.E.-J.Y., J.Z., M.L.M., J.M.K., S.P.H., S.N., G.W., M.L.L., K.E.N., and J.J.Y. gathered data; R.N., R.B.-D., Wentao Yang, N.O., Wenjian Yang, M. Bloom, M.Q., S.N., and G.W. analyzed data; R.N., R.B.-D., Wentao Yang, G.W., K.E.N., and J.J.Y. interpreted data; and all authors critically reviewed the manuscript and agreed to submit the paper for publication.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Jun J. Yang, Hematological Malignancies Program, Comprehensive Cancer Center, Department of Pharmaceutical Sciences, Department of Oncology, St. Jude Children's Research Hospital, MS313, 262 Danny Thomas Place, Memphis, TN 38105-3678; e-mail: jun.yang@stjude.org; and Kim E. Nichols, Hematological Malignancies Program, Comprehensive Cancer Center, Department of Oncology, St. Jude Children’s Research Hospital, MS1170, 262 Danny Thomas Place, Memphis, TN 38105-3678; e-mail: kim.nichols@stjude.org.
