

Key Points
IKAROS novel allelic variants disrupt dimerization of the IKAROS family of transcription factors.
IKAROS dimerization mutants act through a distinctive mechanism and manifest predominantly as hematologic diseases, with limited infections.

Abstract
IKAROS is a transcription factor forming homo- and heterodimers and regulating lymphocyte development and function. Germline mutations affecting the IKAROS N-terminal DNA binding domain, acting in a haploinsufficient or dominant-negative manner, cause immunodeficiency. Herein, we describe 4 germline heterozygous IKAROS variants affecting its C-terminal dimerization domain, via haploinsufficiency, in 4 unrelated families. Index patients presented with hematologic disease consisting of cytopenias (thrombocytopenia, anemia, neutropenia)/Evans syndrome and malignancies (T-cell acute lymphoblastic leukemia, Burkitt lymphoma). These dimerization defective mutants disrupt homo- and heterodimerization in a complete or partial manner, but they do not affect the wild-type allele function. Moreover, they alter key mechanisms of IKAROS gene regulation, including sumoylation, protein stability, and the recruitment of the nucleosome remodeling and deacetylase complex; none affected in N-terminal DNA binding defects. These C-terminal dimerization mutations are largely associated with hematologic disorders, display dimerization haploinsufficiency and incomplete clinical penetrance, and differ from previously reported allelic variants in their mechanism of action. Dimerization mutants contribute to the growing spectrum of IKAROS-associated diseases displaying a genotype-phenotype correlation.
Introduction
IKAROS is a hematopoietic cell-specific zinc finger (ZF) transcription factor that plays a crucial role in lymphocyte differentiation and development.1,2 These effects are mediated through its interaction with the nucleosome remodeling deacetylase (NuRD; Mi-2) and Sin3 histone deacetylase complexes, as well as the polycomb repressive complex 2 proteins.1,3-6 The IKAROS family consists of IKAROS/IKZF1, HELIOS/IKZF2, AIOLOS/IKZF3, EOS/IKZF4, and PEGASUS/IKZF5, each of which has multiple isoforms.7-9 They all contain a conserved N-terminal DNA binding domain (ZF1-3/4) and a C-terminal dimerization domain (ZF5-6). The N-terminal ZFs are required for binding to specific DNA sequences containing the core GGGAA motif, whereas the C-terminal ZFs mediate homo- and heterodimerization between IKAROS isoforms and other IKAROS family members.7
Mouse models have demonstrated that IKAROS is essential for the commitment of early hematopoietic progenitors of the B and T lymphoid lineages, as well as mature lymphoid and myeloid cells.10,11 In humans, somatic IKZF1 mutations leading to functional defects or reduced protein expression were originally observed in patients with B-cell precursor acute lymphoblastic leukemia (ALL) and, rarely, T-cell ALL (T-ALL), and were associated with poor prognosis.12-15 Moreover, germline heterozygous IKZF1 mutations were detected in 0.9% of all B-cell ALL (B-ALL), the most common malignancy in children.12
Recently, we and other investigators reported germline heterozygous IKZF1 allelic variants associated with specific immunological phenotypes. The phenotypes consisted of a common variable immunodeficiency (CVID)-like presentation with progressive B-cell loss, hypogammaglobulinemia, and antibody dysfunction, along with recurrent and severe bacterial infections; B-ALL susceptibility and autoimmune manifestations were also observed.12,16-22 These patients carried heterozygous loss-of-function mutations that primarily affected the DNA binding domain of IKZF1, impairing its function and displaying haploinsufficiency (HI) (ie, the mutant allele did not affect the function of the wild-type [WT] allele). Alternatively, some DNA binding domain loss-of-function mutations acted in a dominant-negative (DN) manner (ie, the mutant allele negatively affected the function of the WT allele). The latter patients presented with more serious clinical and immunological phenotypes, including severe T-cell differentiation and functional defects, as well as an absence of B cells, mild to severe neutropenia, and eosinopenia. Their clinical findings included susceptibility to Pneumocystis jirovecii pneumonia; recurrent bacterial, viral, and/or fungal infection; and predisposition to T-ALL.23
Herein, we describe novel missense and nonsense IKZF1 allelic variants in 4 unrelated families primarily affecting dimerization and acting via a previously unreported mechanism. The affected individuals presented with some distinctive clinical features, including hematopoietic cytopenias presenting as Evans syndrome and hematologic malignancies, including T-ALL and Burkitt lymphoma; other manifestations were B-cell lymphopenia and hypogammaglobinemia, but recurrent or severe infections were not highly prevalent. Our data demonstrate that dimerization defective (DD) mutants acting by HI can also disrupt sumoylation, protein stability, and binding to histone deacetylase 1 (HDAC1), which alters the recruitment of the NuRD complex affecting gene regulation by this transcription factor. These results demonstrate that mutations affecting the dimerization domain, as well as those involving the DNA binding domain, all lead to IKAROS dysfunction. Furthermore, each genotype has a unique mechanism of action likely defining the clinical phenotypes in the growing spectrum of defects producing IKAROS-associated diseases.
Materials and methods
Patients and samples
All patients or their guardians provided written informed consent in accordance with the Declaration of Helsinki under institutional review board–approved protocols of the National Institutes of Health/National Institute of Allergy and Infectious Diseases. Blood from healthy donors (HDs) and patients was obtained under approved protocols, which also allowed for the collection and use of patients’ family history and pedigrees for publication. All procedures were based on standard of care, and established clinical guidelines were followed.
Sequencing
Selected next-generation sequencing results were confirmed by Sanger sequencing in index cases, as well as in all available relatives. Sanger sequencing, whole-exome sequencing, and RNA-sequencing (RNASeq) methods are provided in the supplemental Methods (available on the Blood Web site).
Protein expression, localization, and interaction
IKAROS protein expression, coimmunoprecipitations, immunofluorescence, and electrophoretic mobility shift assay (EMSA) were performed as previously described23 and are detailed in the supplemental Methods.
Luciferase reporter assay
The IKBS1 high-affinity IKAROS binding site (TCAGCTTTTGGGAATACCCTGTCA) was used to assess IKAROS binding activity. Luciferase activity was measured using the Dual-Luciferase Reporter Assay System (Promega), according to the manufacturer’s instructions. Additional information is provided in the supplemental Methods.
Statistical analysis
Statistical analyses were performed with GraphPad Prism 6.0 (GraphPad). Differences were considered significant at P < .05.
Clinical histories
Complete clinical summaries and laboratory results can be found in Table 1 and supplemental Clinical summaries.
Clinical and laboratory feature in patients with heterozygous IKZF1 dimerization defective mutations
| A.I.1 (g-mother) | A.II.1 (father) | A.III.1 (index pt) | B.I.1 (mother) | B.II.1 (index pt) | C.I.1 (g-mother) | C.II.1 (mother) | C.II.2 (aunt) | C.II.3 (aunt) | C.III.1 (index pt) | C.III.2 (sister) | D.I.1 (father) | D.II.1 (index pt) | Reference | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Mutation | p.R213* | p.R213* | p.R213* | p.S427* | p.S427* | p.C467R | p.C467R | p.C467R | p.C467R | p.C467R | p.C467R | p.R502L | p.R502L | % of total lymphocytes |
| Age, y; sex | 74; F | 48; M | 17; M | 74; F | 31; M | 66; F | 44; F | 36; F | 38; F | 18; F | 21; F | 50; M | 15; M | |
| Age at onset, y; diagnosis | 9; ITP | 2.5; T-ALL | 9; ITP | 9; Burkitt lymphoma | ||||||||||
| Lymphocyte phenotyping | ||||||||||||||
| CD3 T, ALC | 812 | 1384 | 970 | 797 | 1348 | 2510 (H) | 1196 | 1821 | 1004 | 1394 | 1556 | 2095 | 4181 (H) | 714-2266 |
| CD3 T, % | 81.2 | 87.6 (H) | 85.8 (H) | 57 (L) | 89.3 (H) | 91.6 (H) | 87.3 (H) | 80.2 | 85.8 (H) | 91.1 (H) | 77.4 | 77.6 | 88.2 (H) | 60-83.7 |
| CD4 T, ALC | 346 (L) | 272 (L) | 421 | 558 | 477 | 1121 | 523 | 742 | 580 | 915 | 824 | 1450 | 2176 (H) | 359-1565 |
| CD4 T, % | 34.7 | 17.2 (L) | 37.3 | 39.9 | 31.6 (L) | 40.9 | 38.2 | 32.7 | 49.6 | 59.8 | 41 | 53.7 | 45.9 | 31.9-62.2 |
| CD4+CD62L+CD45RA+, % | n.d. | 3.8 (L) | 11.6 | n.d. | 3.4 (L) | 3.2 (L) | 7.7 | 4.9 (L) | 14.2 | 20.4 | 8.5 | 18.2 | 25.6 | 7.6-37.7 |
| CD4+CD62L+CD45RA−, % | n.d. | 10.2 (L) | 21.5 | n.d. | 24.7 | 34 (H) | 23 | 25 | 31.3 (H) | 33.9 (H) | 25.4 | 22.8 | 15.1 | 10.4-30.7 |
| CD4+CD62L−CD45RA−, % | n.d. | 3.1 | 4 | n.d. | 3.5 | 3.7 | 6.8 | 2.7 | 4 | 5.2 | 6.6 | 11.7 | 4.6 | 2.3-15.6 |
| CD4+CD62L−CD45RA+, % | n.d. | 0.2 | 0.3 | n.d. | 0 | 0 | 0.6 | 0 | 0.1 | 0.3 | 0.5 | 1.1 | 0.6 | 0-1.5 |
| CD8 T, ALC | 441 | 1063 (H) | 483 | 230 | 808 | 1145 (H) | 564 | 826 | 335 | 412 | 607 | 508 | 1673 (H) | 178-853 |
| CD8, % | 44.1 (H) | 67.3 (H) | 42.7 (H) | 16.5 | 53.5 (H) | 41.8 (H) | 41.2 (H) | 36.4 (H) | 28.6 | 26.9 | 30.2 | 18.8 | 35.3 (H) | 11.2-34.8 |
| CD8+CD62L+CD45RA+, % | n.d. | 26.8 (H) | 20.7 (H) | n.d. | 22 (H) | 18.8 | 11.3 | 16.2 | 16.4 | 20.2 (H) | 16 | 7.2 | 26.2 (H) | 5.7-19.7 |
| CD8+CD62L+CD45RA−, % | n.d. | 6.9 | 6.1 | n.d. | 9.1 | 11.5 (H) | 5.2 | 14 (H) | 7.7 | 1.9 | 6.1 | 3.1 | 2.3 | 1.5-10.3 |
| CD8+CD62L−CD45RA−, % | n.d. | 4.7 | 7.2 | n.d. | 3.2 | 3.1 | 5.4 | 5.2 | 2.6 | 2.3 | 3 | 6 | 3.8 | 1.1-9.2 |
| CD8+CD62L−CD45RA+, % | n.d. | 28.8 (H) | 8.7 (H) | n.d. | 19.2 (H) | 8.4 (H) | 19.3 (H) | 1 | 1.9 | 2.5 | 5.1 | 2.4 | 3 | 0.7-7.8 |
| CD4/CD8 ratio | 0.79 (L) | 0.26 (L) | 0.87 (L) | 2.42 | 0.59 (L) | 0.98 (L) | 0.93 (L) | 0.9 (L) | 1.73 | 2.22 | 1.36 | 2.86 | 1.3 | 1.11-5.17 |
| CD4+CD45RA+CD31+, % | n.d. | n.d. | 8.6 | n.d. | 2.1 | n.d. | 4.8 | n.d. | n.d. | 13.9 | 4.6 | 9.5 | 18.6 | 1.6-20.2 |
| CD4+CD45RA−CXCR5+, % | n.d. | n.d. | 3.4 | n.d. | 5 | n.d. | 7.3 | n.d. | n.d. | 7.6 | 8.5 | 3.2 | 2.9 | 1.8-8.9 |
| CD4+CD25+FOXP3+, % | n.d. | n.d. | 2 | n.d. | 2 | n.d. | 0.6 (L) | n.d. | n.d. | 1.6 | 0.9 (L) | 2.3 | 1.7 | 1.6-4.3 |
| CD20 B, ALC | 76 | 38 (L) | 35 (L) | 247 | 27 (L) | 121 | 92 | 118 | 41 (L) | 44 (L) | 217 | 154 | 351 (H) | 59-329 |
| CD20 B, % | 7.6 | 2.4 (L) | 3.1 | 17.7 | 1.8 (L) | 4.4 | 6.7 | 5.2 | 3.5 | 2.9 (L) | 10.8 | 5.7 | 7.4 | 3-19 |
| CD20+CD27−IgM+ % | n.d. | 78.8 | 84.5 | n.d. | 39.2 (L) | 84.2 | 91.2 | 84.8 | 98.7 (H) | 98.8 (H) | 89.3 | 53.3 (L) | 86.6 | 56.2-91.5 (% of B cells) |
| CD20+CD27+IgM+, % | n.d. | 12.0 | 12.5 | n.d. | 45.19 (H) | 4.3 (L) | 3.9 (L) | 5.7 | 0.5 (L) | 0.9 (L) | 6.9 | 29.9 | 7.9 | 4.8-38.5 (% of B cells) |
| CD20+CD27+IgM−, % | n.d. | 6.6 | 2 (L) | n.d. | 6.5 | 6.1 | 3.2 (L) | 6.2 | 0 (L) | 0.2 (L) | 2.7 (L) | 15.4 | 2.6 (L) | 4.3-19.9 (% of B cells) |
| CD19+CD24++CD38++, % | n.d. | 6.1 | 18.7 | n.d. | 0 (L) | 16.8 | 7.7 | 4.9 | 11.1 | 3.2 (L) | 10.6 | 4.2 (L) | 4.1 (L) | 4.7-18.7 (% of B cells) |
| CD19+CD24−CD38+, % | n.d. | 0.6 | 0.3 | n.d. | 0 (L) | 0.1 (L) | 0.1 (L) | 0.4 | 0 (L) | 0 (L) | 0.1 (L) | 1.5 | 0.7 | 0.3-3.6 (% of B cells) |
| NK, ALC | 106 (L) | 158 | 130 | 353 | 134 | 104 (L) | 85 (L) | 336 | 125 (L) | 95 (L) | 233 | 454 | 209 | 126-729 |
| NK, % | 10.61 | 10 | 11.5 | 25.2 | 8.9 | 3.8 (L) | 6.2 | 14.8 | 10.7 | 6.2 | 11.6 | 16.8 | 4.4 (L) | 6.2-34.6 |
| Immunoglobulins, mg/dL | ||||||||||||||
| IgG | n.d. | 516 (L) | 1001* | 672.7 (L) | 349 (L) | 1147 | n.d. | n.d. | 59.8 (L) | 984* | n.d. | n.d. | 740 | 700-1600 |
| IgA | n.d. | 36 (L) | 49 (L) | 90.5 | 28 (L) | 98 | n.d. | <10 (L) | 12 (L) | n.d. | n.d. | 112 | 70-400 | |
| IgM | n.d. | 15 (L) | 14 (L) | 54.3 | <7 (L) | <10 (L) | n.d. | n.d. | <10 (L) | 6 (L) | n.d. | n.d. | 98 | 40-230 |
| Autoantibodies | n.d. | n.d. | Anti-platelet Ab positive, Coombs negative | n.d. | n.d. | n.d. | n.d. | n.d. | n.d. | Coombs positive | n.d. | n.d. | ANA positive, anti-dsDNA negative, Coombs negative | |
| ANA positive | ||||||||||||||
| Vaccine response | n.d. | n.d. | Progressive loss of protective titers toward pneumococcal and HiB antigens | n.d. | Positive to 1/14 pneumococcal serotypes, nonprotective titers to tetanus toxoid, diphtheria toxoid, measles, mumps, rubella or varicella | Positive to 5/23 pnemococcal serotypes prevaccination and 8/23 postvaccination, tetanus protective | n.d. | n.d. | n.d. | Positive to 2/14 pnemococcal serotypes, protective to diphtheria and tetanus | n.d. | n.d. | n.d. | |
| Clinical history | Asymptomatic | 1 pneumonia (age 25 y), pernicious anemia | ITP, progressive hypogammaglobulinemia | Type 2 diabetes, hypothyroidism | T-ALL, recurrent diarrhea, hypogammaglobulinemia | Pneumonia (×2, starting in her 50s) | Asymptomatic | Asymptomatic | Recurrent URIs, hypogammaglobulinemia, fibromyalgia | ITP, AIHA, hypogammaglobulinemia s/p rituximab | Hashimoto’s | Asymptomatic | Burkitt lymphoma, lymphadenopathy, ITP, neutropenia |
| A.I.1 (g-mother) | A.II.1 (father) | A.III.1 (index pt) | B.I.1 (mother) | B.II.1 (index pt) | C.I.1 (g-mother) | C.II.1 (mother) | C.II.2 (aunt) | C.II.3 (aunt) | C.III.1 (index pt) | C.III.2 (sister) | D.I.1 (father) | D.II.1 (index pt) | Reference | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Mutation | p.R213* | p.R213* | p.R213* | p.S427* | p.S427* | p.C467R | p.C467R | p.C467R | p.C467R | p.C467R | p.C467R | p.R502L | p.R502L | % of total lymphocytes |
| Age, y; sex | 74; F | 48; M | 17; M | 74; F | 31; M | 66; F | 44; F | 36; F | 38; F | 18; F | 21; F | 50; M | 15; M | |
| Age at onset, y; diagnosis | 9; ITP | 2.5; T-ALL | 9; ITP | 9; Burkitt lymphoma | ||||||||||
| Lymphocyte phenotyping | ||||||||||||||
| CD3 T, ALC | 812 | 1384 | 970 | 797 | 1348 | 2510 (H) | 1196 | 1821 | 1004 | 1394 | 1556 | 2095 | 4181 (H) | 714-2266 |
| CD3 T, % | 81.2 | 87.6 (H) | 85.8 (H) | 57 (L) | 89.3 (H) | 91.6 (H) | 87.3 (H) | 80.2 | 85.8 (H) | 91.1 (H) | 77.4 | 77.6 | 88.2 (H) | 60-83.7 |
| CD4 T, ALC | 346 (L) | 272 (L) | 421 | 558 | 477 | 1121 | 523 | 742 | 580 | 915 | 824 | 1450 | 2176 (H) | 359-1565 |
| CD4 T, % | 34.7 | 17.2 (L) | 37.3 | 39.9 | 31.6 (L) | 40.9 | 38.2 | 32.7 | 49.6 | 59.8 | 41 | 53.7 | 45.9 | 31.9-62.2 |
| CD4+CD62L+CD45RA+, % | n.d. | 3.8 (L) | 11.6 | n.d. | 3.4 (L) | 3.2 (L) | 7.7 | 4.9 (L) | 14.2 | 20.4 | 8.5 | 18.2 | 25.6 | 7.6-37.7 |
| CD4+CD62L+CD45RA−, % | n.d. | 10.2 (L) | 21.5 | n.d. | 24.7 | 34 (H) | 23 | 25 | 31.3 (H) | 33.9 (H) | 25.4 | 22.8 | 15.1 | 10.4-30.7 |
| CD4+CD62L−CD45RA−, % | n.d. | 3.1 | 4 | n.d. | 3.5 | 3.7 | 6.8 | 2.7 | 4 | 5.2 | 6.6 | 11.7 | 4.6 | 2.3-15.6 |
| CD4+CD62L−CD45RA+, % | n.d. | 0.2 | 0.3 | n.d. | 0 | 0 | 0.6 | 0 | 0.1 | 0.3 | 0.5 | 1.1 | 0.6 | 0-1.5 |
| CD8 T, ALC | 441 | 1063 (H) | 483 | 230 | 808 | 1145 (H) | 564 | 826 | 335 | 412 | 607 | 508 | 1673 (H) | 178-853 |
| CD8, % | 44.1 (H) | 67.3 (H) | 42.7 (H) | 16.5 | 53.5 (H) | 41.8 (H) | 41.2 (H) | 36.4 (H) | 28.6 | 26.9 | 30.2 | 18.8 | 35.3 (H) | 11.2-34.8 |
| CD8+CD62L+CD45RA+, % | n.d. | 26.8 (H) | 20.7 (H) | n.d. | 22 (H) | 18.8 | 11.3 | 16.2 | 16.4 | 20.2 (H) | 16 | 7.2 | 26.2 (H) | 5.7-19.7 |
| CD8+CD62L+CD45RA−, % | n.d. | 6.9 | 6.1 | n.d. | 9.1 | 11.5 (H) | 5.2 | 14 (H) | 7.7 | 1.9 | 6.1 | 3.1 | 2.3 | 1.5-10.3 |
| CD8+CD62L−CD45RA−, % | n.d. | 4.7 | 7.2 | n.d. | 3.2 | 3.1 | 5.4 | 5.2 | 2.6 | 2.3 | 3 | 6 | 3.8 | 1.1-9.2 |
| CD8+CD62L−CD45RA+, % | n.d. | 28.8 (H) | 8.7 (H) | n.d. | 19.2 (H) | 8.4 (H) | 19.3 (H) | 1 | 1.9 | 2.5 | 5.1 | 2.4 | 3 | 0.7-7.8 |
| CD4/CD8 ratio | 0.79 (L) | 0.26 (L) | 0.87 (L) | 2.42 | 0.59 (L) | 0.98 (L) | 0.93 (L) | 0.9 (L) | 1.73 | 2.22 | 1.36 | 2.86 | 1.3 | 1.11-5.17 |
| CD4+CD45RA+CD31+, % | n.d. | n.d. | 8.6 | n.d. | 2.1 | n.d. | 4.8 | n.d. | n.d. | 13.9 | 4.6 | 9.5 | 18.6 | 1.6-20.2 |
| CD4+CD45RA−CXCR5+, % | n.d. | n.d. | 3.4 | n.d. | 5 | n.d. | 7.3 | n.d. | n.d. | 7.6 | 8.5 | 3.2 | 2.9 | 1.8-8.9 |
| CD4+CD25+FOXP3+, % | n.d. | n.d. | 2 | n.d. | 2 | n.d. | 0.6 (L) | n.d. | n.d. | 1.6 | 0.9 (L) | 2.3 | 1.7 | 1.6-4.3 |
| CD20 B, ALC | 76 | 38 (L) | 35 (L) | 247 | 27 (L) | 121 | 92 | 118 | 41 (L) | 44 (L) | 217 | 154 | 351 (H) | 59-329 |
| CD20 B, % | 7.6 | 2.4 (L) | 3.1 | 17.7 | 1.8 (L) | 4.4 | 6.7 | 5.2 | 3.5 | 2.9 (L) | 10.8 | 5.7 | 7.4 | 3-19 |
| CD20+CD27−IgM+ % | n.d. | 78.8 | 84.5 | n.d. | 39.2 (L) | 84.2 | 91.2 | 84.8 | 98.7 (H) | 98.8 (H) | 89.3 | 53.3 (L) | 86.6 | 56.2-91.5 (% of B cells) |
| CD20+CD27+IgM+, % | n.d. | 12.0 | 12.5 | n.d. | 45.19 (H) | 4.3 (L) | 3.9 (L) | 5.7 | 0.5 (L) | 0.9 (L) | 6.9 | 29.9 | 7.9 | 4.8-38.5 (% of B cells) |
| CD20+CD27+IgM−, % | n.d. | 6.6 | 2 (L) | n.d. | 6.5 | 6.1 | 3.2 (L) | 6.2 | 0 (L) | 0.2 (L) | 2.7 (L) | 15.4 | 2.6 (L) | 4.3-19.9 (% of B cells) |
| CD19+CD24++CD38++, % | n.d. | 6.1 | 18.7 | n.d. | 0 (L) | 16.8 | 7.7 | 4.9 | 11.1 | 3.2 (L) | 10.6 | 4.2 (L) | 4.1 (L) | 4.7-18.7 (% of B cells) |
| CD19+CD24−CD38+, % | n.d. | 0.6 | 0.3 | n.d. | 0 (L) | 0.1 (L) | 0.1 (L) | 0.4 | 0 (L) | 0 (L) | 0.1 (L) | 1.5 | 0.7 | 0.3-3.6 (% of B cells) |
| NK, ALC | 106 (L) | 158 | 130 | 353 | 134 | 104 (L) | 85 (L) | 336 | 125 (L) | 95 (L) | 233 | 454 | 209 | 126-729 |
| NK, % | 10.61 | 10 | 11.5 | 25.2 | 8.9 | 3.8 (L) | 6.2 | 14.8 | 10.7 | 6.2 | 11.6 | 16.8 | 4.4 (L) | 6.2-34.6 |
| Immunoglobulins, mg/dL | ||||||||||||||
| IgG | n.d. | 516 (L) | 1001* | 672.7 (L) | 349 (L) | 1147 | n.d. | n.d. | 59.8 (L) | 984* | n.d. | n.d. | 740 | 700-1600 |
| IgA | n.d. | 36 (L) | 49 (L) | 90.5 | 28 (L) | 98 | n.d. | <10 (L) | 12 (L) | n.d. | n.d. | 112 | 70-400 | |
| IgM | n.d. | 15 (L) | 14 (L) | 54.3 | <7 (L) | <10 (L) | n.d. | n.d. | <10 (L) | 6 (L) | n.d. | n.d. | 98 | 40-230 |
| Autoantibodies | n.d. | n.d. | Anti-platelet Ab positive, Coombs negative | n.d. | n.d. | n.d. | n.d. | n.d. | n.d. | Coombs positive | n.d. | n.d. | ANA positive, anti-dsDNA negative, Coombs negative | |
| ANA positive | ||||||||||||||
| Vaccine response | n.d. | n.d. | Progressive loss of protective titers toward pneumococcal and HiB antigens | n.d. | Positive to 1/14 pneumococcal serotypes, nonprotective titers to tetanus toxoid, diphtheria toxoid, measles, mumps, rubella or varicella | Positive to 5/23 pnemococcal serotypes prevaccination and 8/23 postvaccination, tetanus protective | n.d. | n.d. | n.d. | Positive to 2/14 pnemococcal serotypes, protective to diphtheria and tetanus | n.d. | n.d. | n.d. | |
| Clinical history | Asymptomatic | 1 pneumonia (age 25 y), pernicious anemia | ITP, progressive hypogammaglobulinemia | Type 2 diabetes, hypothyroidism | T-ALL, recurrent diarrhea, hypogammaglobulinemia | Pneumonia (×2, starting in her 50s) | Asymptomatic | Asymptomatic | Recurrent URIs, hypogammaglobulinemia, fibromyalgia | ITP, AIHA, hypogammaglobulinemia s/p rituximab | Hashimoto’s | Asymptomatic | Burkitt lymphoma, lymphadenopathy, ITP, neutropenia |
The B-cell subsets are expressed as the percentage of CD19+ cells. H and L indicate values above/below the normal range, respectively, compared with adult controls in National Institutes of Health Clinical Center.
ALC, absolute lymphocyte count; ANA, antinuclear antibody; dsDNA, double-stranded DNA; F, female; g-mother, grandmother; HiB, Haemophilus influenzae type B; IgA, immunoglobulin A; IgG, immunoglobulin G; IgM, immunoglobulin M; M, male; n.d., not determined; NK, natural killer (cells); pt, patient; URI, upper respiratory infection.
The patient was receiving IgG replacement therapy.
Family A (p.R213*)
Patient A.III.1 is a 17-year-old male who was healthy until 9 years of age when he was diagnosed with immune thrombocytopenia (ITP). Laboratory evaluation showed mild leukopenia, thrombocytopenia, and hypogammaglobulinemia. He did not have a history of recurrent or severe infections. Family history was unremarkable, with the exception of his father (A.II.1) reporting 1 episode of pneumonia in adulthood and pernicious anemia.
Family B (p.S427*)
Patient B.II.1 is a 31-year-old male who was healthy until 2.5 years of age when he was diagnosed and successfully treated for T-ALL. He remained asymptomatic until 27 years of age when he developed recurrent episodes of watery diarrhea. No severe or recurrent infections were reported. Immune evaluation at age 28 showed hypogammaglobulinemia and nonprotective titers to childhood vaccines. Family history was remarkable for his mother (B.I.1) reporting hypothyroidism and type 2 diabetes.
Family C (p.C467R)
Patient C.III.1 is a 18-year-old female who was healthy until 9 years of age when she was diagnosed with ITP. Laboratory studies before treatment with rituximab demonstrated slightly diminished immunoglobulin levels and a poor response to pneumococcal antigens. At age 13, she was diagnosed with Coombs-positive autoimmune hemolytic anemia (AIHA) and relapsing ITP. Family history was remarkable for her sister (C.III.2) reporting Hashimoto’s disease, her maternal aunt (C.II.3) with childhood-onset CVID, and her maternal grandmother (C.I.1) with adult-onset specific antibody deficiency.
Family D (p.R502L)
Patient D.II.1 is a 15-year-old male who was healthy until 9 years of age when he was diagnosed and successfully treated for a disseminated Burkitt lymphoma. At age 12 years, a nonmalignant 5- to 6-cm-diameter intra-abdominal lymph node was diagnosed. Soon after, he developed ITP and neutropenia. Family history was unremarkable.
Results
IKZF1 mutations, familial segregation, transcription, and protein expression
Four individual germline heterozygous mutations in IKZF1 (NM_006060) were identified by whole-exome sequencing in each family’s index patient (ie, c.637C>T (p.R213*) in A.III.1, c.1280C>A (p.S427*) in B.II.1, c.1399T>C (p.C467R) in C.III.1, and c.1505G>T (p.R502L) in D.II.1) (Figure 1A-B); results were confirmed by Sanger sequencing. The index patient’s mutation was detected in other relatives of all 4 families (A.I.1, A.II.1, B.I.1, C.I.1, C.II.1, C.II.2, C.II.3, C.III.2, and D.I.1) (Figure 1A). Amino acids C467 and R502 are conserved through evolution (Figure 1C). None of these variants are reported in the Genome Aggregation Database (supplemental Table 1).
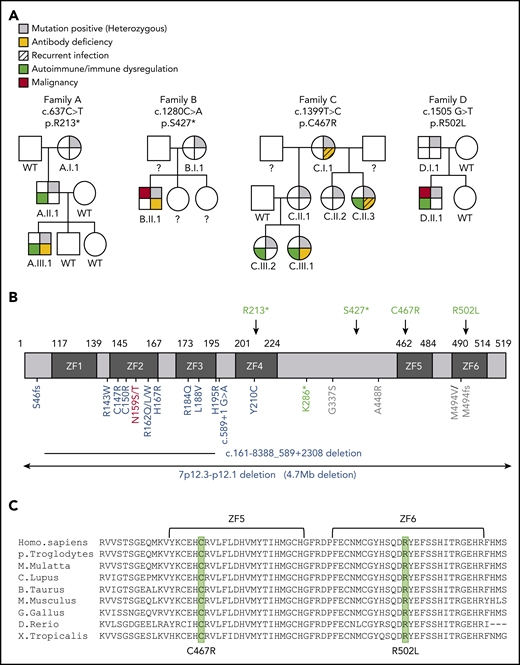
Genetics and pedigrees of families with IKZF1 mutations. (A) Segregation of the IKZF1 mutations and clinical and immunological phenotypes. Squares and circles indicate male and female family members, respectively. Question mark indicates an unscreened individual. (B) Schematic presentation of the structure of IKAROS isoform 1 (NM_006060). Dark gray indicates ZFs, and arrows indicate the site of the mutations. Numbers indicate amino acid location. Previously reported germline IKAROS mutations in CVID or CID patients are indicated below the domain. Blue color indicates haploinsufficient mutations (DNA binding–defective mutations and loss of 1 IKZF1 allele); DN mutations are shown in red; dimerization defective–mutations are shown in green; and gray color indicates the variants whose functions are unclear. (C) Sequence conservation of ZF5 and ZF6 in IKAROS. The mutated amino acid sites found in families C and D (C467 and R502, respectively) are enclosed in the green box.
Genetics and pedigrees of families with IKZF1 mutations. (A) Segregation of the IKZF1 mutations and clinical and immunological phenotypes. Squares and circles indicate male and female family members, respectively. Question mark indicates an unscreened individual. (B) Schematic presentation of the structure of IKAROS isoform 1 (NM_006060). Dark gray indicates ZFs, and arrows indicate the site of the mutations. Numbers indicate amino acid location. Previously reported germline IKAROS mutations in CVID or CID patients are indicated below the domain. Blue color indicates haploinsufficient mutations (DNA binding–defective mutations and loss of 1 IKZF1 allele); DN mutations are shown in red; dimerization defective–mutations are shown in green; and gray color indicates the variants whose functions are unclear. (C) Sequence conservation of ZF5 and ZF6 in IKAROS. The mutated amino acid sites found in families C and D (C467 and R502, respectively) are enclosed in the green box.
Mutant allele transcripts were detected in the index patients and mutation-positive family members tested (Figure 2A). IKAROS expression was evaluated in T and B cells by flow cytometry using a C terminus–directed antibody aimed to detect the WT and the missense forms, but not the C terminus–truncated form, of the protein. In this setting, index patients with missense mutations (C.III.1 and D.II.1) and HDs showed comparable IKAROS levels (Figure 2B). Conversely, patients with nonsense mutations (A.III.1 and B.II.1) demonstrated reduced IKAROS expression, because only 1 copy of the WT protein was expressed and detected (Figure 2B). When an N terminus–directed antibody used to detect the WT and both mutant forms of the protein was tested by western blot, truncated mutant proteins R213* and S427* were discriminated at their predicted molecular weights of 25 kilodalton (kDa) and 45 kDa, respectively (ie, <60-kDa molecular weight of WT [isoform 1] or missense mutated IKAROS), in the nuclear extracts of T-cell blasts of index patients A.III.1 and B.II.1 (Figure 2C). Because the protein expression level of truncated mutants R213* and S427* was lower than WT in T-cell blasts (overexposed blots shown), DD mutant protein stability was evaluated by a cycloheximide chase assay. Compared with each respective sample not treated with cycloheximide, WT IKAROS protein expression was reduced by 32%, whereas DD mutants were 51% to 89% degraded (R213*, 72%; S427*, 70%; C467R, 89%; R502L, 51%; Y462* [laboratory-generated vector lacking ZF5 and ZF6], 80%) (Figure 2D-E). Previously described HI mutant R162Q and DN mutant N159S showed similar degradation levels as WT IKAROS. These results show that, despite their in vitro reduced protein stability that may have contributed to the decreased amount of mutant protein in patients A.III.1 and B.II.1 (Figure 2C), DD mutants are indeed transcribed and expressed in primary and transfected cells.
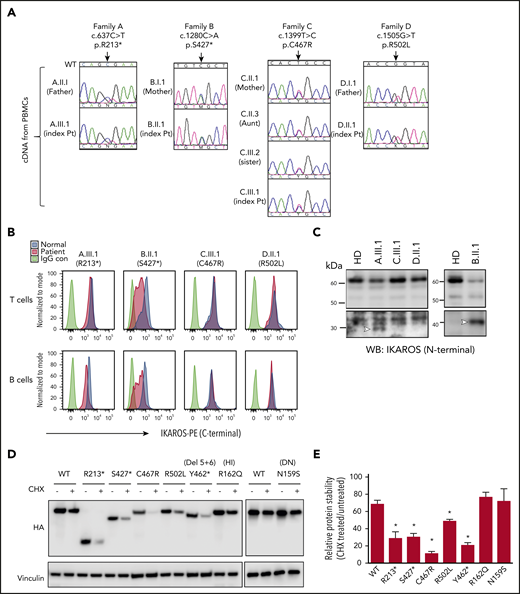
IKAROS transcripts and protein expression in patients. (A) The chromatograms show the sequence of the indicated heterozygous mutation in complementary DNA prepared from peripheral blood mononuclear cells (PBMCs). WT indicates WT of IKAROS’ reference sequence. Arrows indicate the sites of mutations. (B) IKAROS protein expression levels were tested in T and B cells from PBMCs from index patients and a paired HD control. (C) IKAROS expression in nuclear extracts of T-cell blasts. Triangles indicate R213* and S427* mutant proteins. The lower blots were overexposed to visualize the low levels of truncated IKAROS protein (R213* and S427*). Data are representative of 2 independent experiments. (D-E) HEK293T cells were transfected with a vector expressing IKAROS WT or mutant. After 24 hours, cells were treated with cycloheximide (CHX; 20 μg/mL) for an additional 24 hours. (D) Whole-cell lysates were prepared and analyzed by immunoblot analysis using hemagglutinin (HA) antibodies to test IKAROS expression. Vinculin was used as a loading control. (E) The relative IKAROS protein stability was calculated by dividing the CHX-treated sample by the untreated sample (×100) after normalization with vinculin to show the remaining protein amount after CHX treatment. Data are mean ± standard error of the mean of 3 or 4 independent experiments. Data (WT vs each mutant) were analyzed using GraphPad Prism software. *P < .05, unpaired Student t test.
IKAROS transcripts and protein expression in patients. (A) The chromatograms show the sequence of the indicated heterozygous mutation in complementary DNA prepared from peripheral blood mononuclear cells (PBMCs). WT indicates WT of IKAROS’ reference sequence. Arrows indicate the sites of mutations. (B) IKAROS protein expression levels were tested in T and B cells from PBMCs from index patients and a paired HD control. (C) IKAROS expression in nuclear extracts of T-cell blasts. Triangles indicate R213* and S427* mutant proteins. The lower blots were overexposed to visualize the low levels of truncated IKAROS protein (R213* and S427*). Data are representative of 2 independent experiments. (D-E) HEK293T cells were transfected with a vector expressing IKAROS WT or mutant. After 24 hours, cells were treated with cycloheximide (CHX; 20 μg/mL) for an additional 24 hours. (D) Whole-cell lysates were prepared and analyzed by immunoblot analysis using hemagglutinin (HA) antibodies to test IKAROS expression. Vinculin was used as a loading control. (E) The relative IKAROS protein stability was calculated by dividing the CHX-treated sample by the untreated sample (×100) after normalization with vinculin to show the remaining protein amount after CHX treatment. Data are mean ± standard error of the mean of 3 or 4 independent experiments. Data (WT vs each mutant) were analyzed using GraphPad Prism software. *P < .05, unpaired Student t test.
IKAROS homo- and heterodimerization
All 4 DD mutations are predicted to affect ZF5 and/or ZF6, which are essential for homo- or heterodimerization with IKAROS and its family members. Therefore, we examined the ability of the mutant proteins to homodimerize with WT IKAROS and heterodimerize with IKAROS family members AIOLOS and HELIOS. Mutants R213*, S427*, and C467R completely failed to bind WT IKAROS, whereas mutant R502L showed partial binding (∼25% compared with WT/WT binding). Truncated IKAROS mutant proteins R213* and S427* completely failed to bind WT AIOLOS or WT HELIOS, minimal binding (<5%) was detected with the C467R mutant protein, and ∼60% binding was observed with the R502L mutant protein (Figure 3). These results demonstrate that all 4 IKAROS DD mutations disrupt homo- and heterodimerization, either completely (R213*, S427*, and C467R) or partially (R502L).
![Impaired interaction of mutant protein with the IKAROS family. (A-C) 293T cells were transfected with Flag-tagged WT IKAROS, WT AIOLOS, WT HELIOS, or hemagglutinin (HA)-tagged WT IKAROS or mutants (R213*, S427*, C467R, R502L, Y462* [a laboratory-generated mutant with deletion of ZF5 and ZF6]). Immunoprecipitation (IP) was performed using anti-Flag antibodies. Western blot analysis of the IP samples with anti-HA and anti-Flag or AIOLOS or HELIOS antibodies is shown. Five percent of the total volumes of the whole-cellular lysates used for each IP reaction were loaded as input controls. Data are representative of 3 independent experiments. Densitometry analysis of immunoprecipitated HA-IKAROS blots was performed using ImageStudio. EV, empty vector control.](https://ash.silverchair-cdn.com/ash/content_public/journal/blood/137/3/10.1182_blood.2020007292/1/m_bloodbld2020007292f3.png?Expires=1769084592&Signature=GwyUngzu2mhWY8j5VAZjWKcja1kKnzLGLj~nYwWh8STAqze7tOaj5OTPiHF6fEvLMHwRgE5SKviMnfwzwJN3NrA1XpBC78K2zc5tjbVUMvMNb9IivEsqvxrGoSHUUZeMO5DIbKSdQQbjgnYA73cKETQEWGgE~byHSvisqdVoXKiJhKQ0oqJQf2vZV~U6DkgD~OETdOQ7ltyTNkd0D2M-1zf6jcHtPpGEH8TJ1x2aELzOup2fL8Sb7SR28Zvm0U6VezxaV7TsDfFiO6d~NOPY2Ul5qnfy2GFP16vQzGNpoOp0~1D1zARNvFar~tXkcc5OTS8lTnpTarR1RbYxQIP~Mw__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
Impaired interaction of mutant protein with the IKAROS family. (A-C) 293T cells were transfected with Flag-tagged WT IKAROS, WT AIOLOS, WT HELIOS, or hemagglutinin (HA)-tagged WT IKAROS or mutants (R213*, S427*, C467R, R502L, Y462* [a laboratory-generated mutant with deletion of ZF5 and ZF6]). Immunoprecipitation (IP) was performed using anti-Flag antibodies. Western blot analysis of the IP samples with anti-HA and anti-Flag or AIOLOS or HELIOS antibodies is shown. Five percent of the total volumes of the whole-cellular lysates used for each IP reaction were loaded as input controls. Data are representative of 3 independent experiments. Densitometry analysis of immunoprecipitated HA-IKAROS blots was performed using ImageStudio. EV, empty vector control.
Impaired interaction of mutant protein with the IKAROS family. (A-C) 293T cells were transfected with Flag-tagged WT IKAROS, WT AIOLOS, WT HELIOS, or hemagglutinin (HA)-tagged WT IKAROS or mutants (R213*, S427*, C467R, R502L, Y462* [a laboratory-generated mutant with deletion of ZF5 and ZF6]). Immunoprecipitation (IP) was performed using anti-Flag antibodies. Western blot analysis of the IP samples with anti-HA and anti-Flag or AIOLOS or HELIOS antibodies is shown. Five percent of the total volumes of the whole-cellular lysates used for each IP reaction were loaded as input controls. Data are representative of 3 independent experiments. Densitometry analysis of immunoprecipitated HA-IKAROS blots was performed using ImageStudio. EV, empty vector control.
DNA binding and pericentromeric heterochromatin localization
WT or mutant IKAROS proteins were expressed in NIH3T3 cells, which are known to lack endogenous expression of IKAROS or its family members, for the evaluation of pericentric heterochromatin (PC-HC) localization.9,24,25 WT IKAROS exhibited the characteristic punctate staining pattern of PC-HC localization. Complete DD mutants R213*, S427*, Y462*, and C467R exhibited a diffuse distribution in the nucleus without PC-HC localization; partial DD mutant R502L showed a punctate staining pattern similar to that of WT (Figure 4A). These data suggests that partially, but not completely, defective dimerization can support normal PC-HC DNA binding. Coexpressing mutant proteins with WT IKAROS to mimic heterozygosity did not affect WT IKAROS PC-HC targeting, ruling out a DN effect. Likewise, known IKAROS HI mutant R162Q did not have a DN effect; however, previously reported DN mutant N159S did affect WT IKAROS PC-HC targeting (Figure 4B).
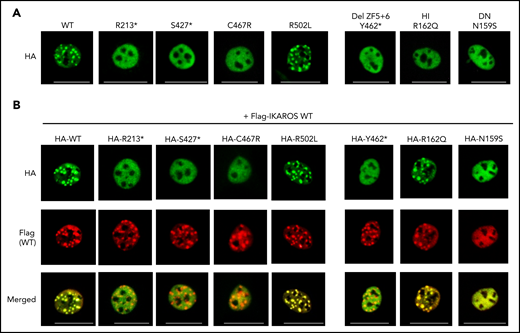
Pericentromeric targeting of the mutant IKAROS. NIH3T3 cells were transfected with a hemagglutinin (HA)-tagged WT or mutant expression vector alone (A) or together (B) with Flag-tagged WT IKAROS. Cells were labeled with anti-HA or together with anti-Flag antibodies, followed by Alexa Fluor 488–conjugated (green) and Alexa Fluor 568–conjugated (red) secondary antibodies. Cells were visualized using a ZOE fluorescence microscope (original magnification, ×175). Data are representative of 3 independent experiments. HI and DN indicate DNA binding–defective haploinsufficient mutation and DN mutation, respectively. Scale bars, 25 μm.
Pericentromeric targeting of the mutant IKAROS. NIH3T3 cells were transfected with a hemagglutinin (HA)-tagged WT or mutant expression vector alone (A) or together (B) with Flag-tagged WT IKAROS. Cells were labeled with anti-HA or together with anti-Flag antibodies, followed by Alexa Fluor 488–conjugated (green) and Alexa Fluor 568–conjugated (red) secondary antibodies. Cells were visualized using a ZOE fluorescence microscope (original magnification, ×175). Data are representative of 3 independent experiments. HI and DN indicate DNA binding–defective haploinsufficient mutation and DN mutation, respectively. Scale bars, 25 μm.
EMSAs were performed to evaluate IKAROS’ ability to specifically bind its DNA target sequences. WT IKAROS recognized DNA target baits and bound as monomers, dimers, and multimers that were discriminated based on their molecular weights. Mutants R213*, S427*, and Y462* showed strong monomer, but no dimer or multimer, binding, suggesting that failed IKAROS homodimerization still allows monomer protein-target binding (Figure 5A). Mutant C467R showed overall reduced binding to all DNA/IKAROS-containing complexes, and mutant R502L showed low to normal binding as a monomer and weak binding as dimers and multimers (Figure 5A). Previously reported HI and DN mutants showed the complete absence of DNA binding as monomers, dimers, or multimers. Coexpression of DD mutants with WT IKAROS did not demonstrate any effect on its DNA binding to target probes, ruling out a DN effect (Figure 5B).
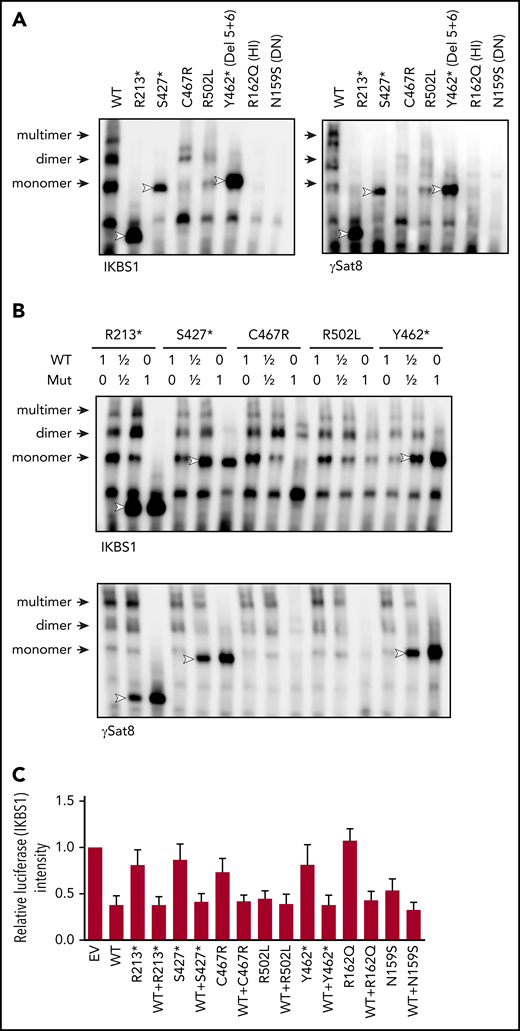
DNA binding and transcription activity of the mutant IKAROS protein. (A-B) EMSAs were performed using nuclear extracts from HEK293T cells transfected with the indicated IKAROS mutation alone (A) or together with WT IKAROS (B). Numbers indicate the ratio of WT and mutant IKAROS DNA used for the cotransfection. The nuclear extracts were allowed to bind to 2 IKAROS probes: IK-bs1, an IKAROS consensus binding sequence, and γ-Sat 8, a sequence from the pericentromeric region of human chromosome 8. IKAROS-containing complexes are indicated with arrows. Triangles indicate mutant IKAROS protein binding to the DNA as a monomer. Data are representative of 3 independent experiments. (C) Four repeats of IKBS1 were inserted into the pGL4.11 vector (pGL4.11-IKBS1) and cotransfected with pcDNA3-HA IKAROS WT or mutants and pRL-TK (Renilla luciferase) as indicated. Twenty hours later, cells were lysed, and luciferase activity was measured using the Dual-Luciferase Reporter Assay System. Firefly luciferase activity was normalized to Renilla luciferase activity, and the results were normalized to the empty vector (EV) control. Each experiment was carried out in duplicate. Data are mean ± standard error of the mean from 3 independent experiments.
DNA binding and transcription activity of the mutant IKAROS protein. (A-B) EMSAs were performed using nuclear extracts from HEK293T cells transfected with the indicated IKAROS mutation alone (A) or together with WT IKAROS (B). Numbers indicate the ratio of WT and mutant IKAROS DNA used for the cotransfection. The nuclear extracts were allowed to bind to 2 IKAROS probes: IK-bs1, an IKAROS consensus binding sequence, and γ-Sat 8, a sequence from the pericentromeric region of human chromosome 8. IKAROS-containing complexes are indicated with arrows. Triangles indicate mutant IKAROS protein binding to the DNA as a monomer. Data are representative of 3 independent experiments. (C) Four repeats of IKBS1 were inserted into the pGL4.11 vector (pGL4.11-IKBS1) and cotransfected with pcDNA3-HA IKAROS WT or mutants and pRL-TK (Renilla luciferase) as indicated. Twenty hours later, cells were lysed, and luciferase activity was measured using the Dual-Luciferase Reporter Assay System. Firefly luciferase activity was normalized to Renilla luciferase activity, and the results were normalized to the empty vector (EV) control. Each experiment was carried out in duplicate. Data are mean ± standard error of the mean from 3 independent experiments.
Gene transcription regulation by IKAROS DD mutations was assessed by a luciferase assay (vector carrying the IKAROS consensus sequence IKBS1). Ectopic expression of WT IKAROS repressed ∼60% of basal transcriptional activity (Figure 5C). Complete dimerization-defective mutants R213*, S427*, Y462*, and C467R minimally repressed the basal transcriptional activity; partial DD mutant R502L repressed levels similar to WT IKAROS. When WT and DD IKAROS mutants were coexpressed, luciferase activity was repressed to the levels of WT alone, suggesting no DN effect by the DD mutants. Of note, DNA binding–defective HI mutant R162Q completely failed to repress the basal transcriptional activity, but DN mutant N159S repressed the transcriptional activity by 46%, despite its inability to bind to its specific DNA binding sites, highlighting the intrinsic limitations of this type of study (Figure 5A,C).
Altogether, because complete DD mutants R213*, S427*, and C467R have lost PC-HC localization (Figure 4A), binding their target sequence as monomers seems insufficient to localize to PC-HC or repress transcription, suggesting that binding as dimers and/or multimers is required for the pericentromeric localization and transcription regulation.
Posttranslational modification and transcriptional regulation
Sumoylation, a posttranslational process that involves the conjugation of small ubiquitin-like modifiers (SUMOs; 12 kDa), is a major regulator of protein functions, including cell cycling, cell survival, DNA repair, protein stability, nuclear transport, and transcription.26 IKAROS harbors 4 SUMO acceptor sites (K58, K240, K425, K459), and its sumoylation disrupts the interaction between IKAROS and corepressors of the NuRD complex.27 We detected multiple shifted bands of single and multi-sumoylated IKAROS WT protein (Figure 6A). Complete DD mutants (R213*, S427*, C467R, and Y462*) showed markedly reduced sumoylated proteins (Figure 6A), either by missed sumoylation sites (eg, R213*, S427*) and/or likely structural/functional defects associated with each particular DD mutation (eg, C467R, Y462*). Partial DD mutant R502L and DNA binding–defective mutants HI R162Q and DN N159S did not affect sumoylation. These data suggest that IKAROS dimerization (fully or partially preserved) is necessary for IKAROS sumoylation.
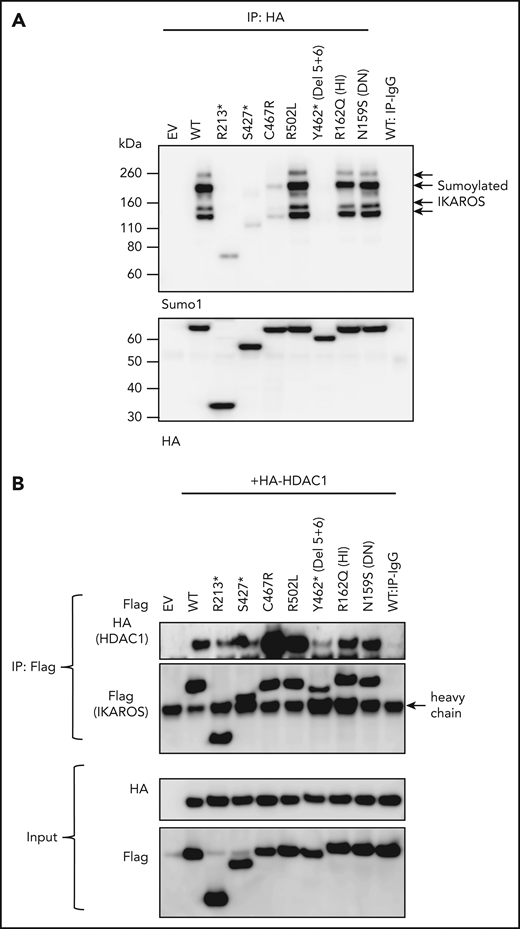
Sumoylation and HDAC1 binding of IKAROS WT and mutant proteins. (A) HEK293T cells were cotransfected with GFP-sumo1 and hemagglutinin (HA)-tagged IKAROS WT or mutant. Protein extracts were immunoprecipitated with anti-HA antibodies and probed with anti-Sumo1 antibodies to see the sumoylation, as well as with anti-HA antibodies to see the immunoprecipitated IKAROS. Arrows near 100 to 200 kDa indicate single or multiple sumoylated IKAROS protein. (B) HEK293T cells were cotransfected with Flag-tagged IKAROS WT or mutant and HA-tagged HDAC1. Protein lysates were immunoprecipitated with antibodies to the Flag epitope. Western blot analysis of the immunoprecipitation (IP) samples with anti-HA (for HDAC1) and anti-Flag (for IKAROS) antibodies is shown. Five percent of the total volume of the whole-cellular lysates used for IP reactions was loaded as input controls. Data shown are representative of 3 independent experiments.
Sumoylation and HDAC1 binding of IKAROS WT and mutant proteins. (A) HEK293T cells were cotransfected with GFP-sumo1 and hemagglutinin (HA)-tagged IKAROS WT or mutant. Protein extracts were immunoprecipitated with anti-HA antibodies and probed with anti-Sumo1 antibodies to see the sumoylation, as well as with anti-HA antibodies to see the immunoprecipitated IKAROS. Arrows near 100 to 200 kDa indicate single or multiple sumoylated IKAROS protein. (B) HEK293T cells were cotransfected with Flag-tagged IKAROS WT or mutant and HA-tagged HDAC1. Protein lysates were immunoprecipitated with antibodies to the Flag epitope. Western blot analysis of the immunoprecipitation (IP) samples with anti-HA (for HDAC1) and anti-Flag (for IKAROS) antibodies is shown. Five percent of the total volume of the whole-cellular lysates used for IP reactions was loaded as input controls. Data shown are representative of 3 independent experiments.
The N- and C-terminal IKAROS regions are associated with the NuRD complex, regulating the repression of transcriptional activity via HDAC-dependent or -independent mechanisms. IKAROS sumoylation also affects its interaction with the NuRD complex.27 Mutants lacking the dimerization domain (R213*, S427*, Y462*) showed markedly decreased interaction with HDAC1 (a core subunit of the NuRD complex); missense mutants (C467R, R502L) showed markedly increased HDAC1 binding (Figure 6B). No HDAC1 binding defect was detected with HI mutant R162Q or DN mutant N159S. When we tested the interaction between IKAROS and the C-terminal binding protein, an HDAC-independent corepressor known to associate with IKAROS through a PEDLS motif located at the IKAROS N terminus domain,28 truncated and missense IKAROS DD mutants showed similar levels of binding (supplemental Figure 1). This result suggests that IKAROS-HDAC1 interaction is likely independent of transcriptional regulators binding to the N terminus of IKAROS, such as C-terminal binding protein, and is primarily dependent on the structural/functional integrity of the IKAROS C-terminal region. These results also suggest that dysregulated HDAC1 recruitment by DD mutants is unlikely to be dependent upon PC-HC localization or sumoylation: mutants C467R and R502L, presenting opposite effects on these functions, showed similarly increased HDAC1 binding. Different structural/functional defects associated with each particular DD mutation likely determine their dysregulated HDAC1 binding through the C terminus region of the protein.
The effect of the DD variants on gene transcription was evaluated by RNASeq analysis on T-cell blasts and compared with HDs and patients with DNA binding defective HI (H167R) and DN (N159S) allelic variants (1 of each). Correlation analysis with unsupervised clustering showed that gene expression signatures in all allelic variants clustered together and separated from HDs (Figure 7A). An allelic variant–focused evaluation confirmed that compared with HDs, each mutation type was associated with a particular set of updysregulated and downdysregulated genes: 42 in DD patients (12 upregulated, 30 downregulated), 63 in the DNA binding HI patients (19 upregulated, 44 downregulated), and 534 in the DNA binding DN patients (315 upregulated, 219 downregulated). A more discrete number of genes was commonly dysregulated among all allelic variants (Figure 7B-C). Ingenuity pathway analysis (IPA) showed that the functions related to the development of malignant tumor and differentiation of leukemic cell lines were activated in DD patient cells, whereas functions associated with infection, inflammatory response, T-cell homeostasis, development, and differentiation were not (Figure 7D; supplemental Figure 2). Interestingly, although the malignant tumor–related pathway is largely upregulated in DD patients, fewer canonical pathways were affected compared with HI and DN patients, suggesting that the different regulation of genes, at least in part, is associated with different clinical and immunological phenotypes in the 3 groups of IKAROS allelic variants.
![Correlation analysis of targeted RNASeq data on T-cell blasts from HD controls (n = 5) and patients with DD, HI, or DN mutations. (A) RNASeq was performed using RNA extracted from enriched blasted T cells. Scale bar indicates Pearson correlation coefficient for variance stabilized data for differentially expressed messenger RNA. (B) Venn diagram analysis comparing lists of differentially expressed genes for each IKAROS study group (ie, DD [n = 4], HI (n = 1), DN (n = 1)] vs HD controls (n = 5). (C) Table of differentially expressed genes for the DD study group only as well as differentially expressed genes that are common across groups. Genes overexpressed compared with HD controls are in red text; genes underexpressed compared with HD controls are in blue text. (D) IPA Diseases and Functions Analysis heat map showing a comparison of the predicted effected cellular processes and biological functions based on gene expression. The activation z score represents the bias in gene regulation that predicts whether the biological function exists in an activated (orange) or inactivated (blue) state.](https://ash.silverchair-cdn.com/ash/content_public/journal/blood/137/3/10.1182_blood.2020007292/1/m_bloodbld2020007292f7.png?Expires=1769084592&Signature=lnfOwKIaPqc8-oaoKHqkRs2S8WnAjs-VxSQl26Aaj7edQViZjrellr2FluxXjOqU~U-RTqpFDb26pwuO9HP7oXuR1dmZOqcvbr9VC3syRER2XPj95gI0OqT-6jZnvA0bajBwKWOO1zvnhlI1Ozcvp~Cww953k0c21IkIzZFBZan80veyHRb5I-eVGJa3eLqu8UVrR1WH2LdPXIpskCZ8Aim1vJQqoaZNkDI6qqUT13YZ6pEWdJzCLtnmAxX~VW35uC3eZEflzYYfswAcjB9oU~Jn51igc2nSu3bk4xWAnkfLWoUpLq6eY1AekmVm74DHR~Lw5Bjm~iRAJv-cINyVPw__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
Correlation analysis of targeted RNASeq data on T-cell blasts from HD controls (n = 5) and patients with DD, HI, or DN mutations. (A) RNASeq was performed using RNA extracted from enriched blasted T cells. Scale bar indicates Pearson correlation coefficient for variance stabilized data for differentially expressed messenger RNA. (B) Venn diagram analysis comparing lists of differentially expressed genes for each IKAROS study group (ie, DD [n = 4], HI (n = 1), DN (n = 1)] vs HD controls (n = 5). (C) Table of differentially expressed genes for the DD study group only as well as differentially expressed genes that are common across groups. Genes overexpressed compared with HD controls are in red text; genes underexpressed compared with HD controls are in blue text. (D) IPA Diseases and Functions Analysis heat map showing a comparison of the predicted effected cellular processes and biological functions based on gene expression. The activation z score represents the bias in gene regulation that predicts whether the biological function exists in an activated (orange) or inactivated (blue) state.
Correlation analysis of targeted RNASeq data on T-cell blasts from HD controls (n = 5) and patients with DD, HI, or DN mutations. (A) RNASeq was performed using RNA extracted from enriched blasted T cells. Scale bar indicates Pearson correlation coefficient for variance stabilized data for differentially expressed messenger RNA. (B) Venn diagram analysis comparing lists of differentially expressed genes for each IKAROS study group (ie, DD [n = 4], HI (n = 1), DN (n = 1)] vs HD controls (n = 5). (C) Table of differentially expressed genes for the DD study group only as well as differentially expressed genes that are common across groups. Genes overexpressed compared with HD controls are in red text; genes underexpressed compared with HD controls are in blue text. (D) IPA Diseases and Functions Analysis heat map showing a comparison of the predicted effected cellular processes and biological functions based on gene expression. The activation z score represents the bias in gene regulation that predicts whether the biological function exists in an activated (orange) or inactivated (blue) state.
To evaluate IKAROS-specific NuRD- and Sin3-related gene transcription, a polymerase chain reaction gene array methodological approach was tested in patients for all known IKAROS allelic variants. Patients carrying DD (complete and partial), HI, and DN DNA binding–defective variants could be discriminated upon analysis. Among the NuRD- and Sin3-related gene transcripts, genes in which mutations have been described to be associated with T-ALL and Burkitt lymphoma in the COSMIC database (ie, CREBBP, NCOR1, NCOR2) were dysregulated in DD patients. Moreover, known cancer-associated genes (eg, CHD, MTA, NCOR, HDAC)29-32 and genes associated with immune dysregulation or autoimmune diseases (ie, ARID4A, histone H4 [HIST4H4, HIST1H4I], MTA2)33-35 were also dysregulated in DD patients (supplemental Figure 3). Taken together, these results suggest that DD mutations dysregulate IKAROS function and transcription through a mechanism different from that of DNA binding–defective mutations, and a subset of the dysregulated genes is closely associated with the hematologic manifestations presented by DD patients.
Baseline lymphocyte phenotype and immune functions were evaluated in DD patients (Table 1), and results were compared with previously reported allelic variants. The overall B-cell and T-cell immunophenotype was closer to DNA binding–defective HI patients (ie, progressive B-cell lymphopenia, elevated CD8+ T cells) than to DN patients (ie, early absence of B cells, severe T-cell maturation and functional defects).16,23,36 However, distinctive immunophenotypic features could also be identified between DD and HI allelic variants. Dimerization defects displayed a more limited impact on B cells than did the other allelic variants, because B cells were ≤2% in 1 of 13 (7.6%) DD patients compared with 16 of 26 (61.5%) HI patients all 7 (100%) DN patients.16,23 Moreover, CD8+ T cells were increased in 14 of 26 (54%) HI patients, but this feature was less prevalent in the DD (3/13; 23%) or DN (2/7; 29%) cases studied. Furthermore, although patients carrying DN IKAROS mutations had defects in memory T-cell differentiation and impaired T-cell activation and proliferation, all 4 DD index patients had normal percentages of memory T cells and normal T-cell proliferation (Table 1; supplemental Figure 4). When B-cell function was tested in the 4 index patients, B-cell receptor– or CD40L-induced B-cell proliferation was comparable to the normal controls in 3 patients (A.III.1, C.III.1, D.II.1) and slightly/moderately decreased in 1 patient (B.II.1). To further evaluate B-cell functions, we tested immunoglobulin secretion from naive B cells and total peripheral blood mononuclear cells (PBMCs). Although markedly decreased immunoglobulin G (IgG) generation was observed in naive B cells and total PBMCs from index patients B.II.1 and C.III.1 (A.III.1 was not tested because of limited sample availability), slightly low and normal levels of IgG secretion were detected in patient D.II.1’s naive B cells and PBMCs, respectively (supplemental Figure 5). This result correlates with the serum immunoglobulin levels in index patients (B.II.1, C.III.1, and D.II.1) and explains the milder impact of B-cell function in family D. Interestingly, variable results were observed among family members carrying the same mutations, reinforcing the variable penetrant and expressivity impact of IKZF1 DD mutants.
Discussion
We report 13 individuals in 4 unrelated families carrying 4 novel germline heterozygous IKZF1 mutations primarily affecting dimerization. We demonstrated that DD mutants were completely or partially incapable of binding to WT IKAROS, AIOLOS, and HELIOS. In addition to the dimerization defect, several other downstream functions were affected. These mutant proteins could not bind to their consensus sequences in target DNA as dimers or multimers, but 2 of them (ie, R213* and S427*) were still able to bind as monomers, and all but mutant R502L failed to localize to PC-HC, where IKAROS recruits its target genes for transcription regulation. Of note, the 4 mutants showed different degrees of impact, depending on the function evaluated; the nonsense mutations were the most deleterious, and R502L was the least deleterious, based on the experiments performed. Using a structural/protein modeling analysis, it is predicted that the polar basic to nonpolar missense change associated with mutation R502L within the α helix of the IKAROS C2H2-6 domain may abolish cation-π interactions, thereby destabilizing the IKAROS C2H2 α helix and disrupting the homodimerization interface, as the other DD mutants also do (data not shown). From a genotype/phenotype perspective, the R502L dichotomy between reduced biologic impact and a still aggressive clinical presentation is likely a matter of functional threshold (or gene dosage) associated with the intrinsic biology of DD HI. The performance of R502L, as seen with the other DD mutants, falls under the HI range of activity for multiple IKAROS functions. Once this threshold is surpassed (by a little or a lot), the function seems compromised and results in a biologic and, in turn, clinical phenotype. More difficult to define is the exact HI range or threshold for each IKAROS function, particularly considering that this transcription factor includes ≥17 isoforms acting by homo- or heterodimerization with other family members. Nonetheless, mutant protein R502L was still able to generate a robust biologic and clinical impact, despite a reduced functional effect of PC-HC binding and sumoylation. The R502L mutant, associated with hematologic cytopenias and malignancy, exhibited reduced IKAROS homo- or heterodimerization (∼25% and ∼60% of WT, respectively) and decreased protein stability, affected IKAROS dimer/multimer binding to DNA consensus sequences, and dysregulated general and NuRD/Sin3-related specific gene transcription and HDAC1 binding.
In fact, all DD mutants altered all these functions, highlighting their common underlying pathophysiologic mechanisms. Moreover, these features also distinguish them from the HI and DN allelic variants that share defective DNA binding but do not display any of the DD-related defects mentioned above (Table 2).
Summary of various effects of germline heterozygous IKZF1 mutations
| Functional test | WT | R213* (DD) | S427* (DD) | C467R (DD) | R502L (DD) | Y462* (del ZF5+6) | R162Q (HI) | N159S (DN) |
|---|---|---|---|---|---|---|---|---|
| Dimerization | +++ | − | − | − | + | − | +++ | +++ |
| PC-HC (WT or mutant alone) | +++ | − | − | − | +++ | − | − | − |
| PC-HC (coexpression with WT) | ||||||||
| Mut-Mut | NA | − | − | − | +++ | − | − | − |
| WT-Mut | NA | − | − | − | +++ | − | ++ | − |
| WT-WT | +++ | ++ | ++ | ++ | +++ | ++ | ++ | − |
| DNA binding | ||||||||
| Monomer | +/++ | +++ | +++ | + | ++ | +++ | − | − |
| Multimer | +++ | − | − | + | + | − | − | − |
| Sumoylation | +++ | + | + | + | +++ | + | +++ | +++ |
| HDAC1 binding | + | +/− | +/− | +++ | ++ | +/− | + | + |
| Protein stability (CHX treated) | +++ | + | + | + | ++ | + | +++ | +++ |
| T/B/myeloid abnormalities | NA | T−/B++/M− | NA | T−/B+++/M− | T+++/B+++/M+++ | |||
| Infections | NA | −/+ | NA | +++ | +++ | |||
| Autoimmune cytopenias | NA | ++/+++ | NA | + | − | |||
| Hematologic malignancies | NA | ++/+++ | NA | + | +/++ | |||
| Functional test | WT | R213* (DD) | S427* (DD) | C467R (DD) | R502L (DD) | Y462* (del ZF5+6) | R162Q (HI) | N159S (DN) |
|---|---|---|---|---|---|---|---|---|
| Dimerization | +++ | − | − | − | + | − | +++ | +++ |
| PC-HC (WT or mutant alone) | +++ | − | − | − | +++ | − | − | − |
| PC-HC (coexpression with WT) | ||||||||
| Mut-Mut | NA | − | − | − | +++ | − | − | − |
| WT-Mut | NA | − | − | − | +++ | − | ++ | − |
| WT-WT | +++ | ++ | ++ | ++ | +++ | ++ | ++ | − |
| DNA binding | ||||||||
| Monomer | +/++ | +++ | +++ | + | ++ | +++ | − | − |
| Multimer | +++ | − | − | + | + | − | − | − |
| Sumoylation | +++ | + | + | + | +++ | + | +++ | +++ |
| HDAC1 binding | + | +/− | +/− | +++ | ++ | +/− | + | + |
| Protein stability (CHX treated) | +++ | + | + | + | ++ | + | +++ | +++ |
| T/B/myeloid abnormalities | NA | T−/B++/M− | NA | T−/B+++/M− | T+++/B+++/M+++ | |||
| Infections | NA | −/+ | NA | +++ | +++ | |||
| Autoimmune cytopenias | NA | ++/+++ | NA | + | − | |||
| Hematologic malignancies | NA | ++/+++ | NA | + | +/++ | |||
All mutations, with the exception of Y462* (a laboratory-generated mutant to test the effect of deletion of ZF5 and ZF6), were found in patients. R162Q and N159S are previously reported DNA binding–defective IKZF1 mutations acting by HI or DN effect, respectively. +/++/+++ indicates the strength of each IKAROS function test; +++ is the strongest; and − is not detected (all vs WT).
CHX, cycloheximide; NA, not applicable.
Posttranslational modification of transcription factors has been proposed to modulate activation or repression of numerous genes during lymphocyte development.27,37 IKAROS is required to facilitate chromosome accessibility on target genes, where it recruits the NuRD complex for gene-expression regulation. Sumoylation, a posttranslational change affecting IKAROS, has been shown to interfere with the recruitment of gene transcription corepressors.27 Impaired DNA binding of IKAROS is associated with release of the NuRD complex from IKAROS target genes. Released NuRD complexes cause lymphocyte maturation arrest, which promotes leukemogenesis.38 Our study shows that germline mutants that lack the IKAROS dimerization domain (R213*, S427*) result in a marked reduction in sumoylation and HDAC1 binding. These findings indicate that IKAROS’ N-terminal domain is involved in recruitment of HDAC1 but depends on the C-terminal domain to be functionally intact (Figure 6). In contrast, missense mutants that maintain the IKAROS dimerization domain but still demonstrate defective dimerization (C467R and R502L) show markedly increased binding to HDAC1, suggesting that the C-terminal domain can recruit more HDAC1 when not occupied by IKAROS family members. To address how dimerization defects and alteration of HDAC1 recruitment affect transcription activity, we tested luciferase activity using a pGL4.11 vector containing IKBS1 repeats. Although complete DD mutants R213*, S427*, C467R, and Y462* and HI mutant R162Q failed to repress the transcription activity, partial DD mutant R502L did not show any defect in repression of transcription activity. This shows that binding as polymers is more important for the transcription repression than is the alteration of NuRD/HDAC1 complex recruitment, which was increased by C467R and R502L but decreased by R213* and S427*. Based on our experience with this technique, we acknowledge that luciferase results using an extrachromosomal reporter plasmid containing a small part of the IKAROS binding region may not accurately reproduce long-distance interactions that may be essential for IKAROS function, chromatin assembly, or recruitment of NuRD complex. Therefore, the detailed mechanisms by which altered NuRD complex recruitment of DD mutants affect transcription repression need to be further studied for full elucidation.
When we assessed gene-level transcription by RNASeq, we found that all IKAROS allelic variants tested (complete and partial DD mutants and DNA binding–defective HI and DN mutants) shared an abnormal gene-expression pattern. However, comparing HD vs each IKAROS mutation type revealed that each allelic variant dysregulated a unique set of genes. IPA revealed that, although functions associated with malignant tumor development were largely dysregulated in DD and DN patients, functions related to T-cell development and differentiation were preferentially dysregulated in HI and DN patients. These results suggest a correlation between genotypes and clinical/immunological phenotypes, because hematologic malignancies were more prevalent in DD (2/13) and DN (1/8) patients compared with HI patients (3/59), whereas abnormal T-cell lineage differentiation and/or function was reported more frequently in DN (8/8) and HI (13/26) patients but was not that prevalent in DD mutation carriers (3/13).16,36 Moreover, we performed an IKAROS-associated NuRD- and Sin3-related gene transcription evaluation using a different technical approach to achieve a more in-depth assessment. This polymerase chain reaction gene array–based analysis further discriminated the mutation types at the transcriptomic level and linked the DD mutants to the patients’ clinical presentations (hematologic cytopenias due to immunedysregulation and malignancies) (Figure 7; supplemental Figure 3). Our data show that DD mutations affect the chromatin-remodeling complex of particular genes and modify the chromatin landscape, likely leading to oncogenesis and a break in immunological tolerance that is highly compatible with the clinical phenotype described in our patients.
In terms of clinical presentations, 4 of 13 mutation-positive individuals reported herein were asymptomatic (A.I.1, C.II.1, C.II.2, and D.I.1), and 1 (B.I.1) presented with hypothyroidism and type 2 diabetes, symptoms questionably related to the IKAROS defect. On the other hand, 2 presented with tissue-specific autoimmunity (ie, pernicious anemia in A.II.1 and Hashimoto’s disease in C.III.2), and 2 presented with recurrent infections associated with late-onset specific antibody deficiency (C.I.1) or early-onset CVID-like disease (C.II.3). Interestingly, the 4 index cases (A.III.1, B.II.1, C.III.1, and D.II.1) presented with hematologic cytopenias or malignancies but no, or very limited, history of infection. Manifestations in the index patients started during their first decade of life as T-ALL, Burkitt lymphoma, or cytopenias resembling Evans syndrome, a rare autoimmune disorder of AIHA, ITP, and/or autoimmune neutropenia. A recent study demonstrated that 49 of 80 pediatric patients with Evans syndrome had germline variants in primary immunodeficiency–causing genes.39 Although that study did not provide biologic validation for the variants found, 29 were linked to mutations that had been previously proven deleterious, whereas 20 were not. Among the latter, 2 patients with IKZF1 mutations and 1 with a IKZF2 mutation presented with Evans syndrome during childhood.39 Our study reinforces the genetic and clinical relationship between IKAROS and Evans syndrome as recently reported, as well as provides biologic validation of this genotype-phenotype correlation describing a novel dimerization-defective mechanism for IKAROS-associated diseases. Recently, other germline heterozygous mutations predicted to negatively affect IKAROS’ dimerization domain (K286*, D186fs, M306*, and C394*) have been reported.12,21 Although the intrinsic dimerization defect and mechanism of action were outside the scope of those studies, these mutants appeared to exhibit incomplete clinical penetrance and were primarily associated with hematologic manifestations (ie, B-ALL and different degrees of B-cell deficiency); recurrent infections likely occurred in only 1 of those cases.12,21
All germline heterozygous IKZF1 allelic variants reported to date (ie, HI, DN) and herein (ie, DD) are associated with certain clinical phenotypes. Although patients with DN mutations present with a T- and B-cell combined immunodeficiency (CID) phenotype that is primarily characterized by opportunistic infections (7/7),23 patients carrying HI mutations primarily present with a CVID picture of increased susceptibility to B-cell immunity–controlled infections (∼60%)16-22,40 ; patients with DD mutations most typically present with hematologic dyscrasias (38%, 5/13; cytopenias in 4, malignancies in 2). Although the clinical presentations of the 3 allelic variants cannot be completely differentiated from each other, they largely segregate with a particular pathophysiologic mechanism (HI, DN, or DD) affecting IKAROS function.
Similar to patients carrying HI defects, but in contrast to those with DN allelic variants, we detected virtually asymptomatic DD mutation–positive relatives of the index cases in ≥3 of the 4 extended families evaluated. Some of the asymptomatic relatives showed signs that could be interpreted as immunologic penetrance, despite the lack of symptoms (eg, B.I.1 had slightly diminished IgG levels; C.II.1 had reduced switched and nonswitched memory B cells). This type of presentation with incomplete immunologic and/or clinical penetrance is common in autosomal-dominant primary immunodeficiencies.16,41 Moreover, although all index cases clinically manifested symptoms within the first decade of life, late-onset disease, rather than nonclinical penetrance, cannot be ruled out.
In summary, germline heterozygous IKAROS dimerization defects acting through HI work via a unique pathophysiologic mechanism and may manifest during childhood with hematologic diseases consisting of cytopenias (ITP, AIHA, and/or neutropenia)/Evans syndrome and malignancies (T-ALL and Burkitt lymphoma), with limited infections and what appears to be incomplete clinical penetrance. These findings, together with previous reports of DNA binding HI mutations presenting primarily as CVID-like disease and DN defects presenting as CID, expand the genotype-phenotype spectrum of IKAROS-associated diseases with underlying novel pathophysiology mechanisms and distinctive immunological and clinical features.
Data sharing requests should be sent to Hye Sun Kuehn (hyesun.kuehn@nih.gov.
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Acknowledgments
The authors thank the patients and their families for contributions to the study.
This work was supported by the National Institutes of Health/Intramural Research Program, National Institutes of Health Clinical Center.
The content of this article does not necessarily reflect the views or policies of the US Department of Health and Human Services, nor does mention of trade names, commercial products, or organizations imply endorsement by the US government.
Authorship
Contribution: H.S.K. and S.D.R. designed the project and wrote the manuscript; C.C.-R., S.L., J.R., A.T., W.A.G., S.S., Z.J., E.G., K.B., T.A.F., J.V., and S.C. collected biologic and clinical data from patients and analyzed data; H.S.K., S.C.M., and S.G. performed experiments and analyzed data; J.E.N. and J.S. performed RNASeq and analyzed data; J.E.N., J.S., R.J.H., and T.S. performed next-generation sequencing and/or analyzed data; M.G., M.H., and L.L. assisted with the informed consent process and arranged for sample delivery; and E.G., J.R., C.C.-R., and S.C. provided samples and clinical information and analyzed data.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Hye Sun Kuehn, Immunology Service, Department of Laboratory Medicine, National Institutes of Health Clinical Center, Building 10, Room 2C306, 10 Center Dr, MSC1508, Bethesda, MD 20892; e-mail: hyesun.kuehn@nih.gov; and Sergio D. Rosenzweig, Immunology Service, Department of Laboratory Medicine, National Institutes of Health Clinical Center, Building 10, Room 2C306, 10 Center Dr, MSC1508, Bethesda, MD 20892; e-mail: srosenzweig@cc.nih.gov.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal