

Key Points
A DNA damage sensitizing gene SLFN11 promotes stalled fork degradation caused by DNA2 and MRE11 nucleases by inhibiting RAD51 accumulation.
Suppression of SLFN11 in Fanconi anemia cells attenuates FA phenotypes such as chromosomal breakages or cell cycle arrest on DNA damage.

Abstract
Fanconi anemia (FA) is a hereditary disorder caused by mutations in any 1 of 22 FA genes. The disease is characterized by hypersensitivity to interstrand crosslink (ICL) inducers such as mitomycin C (MMC). In addition to promoting ICL repair, FA proteins such as RAD51, BRCA2, or FANCD2 protect stalled replication forks from nucleolytic degradation during replication stress, which may have a profound impact on FA pathophysiology. Recent studies showed that expression of the putative DNA/RNA helicase SLFN11 in cancer cells correlates with cell death on chemotherapeutic treatment. However, the underlying mechanisms of SLFN11-mediated DNA damage sensitivity remain unclear. Because SLFN11 expression is high in hematopoietic stem cells, we hypothesized that SLFN11 depletion might ameliorate the phenotypes of FA cells. Here we report that SLFN11 knockdown in the FA patient-derived FANCD2-deficient PD20 cell line improved cell survival on treatment with ICL inducers. FANCD2−/−SLFN11−/− HAP1 cells also displayed phenotypic rescue, including reduced levels of MMC-induced chromosome breakage compared with FANCD2−/− cells. Importantly, we found that SLFN11 promotes extensive fork degradation in FANCD2−/− cells. The degradation process is mediated by the nucleases MRE11 or DNA2 and depends on the SLFN11 ATPase activity. This observation was accompanied by an increased RAD51 binding at stalled forks, consistent with the role of RAD51 antagonizing nuclease recruitment and subsequent fork degradation. Suppression of SLFN11 protects nascent DNA tracts even in wild-type cells. We conclude that SLFN11 destabilizes stalled replication forks, and this function may contribute to the attrition of hematopoietic stem cells in FA.
Introduction
The Schlafen (SLFN) family was first described as a set of homologous genes that are involved in T-cell development and inhibit cell growth.1 These genes are found only in mammals and a small number of nonmammalian species.2 A member of the SLFN family, SLFN11, is a putative DNA/RNA helicase ubiquitously expressed in the human body.3 Notably, its expression is often lost in primary cancers and in commonly used cancer cell lines by epigenetic silencing.4,5 Recent studies indicated that SLFN11 expression in cancer correlate with a favorable response to widely used anticancer drugs, such as irinotecan, cisplatin, etoposide, and poly(ADP-ribose) polymerase inhibitors, with a better prognosis for the patients.5-7 SLFN11 expression has the strongest association among the DNA repair proteins with the sensitivities to DNA damaging cancer chemotherapy drugs but not to non-DNA damaging agents.8 It has also been reported that SLFN11 associates with replication protein A (RPA) and negatively regulates RPA loading to chromatin, thereby disfavoring homologous recombination (HR) repair.9 Another study found that SLFN11 accumulates at stalled replication forks and blocks replication.10 SLFN11 may also affect protein translation by cleavage of a specific group of tRNAs, resulting in the abrogation of ATR kinase expression during the DNA damage response.11,12 These studies define SLFN11 as a guardian of the genome that controls cell fate decisions in response to DNA damage and replication stress. However, how SLFN11 exerts this function remains unclear.
Fanconi anemia (FA) is a rare hereditary disorder that is caused by mostly recessive mutations in any 1 of 22 FA genes identified thus far (FANCA-W), leading to hematopoietic stem cell failure and cancer predisposition.13,14 FA proteins act in the common FA pathway to repair interstrand crosslink (ICL) damage, and therefore FA cells are hypersensitive to ICL-inducing agents such as mitomycin C, cisplatin, and formaldehyde. In FA cells, ICL damage induces abnormally high levels of cell death, chromosome breakage, and checkpoint activation, with more cells arrested in G2 phase. It has been postulated that DNA damage caused by endogenous aldehydes results in senescence or cell death,13 leading to hematopoietic stem cell failure in FA. Alternatively, imprecise repair of such damage results in mutations or translocations that can lead to oncogenesis or leukemogenesis.
Besides ICL repair, some of the FA proteins including FANCA, FANCD1 (BRCA2), FANCD2, FANCR (RAD51), and FANCS (BRCA1) have a protective role in fork stability during replication stress.14-18 In contrast to overt replication blockage by ICLs, replication stress is a condition in which progression of DNA replication is halted by any kind of obstacle such as secondary DNA structures, conflicts with transcription, or oncogene activation.15 During prolonged replication stress experimentally induced by hydroxyurea (HU) treatment, which depletes deoxynucleotides, stalled replication forks are remodeled/reversed to form a 4-way junction structure (so-called “chicken foot”) by the actions of remodeling factors (eg, SMARCAL1, ZRANB3, HLTF) and RAD51.16-18 RAD51 and other FA proteins protect the reversed fork from degradation by DNA2 or MRE11 nuclease activities.19-22 The reversed fork and its processing are critical for restart of DNA replication, but excessive degradation results in genome instability. Although this subset of FA proteins also functions in the core machinery of HR and ICL repair, HR and fork protection are considered to be 2 distinct functions based on the existence of separation of function mutations.20,21 It is therefore an interesting question how these 2 FA protein functions interplay to prevent the pathophysiology of FA, such as the attrition of hematopoietic stem cells.
Given that SLFN11 gene expression promotes cell killing by ICL inducers, and FA cells display hypersensitivity to ICLs, we hypothesized that SLFN11 depletion could reverse the FA phenotypes. Indeed, loss of SLFN11 in FA cells partially restored ICL tolerance, which was associated with less pronounced mitomycin C (MMC)-induced chromosome breakage, as well as loss of CHK1 phosphorylation and cell cycle arrest in G2 phase. Surprisingly, we discovered that HU- or MMC-induced fork degradation in FANCD2−/− cells was also prevented by SLFN11 depletion. The fork degradation in wild-type cells was also mitigated. Moreover, we uncovered that the ATPase domain of SLFN11 is essential for promoting stalled fork degradation in FA cells because it affects RAD51 recruitment to the reversed fork.
Methods
Cell culture and reagents
PD20 cells and PD20 complemented with FANCD2 (kind gifts from Toshiyasu Taniguchi, Tokai University School of Medicine) were cultured in α−minimum essential medium (Nacalai Tesque) supplemented with 20% fetal calf serum (Thermo Fisher Scientific). HAP1 cells and derivatives were cultured in Iscove modified Dulbecco medium (Nacalai Tesque) supplemented with 10% fetal calf serum (Thermo Fisher Scientific). hTERT RPE-1, HCT116, or K562 and Jurkat cells were cultured in Dulbecco’s modified Eagle medium/Ham’s F-12 (Nacalai Tesque), McCoy’s 5a, or RPMI 1640, respectively, all of them supplemented with 10% fetal calf serum (Thermo Fisher Scientific). U2OS and HeLa cells were maintained in Dulbecco’s modified Eagle medium supplemented with 10% fetal calf serum (Thermo Fisher Scientific). Cells were treated with cis-diamminedichloridoplatinum(II) (CDDP; Nippon-kayaku), MMC (Kyowa-hakkou Kirin), hydroxyurea (HU) (MilliporeSigma), formaldehyde (Polysciences), CHK1 inhibitor (LY2606368; Cayman Chemical), Mirin (MilliporeSigma), and dimethyl sulfoxide (Nacalai Tesque) at the indicated concentrations. γ-Ray irradiation was performed with a 137Cs γ Cell Exactor (Nordion).
Antibodies
The following antibodies were obtained from commercial sources: rabbit polyclonal anti-FANCD2 (Novus); rabbit polyclonal anti-FANCA (Bethyl Laboratories); mouse monoclonal anti-SLFN11 (E4, Santa Cruz Biotechnology); rabbit polyclonal anti-DNA2 (Abcam); rabbit polyclonal anti–phospho-CHK1 (Ser345; Cell Signaling); mouse monoclonal anti–5-bromo-2′-deoxyuridine (BrdU) (anti-IdU; 347580; BD Biosciences); rat polyclonal anti-BrdU (anti-CldU; ab6326; Abcam); mouse monoclonal anti-RPA2 (9H8; Abcam); mouse monoclonal anti–DDDDK-tag (anti-FLAG, MBL); mouse monoclonal anti-tubulin (T5168; MilliporeSigma); and Alexa Fluor 594-conjugated anti-mouse IgG and Alexa Fluor 488–conjugated anti-mouse, rabbit, or anti-rat IgG antibodies (Molecular Probes). Rabbit polyclonal anti-RAD51 was a kind gift from Hitoshi Kurumizaka.
Generation of genome-edited cell lines and lentiviral transduction
Primers used in this study are listed in supplemental Table 1 available on the Blood Web site. For genome editing in HAP1 cells, the gRNA oligonucleotides for disruption of FANCD2,23 FANCA, or SLFN11 were cloned into a clustered regularly interspaced palindromic repeats (CRISPR) vector. The respective guide RNA (gRNA) oligonucleotides with a proto-spacer adjacent motif sequence were also cloned into pMK174 loxP-puro plasmid, which was used in the obligate ligation-gated recombination (ObLiGaRe) method as previously described.24 For genome editing in hTERT RPE-1 cells, the same gRNA targeting FANCD2 exon 11 as above was delivered. Two independent RPE1 FANCD2−/− cell lines were generated, designated clones #2B7 and #2C10. To express SLFN11-FLAG under tetracycline-controlled transcriptional activation, lentiviral plasmid CSIV-TRE-RfA-UbC-KT (a gift from Makoto Nakanishi and Hiroyuki Miyoshi) was used. SLFN11 expression was induced by treatment with 2 μg/mL doxycycline for >48 hours.
Small interfering RNA (siRNA) transfection
Transfection and cotransfection were carried out using Lipofectamine RNAiMax (Thermo Fisher Scientific). The individual siRNA duplexes used were as follows: siSLFN11 (SilencerSelect, s40702 and s40704; Thermo Fisher Scientific); siDNA2 (5′CAUCCAAUAUUUUCCCGUA-3′; MilliporeSigma)25 ; and Luciferase Control (ctrl; 5′UCGAAGUAUUCCGCGUACGTT-3′; MilliporeSigma).26
Immunoblotting and immunohistochemistry
Immunoblotting or detection of the RPA or RAD51 foci were done as previously described.27 For quantification of SLFN11-FLAG foci, cells were preextracted with 0.5% Triton X-100/CSK buffer (100 mM NaCl, 300 mM sucrose, 3 mM MgCl2, 10 mM piperazine-1,4-bis [2-ethanesulfonic acid]) for 1 minute on ice, and fixed with 4% paraformaldehyde for 30 minutes at room temperature. After blocking with 2% bovine serum albumin (BSA)/phosphate-buffered saline (PBS), the fixed cells were stained with a primary antibody diluted in 2% BSA/PBS for 1 hour at room temperature. Quantification of the RAD51, RPA, and FLAG foci was determined using IN Cell Investigator 1.6 software (GE Healthcare).
In situ proximity ligation assay for single-stranded DNA–protein interaction
For detection of nascent single-stranded DNA (ssDNA)–protein interaction, cells were labeled with 100 μM IdU for 30 minutes. After HU (4 mM, 5 hours) treatment, cells were washed with PBS 3 times and fixed with PBS containing 3% paraformaldehyde, 2% sucrose, and 0.5% Triton-X-100 for 30 minutes on ice and then permeabilized with 0.5% Triton X-100/PBS for 5 minutes at room temperature. After blocking with 2% BSA/PBS, cells were stained with the indicated primary antibody diluted in 2% BSA/PBS for 1 hour at room temperature. Proximity ligation assay (PLA) was performed with reagents from DuoLink Biosciences. Images were captured and quantified using an IN Cell Analyzer 2000 (GE Healthcare).
Cell growth assays
A total of 1.0 × 104 cells/mL were plated in 6-well plates and counted every 24 hours. Growth curves were made from 3 independent assays.
Cell survival assays
Cell survival assay was performed as previously described with some modification.27 Briefly, a defined number of HAP1 cells were treated in triplicate with the indicated concentrations of the DNA damaging agents for 24 hours. Cells were permitted to recover for 24 hours after treatment and then were plated with serial dilutions. After 7 to 10 days of incubation, cells were fixed and stained. A survival assay for PD20 cells was performed as previously described.28 In brief, PD20 fibroblasts were transfected with siRNAs, and 48 hours later, they were treated with the DNA damaging agents. After about 7 days, cells were fixed with 10% methanol (v/v)/0% acetic acid (v/v) and then stained for 5 to 10 minutes with 1% crystal violet in methanol. The absorbed dye was solubilized with methanol containing 1% sodium dodecyl sulfate, and the optical density was measured at 595 nm and normalized to the value without DNA damage agents.
Cell cycle analysis
PD20 cells or HAP1 cells were transfected with indicated siRNA. Forty-eight hours later, they were treated with 1 to 10 ng/mL MMC or 20 μM formaldehyde, as shown in the legends. For the BrdU pulse-label experiments, cells were cultured with 10 μM BrdU for 30 minutes and then fixed by 70% ethanol. Cells were permeabilized with 2 N HCl/0.5% Triton X-100 for 30 minutes and stained with mouse anti-BrdU antibody (1:100; BD Biosciences; 347580) followed by incubation with secondary Alex Fluor 488 anti-mouse IgG antibody, and propidium iodide (PI; 5 μg/mL) in PBS/1% fetal bovine serum. For PI staining, cells were fixed by 70% ethanol and stained with PBS containing PI (5 μg/mL). The analysis was done using a FACSCantoII Flow Cytometer (BD Biosciences).
DNA fiber assay
DNA fiber assays were carried out as described previously29 with some modification. Briefly, cells were cultured in the presence of 25 μM IdU for the indicated periods, washed three times with PBS, and then cultured in the presence of 250 μM CldU. The cells were trypsinized and resuspended in ice-cold PBS or 70% ethanol at a concentration of 7.5 × 105 cells/mL, and then 2 μL cell suspension was spotted onto an amino silane-coated glass slide. The cell suspension was briefly dried, and then 12 μL lysis solution (50 mM EDTA and 0.5% sodium dodecyl sulfate in 200 mM Tris-HCl, pH 7.5) was applied to the cells and gently mixed. DNA fibers on the slides were fixed in Carnoy’s solution for 10 minutes and then denatured in 2.5 M HCl for 60 minutes. The slides were immunostained with mouse anti-BrdU antibody (1:250; BD Biosciences; 347580) for IdU and rat anti-BrdU antibody (1:250; Abcam; ab6326) for CldU detection. The secondary antibodies used were Alexa Fluor 594- and 488-conjugated anti-mouse and -rat IgG antibodies (Thermo Fisher Scientific), respectively. Replication tract lengths were measured for more than 450 CldU-labeled DNA fibers (in 3 independent experiments) using an IN Cell Analyzer 2000 (GE Healthcare).
Chromosome breakage analysis
Cells were plated onto a 10-cm dish and treated with 40 ng/mL MMC for 24 hours. After the treatment, the cells were incubated with 0.1 mg/mL colcemid for 2 hours and harvested into a hypotonic solution (0.075 M KCl) and then fixed with Carnoy’s solution (3:1 methanol:acetic acid). Metaphase spreads were prepared by dropping the cells onto a clean glass slide and staining with Wright-Giemsa stain. Fifty metaphases were scored using a BZ-9000 Fluorescence Microscope (Keyence).
Statistical analysis
Prism (GraphPad Software) was used to perform unpaired, 2-tailed Student t tests.
Results
Fanconi anemia phenotypes in FANCD2 mutant cell line PD20 are rescued by SLFN11 knockdown
Many human cell lines often do not express SLFN11, likely by epigenetic silencing.4,5 However, normal tissues in the human body express SLFN11, and in particular, hematopoietic stem cells and progenitors display relatively higher levels of SLFN11 expression (Gene Expression Commons. https://gexc.riken.jp; supplemental Figure 1A).30 SLFN11 blocks replication in response to replication stress, and FA cells experience replication stress exacerbated by defective stabilization of stalled replication forks. We hypothesized that hypersensitivity to ICL inducers in cells deficient in the FA pathway could be reversed by SLFN11 depletion. To evaluate the cellular phenotypes of FA in the absence of SLFN11, we screened several cell lines by Western blot analysis and found that HAP1, Jurkat, and PD20 cell lines expressed SLFN11. SLFN11 expression was not detected in K562, HeLa, U2OS, hTERT RPE-1, and HCT116 cells (Figure 1A-B).
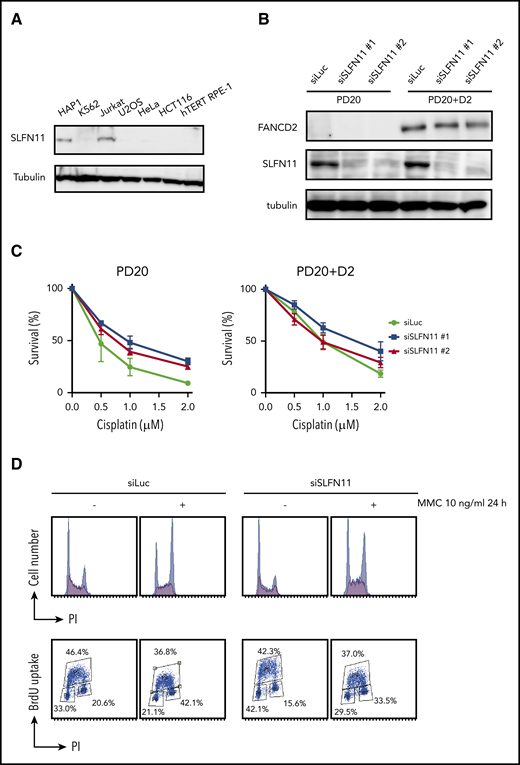
SLFN11 depletion by siRNA reverses cisplatin sensitivity in FANCD2 deficient PD20 cells. (A) SLFN11 expression in human cell lines as determined by western blotting. (B) SLFN11 expression in PD20 cells treated with siSLFN11#1 and #2. siLuc, siLuciferase. (C) Cell survival assay of PD20 and PD20 complemented with FANCD2. Cells were treated with 2 different siSLFN11. Means ± standard error of the mean (SEM) of 3 independent experiments are shown. (D) Cell cycle profile. Cells were transfected with siRNA oligos and 48 hours later treated with 10 ng/mL MMC for an additional 24 hours. Cells were then fixed in 70% ethanol, stained with anti-BrdU and PI, and analyzed using a FACS Canto II flow cytometer (BD Biosciences).
SLFN11 depletion by siRNA reverses cisplatin sensitivity in FANCD2 deficient PD20 cells. (A) SLFN11 expression in human cell lines as determined by western blotting. (B) SLFN11 expression in PD20 cells treated with siSLFN11#1 and #2. siLuc, siLuciferase. (C) Cell survival assay of PD20 and PD20 complemented with FANCD2. Cells were treated with 2 different siSLFN11. Means ± standard error of the mean (SEM) of 3 independent experiments are shown. (D) Cell cycle profile. Cells were transfected with siRNA oligos and 48 hours later treated with 10 ng/mL MMC for an additional 24 hours. Cells were then fixed in 70% ethanol, stained with anti-BrdU and PI, and analyzed using a FACS Canto II flow cytometer (BD Biosciences).
PD20 is a prototypic FANCD2-deficient cell line derived from SV40-immortalized fibroblasts of a FANCD2-mutated patient and maintains typical cellular phenotypes of FA, such as a severe sensitivity to ICL agents and prolonged G2 arrest induced by MMC. The higher accumulation of cells at G2 phase in the continuous presence of MMC (10 ng/mL, 24-48 hours) is one of the hallmark phenotypes in FA. siSLFN11 treatment successfully abrogated SLFN11 expression in PD20 and in PD20 complemented with FANCD2 (Figure 1B). As expected, SLFN11 depletion decreased cellular sensitivities to cisplatin, MMC, and formaldehyde, and G2 arrest after MMC treatment in PD20 cells (Figure 1C-D; supplemental Figure 1B). The FANCD2-complemented PD20 cells mildly increased cisplatin tolerance after SLFN11 depletion (Figure 1C).
SLFN11 knockout partially restores ICL tolerance in FANCD2−/− HAP1 cells
To confirm the PD20 results in other cells, we decided to generate isogenic knockout cell lines from diploid HAP1 cells using the Obligate ligation-gated recombination (ObLiGaRe) method (supplemental Figure 2A-B). Although HAP1 cells were originally derived from human haploid leukemia, it is known that they rather quickly convert to diploid status. The ObLiGaRe is a gene disruption protocol based on the efficient integration into the CRISPR-disrupted target locus by nonhomologous end joining (NHEJ) of a plasmid harboring a drug resistance gene cassette and the CRISPR gRNA sequence.24 First, we knocked out FANCD2 or FANCA in diploid HAP1 cells by integrating a puromycin resistance gene cassette. Then, both copies of SLFN11 were disrupted using a blastocidin S resistance gene cassette (supplemental Figure 2A-B). FANCD2−/−SLFN11−/− cells seemed to grow slightly faster than FANCD2−/− cells, which proliferated significantly slower than wild type (Figure 2A-B). In the wild-type background, SLFN11 deletion also made cells grow modestly faster. This observation may be related with the previously reported antiproliferative effects of SLFN family genes.1 Consistent with the observation in PD20, the SLFN11 disruption partially rescued cisplatin, MMC, and formaldehyde sensitivities in FANCD2−/− or FANCA−/− HAP1 cells (Figure 2C; supplemental Figure 2C). Additionally, we observed that SLFN11 knockout was also able to partially reverse HU sensitivity, but not ionizing irradiation (IR) sensitivity, in FANCD2−/− HAP1 cells (Figure 2C). It is interesting to note that the SLFN11 disruption conferred a slight resistance to CDDP, MMC, or HU exposure, but not IR, in parental HAP1 cells (Figure 2C). Furthermore, we complemented FANCD2−/−SLFN11−/− double knockout cells with a lentivirus-encoded, doxycycline-inducible SLFN11. Because SLFN11 is a putative DNA/RNA helicase, we also expressed a version of SLFN11 with mutations in the ATPase domain, as previously described9 (K605/D688A, Figure 2D-E). We found that reexpression of wild-type SLFN11, but not the ATPase-dead mutant, could enhance MMC and formaldehyde sensitivity (Figure 2F).
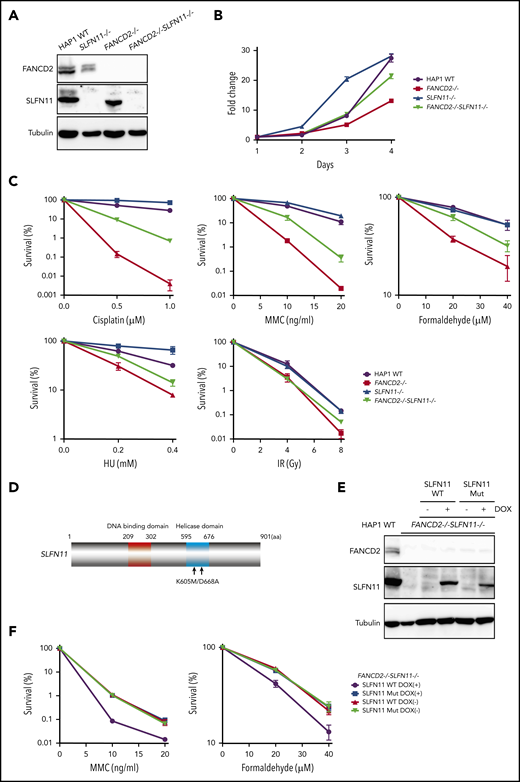
SLFN11 disruption partially reverses drug sensitivity in FANCD2−/−HAP1 cells. (A) FANCD2 and SLFN11 expression in knockout HAP1 cell lines. WT, wild type. (B) Cell growth curve of the indicated knockout HAP1 cell lines. Fold proliferation was calculated in 3 independent experiments. Means ± standard error of the mean (SEM) are shown. (C) Clonogenic cell survival assays in response to indicated doses of various genotoxic agents. MMC, mitomycin C. Means ± SEM of 3 independent experiments are shown. (D) A schematic of SLFN11 protein structure. Positions of the mutations in the ATPase domain are shown. (E) Expression of DOX-induced SLFN11 wild-type (WT) or ATPase dead mutant (Mut) protein in the FANCD2−/−SLFN11−/− HAP1 knockout cell line. Lentivirally transduced cells were selected and treated for 48 hours with doxycycline (DOX). (F) Clonogenic cell survival assays in FANCD2−/−SLFN11−/− cells transduced with SLFN11 wild-type or an ATPase dead mutant. DOX (2 μg/mL)-treated cells were exposed to the indicated doses of MMC or formaldehyde for 24 hours, and colonies were counted 5 to 7 days later. Means ± SEM of 3 independent experiments are shown.
SLFN11 disruption partially reverses drug sensitivity in FANCD2−/−HAP1 cells. (A) FANCD2 and SLFN11 expression in knockout HAP1 cell lines. WT, wild type. (B) Cell growth curve of the indicated knockout HAP1 cell lines. Fold proliferation was calculated in 3 independent experiments. Means ± standard error of the mean (SEM) are shown. (C) Clonogenic cell survival assays in response to indicated doses of various genotoxic agents. MMC, mitomycin C. Means ± SEM of 3 independent experiments are shown. (D) A schematic of SLFN11 protein structure. Positions of the mutations in the ATPase domain are shown. (E) Expression of DOX-induced SLFN11 wild-type (WT) or ATPase dead mutant (Mut) protein in the FANCD2−/−SLFN11−/− HAP1 knockout cell line. Lentivirally transduced cells were selected and treated for 48 hours with doxycycline (DOX). (F) Clonogenic cell survival assays in FANCD2−/−SLFN11−/− cells transduced with SLFN11 wild-type or an ATPase dead mutant. DOX (2 μg/mL)-treated cells were exposed to the indicated doses of MMC or formaldehyde for 24 hours, and colonies were counted 5 to 7 days later. Means ± SEM of 3 independent experiments are shown.
SLFN11 depletion lowers the number of ICL-induced chromosomal breaks and averts cell cycle arrest in FANCD2−/− HAP1 cells
We next analyzed the prolonged G2 arrest phenotype in the knockout HAP1 cell lines. Interestingly, similarly to the siSLFN11 experiment in PD20, the MMC- or formaldehyde-induced G2 phase arrest was significantly reduced by the SLFN11 knockout in FANCD2−/− HAP1 cells (Figure 3A; supplemental Figure 3A). In line with this observation, the levels of phosphorylated CHK1 on Serine 345 at 24 to 48 hours after MMC treatment were markedly reduced compared with FANCD2−/− cells (Figure 3B), suggesting earlier shutting off of the checkpoint signaling. This reduction of CHK1 phosphorylation was milder at earlier time points (ie, 2-6 hours) after MMC (supplemental Figure 3B). The MMC-induced CHK1 phosphorylation levels were reduced in SLFN11−/− cells compared with wild-type cells (Figure 3B).
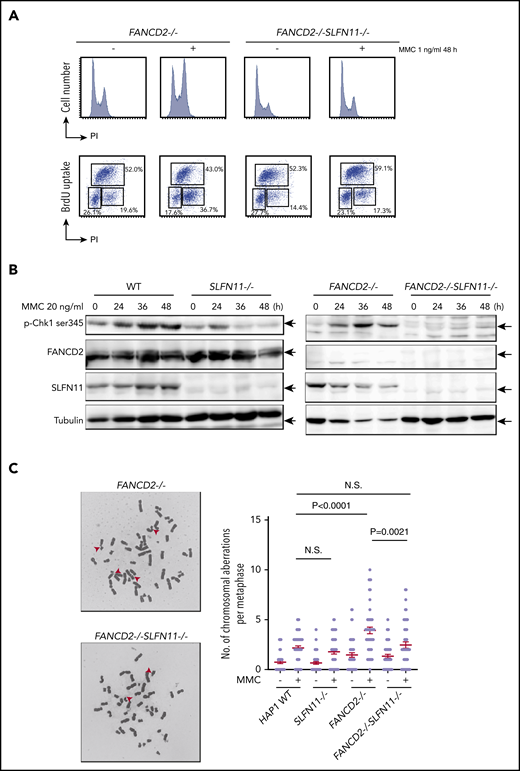
MMC-induced G2/M checkpoint is partially obviated by SLFN11 disruption. (A) Cell cycle profile. FANCD2−/− and FANCD2−/−SLFN11−/− cells were treated with 1 ng/mL MMC for 48 hours and pulse-labeled with BrdU. Cells were then fixed in 70% ethanol, stained with either PI alone (i) or with anti-BrdU and PI (ii), and analyzed using a FACS Canto II flow cytometer (Becton-Dickinson). (B) CHK1 activation by Western blotting. CHK1 phosphorylation on Serine 345 was examined at the indicated time points during 20 ng/mL MMC exposure in wild type, SLFN11−/−, FANCD2−/−, and FANCD2−/−SLFN11−/− HAP1 cells. The loading was equalized based on the amount of protein, and the weaker tubulin bands in FANCD2−/− cells were likely caused by the toxicity of MMC treatment. (C) Chromosome breakage analysis. Cells were subjected to 40 ng/mL MMC exposure for 24 hours. Each dot represents the number of chromosomal breaks in a single metaphase, and means ± SEM are shown. More than 50 metaphases were scored in each condition. Representative images of metaphases after MMC treatment are shown. P values were calculated by unpaired, 2-tailed Student t test.
MMC-induced G2/M checkpoint is partially obviated by SLFN11 disruption. (A) Cell cycle profile. FANCD2−/− and FANCD2−/−SLFN11−/− cells were treated with 1 ng/mL MMC for 48 hours and pulse-labeled with BrdU. Cells were then fixed in 70% ethanol, stained with either PI alone (i) or with anti-BrdU and PI (ii), and analyzed using a FACS Canto II flow cytometer (Becton-Dickinson). (B) CHK1 activation by Western blotting. CHK1 phosphorylation on Serine 345 was examined at the indicated time points during 20 ng/mL MMC exposure in wild type, SLFN11−/−, FANCD2−/−, and FANCD2−/−SLFN11−/− HAP1 cells. The loading was equalized based on the amount of protein, and the weaker tubulin bands in FANCD2−/− cells were likely caused by the toxicity of MMC treatment. (C) Chromosome breakage analysis. Cells were subjected to 40 ng/mL MMC exposure for 24 hours. Each dot represents the number of chromosomal breaks in a single metaphase, and means ± SEM are shown. More than 50 metaphases were scored in each condition. Representative images of metaphases after MMC treatment are shown. P values were calculated by unpaired, 2-tailed Student t test.
Importantly, MMC-induced chromosome breakage levels were also decreased in FANCD2−/−SLFN11−/− cells compared with FANCD2−/− cells (Figure 3C) or in FANCA−/−SLFN11−/− cells relative to FANCA−/− cells (supplemental Figure 3C). Thus, it is likely that SLFN11 exacerbates genome damage that prompts CHK1 activation and prolonged G2 phase accumulation in FANCD2−/− cells. We also observed that ICL sensitivity was less effectively ameliorated by a CHK1 inhibitor (LY2606368) in FANCD2−/−SLFN11−/− cells compared with FANCD2−/− cells (supplemental Figure 3D), suggesting that MMC-induced CHK1 activation is prevented in FANCD2−/−SLFN11−/− cells. This is in agreement with a previous report that treatment with CHK1 inhibitor rescued the ICL sensitivity in FA cells.31
SLFN11 enhances fork degradation in a manner dependent on DNA2 and MRE11
How does SLFN11 deficiency rescue FA phenotypes in FANCD2-deficient cells? It was reported that RAD51, BRCA1, BRCA2, and FANCD2 protect stalled replication forks from degradation by nucleases like DNA2 or MRE11 in cells treated with HU, and a stabilized replication fork confers chemo-resistance in BRCA-deficient cells.32 Consistently, it was also reported that DNA2 depletion reverses ICL sensitivity in FANCD2−/− and FANCL−/− cells.33,34 From these results, we hypothesized that SLFN11 deficiency may promote fork stability. To test this, we monitored the length of nascent DNA tracts in FANCD2-deficient cells with or without SLFN11 expression by the DNA fiber assay using the standard and accepted protocol as depicted in Figure 4A. Cells were sequentially pulse-labeled with IdU and CldU and then exposed to HU to stall active replication forks. The tract length of CldU-labeled DNA serves as an index of replication fork degradation. Intriguingly, we found that SLFN11 depletion by siRNA reversed the shortened tract length in PD20 cells treated with HU (Figure 4B). SLFN11 knockout also reversed nascent tract lengths in HU- or MMC-treated FANCD2−/− HAP1 cells (Figure 4C-D). We further carried out DNA fiber assays in FANCD2−/−SLFN11−/− HAP1 cells complemented with wild-type SLFN11 or the SLFN11 ATPase-dead mutant. We found that wild-type SLFN11 reexpression promoted fork degradation, but the ATPase dead mutant promoted fork degradation to a much lesser extent (Figure 4E). Next, we tested whether the protection from fork degradation conferred by SLFN11 depletion resulted in increased replication restart events after replication stress was removed. We used the experimental protocol to detect restart events as CldU-labeled tracts after HU treatment. Indeed, we found that the drastically decreased percentage of replication restart events in FANCD2−/− cells was nearly normalized in FANCD2−/−SLFN11−/− cells (Figure 4F). It is also notable that the fork degradation was also mildly mitigated in wild-type cells when SLFN11 was inactivated, and this resulted in higher fork restart events (Figure 4C,D,F).

SLFN11 deficiency prevents replication fork degradation. (A) A schema of the experimental protocol. (a) Overview of the experiment. Cells are sequentially pulse-labeled by IdU and CldU to visualize nascent DNA tracts and further treated with HU or MMC. The shortened length of the CldU tracts indicates the degree of fork degradation. (b) The stalled forks are remodeled and reversed into a 4-way junction form, which is a target of the nucleases. (c) The DNA fiber protocol. Pulse-labeled cells were fixed, spotted on a slide glass, and lyzed. The slide glass is tilted to spread DNA fibers. Finally, the IdU and CldU tracts are detected by specific antibodies and visualized by an In Cell Analyzer. (B) DNA fiber analysis to quantify fork degradation/protection. CldU tract length was measured in PD20 cells in which SLFN11 expression was knocked down by siRNA transfection with or without HU treatment (4 mM, 5 hours). The tract lengths were quantified in 3 independent experiments, and the cumulative results of more than 450 fibers with means ± standard error of the mean (SEM) are shown. P values were calculated by unpaired, 2-tailed Student t tests. HAP1 cells with the indicated genotypes were analyzed for fork degradation/protection in the presence of HU (C) or MMC (D). (E) HAP1 FANCD2−/−SLFN11−/− cells complemented with wild-type or an ATPase-dead SLFN11 mutant were analyzed by the DNA fiber assay. (F) Quantification of restarting forks in HAP1 FANCD2−/−SLFN11−/− cells. The percentage of restarting forks was calculated in the cumulative results of more than 300 fibers from triplicate experiments. Means ± SEM are shown. P values were calculated by unpaired, 2-tailed Student t tests.
SLFN11 deficiency prevents replication fork degradation. (A) A schema of the experimental protocol. (a) Overview of the experiment. Cells are sequentially pulse-labeled by IdU and CldU to visualize nascent DNA tracts and further treated with HU or MMC. The shortened length of the CldU tracts indicates the degree of fork degradation. (b) The stalled forks are remodeled and reversed into a 4-way junction form, which is a target of the nucleases. (c) The DNA fiber protocol. Pulse-labeled cells were fixed, spotted on a slide glass, and lyzed. The slide glass is tilted to spread DNA fibers. Finally, the IdU and CldU tracts are detected by specific antibodies and visualized by an In Cell Analyzer. (B) DNA fiber analysis to quantify fork degradation/protection. CldU tract length was measured in PD20 cells in which SLFN11 expression was knocked down by siRNA transfection with or without HU treatment (4 mM, 5 hours). The tract lengths were quantified in 3 independent experiments, and the cumulative results of more than 450 fibers with means ± standard error of the mean (SEM) are shown. P values were calculated by unpaired, 2-tailed Student t tests. HAP1 cells with the indicated genotypes were analyzed for fork degradation/protection in the presence of HU (C) or MMC (D). (E) HAP1 FANCD2−/−SLFN11−/− cells complemented with wild-type or an ATPase-dead SLFN11 mutant were analyzed by the DNA fiber assay. (F) Quantification of restarting forks in HAP1 FANCD2−/−SLFN11−/− cells. The percentage of restarting forks was calculated in the cumulative results of more than 300 fibers from triplicate experiments. Means ± SEM are shown. P values were calculated by unpaired, 2-tailed Student t tests.
Previous studies implicated nucleases such as DNA2 and MRE11 in fork degradation during HU treatment.19-22 We found that DNA2 knockdown or treatment with the MRE11 inhibitor Mirin could increase the CldU tract length in FANCD2−/− cells but could not further increase the tract length in FANCD2−/−SLFN11−/− cells (supplemental Figure 4A-C). As expected, DNA2 depletion reversed the sensitivity to ICL reagents in FANCD2−/− HAP1 cells (supplemental Figure 4D). Taken together, these results indicate that SLFN11 promotes fork degradation via its ATPase domain in combination with the nuclease activities that include DNA2 and MRE11.
The above-described data have clearly established that loss of SLFN11 expression impacts stalled fork degradation. However, most of the commonly used cell lines lack expression of SLFN11, and it is unclear whether exogenous introduced SLFN11 exerts a similar function. The hTERT RPE-1 cell line was derived from normal retinal epithelium and was found not to endogenously express SLFN11 (Figure 1A). We generated FANCD2−/− hTERT1 RPE-1 cells (supplemental Figure 5A) and achieved the expression of SLFN11 in this cell line using a doxycycline (DOX)-inducible system (Figure 5A). Interestingly, exogenous SLFN11 expression modestly decreased cell growth (Figure 5B). We also found that similar to other cell lines, FANCD2 knockout in hTERT RPE-1 cells rendered the forks vulnerable to degradation during prolonged HU treatment. Furthermore, SLFN11 expression slightly accelerated fork degradation in those cells (Figure 5C).
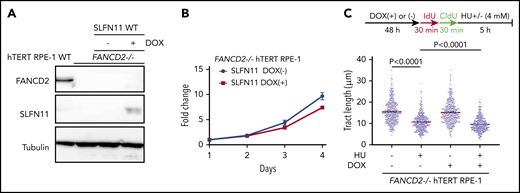
Effects of exogenously expressed SLFN11 in FANCD2−/− hTERT RPE-1 cells. (A) DOX-induced expression of wild-type SLFN11 in lentivirally transduced FANCD2−/− hTERT RPE-1 cells (clone #2B7). Cells were stimulated or not with doxycycline (2 μg/mL) for 48 hours. WT, wild-type hTERT RPE-1 cells. (B) Growth curve of FANCD2−/− hTERT RPE-1 cells transduced with wild-type SLFN11. Means ± standard error of the mean (SEM) of triplicate cultures with or without DOX are shown. (C) DNA fiber analysis to quantify fork degradation/protection. The CldU tract length was measured in FANCD2−/− RPE1 cells with or without expression of wild-type SLFN11. Cells were stimulated or not by HU treatment. Means ± SEM of more than 450 fibers were quantified from 3 independent experiments. P values were calculated by unpaired, 2-tailed Student t tests.
Effects of exogenously expressed SLFN11 in FANCD2−/− hTERT RPE-1 cells. (A) DOX-induced expression of wild-type SLFN11 in lentivirally transduced FANCD2−/− hTERT RPE-1 cells (clone #2B7). Cells were stimulated or not with doxycycline (2 μg/mL) for 48 hours. WT, wild-type hTERT RPE-1 cells. (B) Growth curve of FANCD2−/− hTERT RPE-1 cells transduced with wild-type SLFN11. Means ± standard error of the mean (SEM) of triplicate cultures with or without DOX are shown. (C) DNA fiber analysis to quantify fork degradation/protection. The CldU tract length was measured in FANCD2−/− RPE1 cells with or without expression of wild-type SLFN11. Cells were stimulated or not by HU treatment. Means ± SEM of more than 450 fibers were quantified from 3 independent experiments. P values were calculated by unpaired, 2-tailed Student t tests.
RAD51 loading onto resected forks is inhibited by SLFN11 in FANCD2−/− HAP1 cells
Previous studies showed that SLFN11 is recruited to stalled replication forks and forms foci.9,10 We confirmed this observation in HU-treated hTERT RPE-1 cells expressing exogenous wild-type SLFN11-FLAG protein by immunohistochemical analysis, using anti-FLAG antibody. Interestingly, levels of SLFN11 foci were higher in FANCD2−/− cells compared with the wild-type background, consistent with the role of SLFN11 in fork degradation in FANCD2−/− cells (supplemental Figure 5B-C).
Because it was proposed that SLFN11 may negatively affect chromatin loading or retention of RPA, thereby reducing HR activity,9 we next examined RPA and RAD51 foci formation in our knockout cell lines by immunofluorescence analysis. We found that RPA and RAD51 foci increased remarkably after MMC treatment (100 ng/mL, 24 hours) in FANCD2−/−SLFN11−/− cells compared with FANCD2−/− cells (Figure 6A) or SLFN11−/− compared with wild-type cells (supplemental Figure 6A). Thus, we hypothesized that SLFN11 may enhance replication fork degradation by impeding RAD51 recruitment to the stalled and reversed fork. Indeed, both RPA and RAD51 foci formation after treatment with HU (4 mM, 5 hours) were enhanced by SLFN11 disruption in both FANCD2−/− (Figure 6B) and wild-type cells (supplemental Figure 6B).
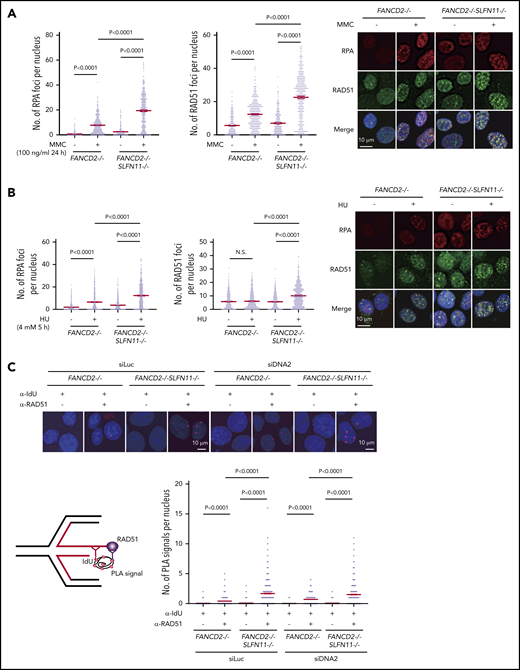
RPA and RAD51 recruitment to stalled forks in FANCD2−/−SLFN11−/−HAP1 cells. (A) RPA and RAD51 foci formation in FANCD2−/− and FANCD2−/−SLFN11−/− HAP1 cells treated or not with MMC (100 ng/mL, 24 hours). Means ± standard error of the mean (SEM) are shown. More than 300 nuclei were examined. Representative results from 2 independent experiments are shown. (B) RPA and RAD51 foci formation in FANCD2−/− and FANCD2−/−SLFN11−/− HAP1 cells on HU treatment (4 mM, 5 hours). More than 300 nuclei were examined. Representative results from 2 independent experiments are shown. (C) PLA between the nascent DNA (IdU tract) and RAD51. Cells were transfected with control or siRNA targeting DNA2 and then 48 hours later, cells were pulsed for 30 minutes with IdU and then treated with HU (4 mM, 5 hours). PLA was performed in a native condition as described previously.36 PLA performed with anti-IdU alone served as a negative control. Means ± SEM with representative images from 2 independent experiments are shown. P values were calculated by unpaired, 2-tailed Student t tests. A schematic illustration of the PLA assay is shown.
RPA and RAD51 recruitment to stalled forks in FANCD2−/−SLFN11−/−HAP1 cells. (A) RPA and RAD51 foci formation in FANCD2−/− and FANCD2−/−SLFN11−/− HAP1 cells treated or not with MMC (100 ng/mL, 24 hours). Means ± standard error of the mean (SEM) are shown. More than 300 nuclei were examined. Representative results from 2 independent experiments are shown. (B) RPA and RAD51 foci formation in FANCD2−/− and FANCD2−/−SLFN11−/− HAP1 cells on HU treatment (4 mM, 5 hours). More than 300 nuclei were examined. Representative results from 2 independent experiments are shown. (C) PLA between the nascent DNA (IdU tract) and RAD51. Cells were transfected with control or siRNA targeting DNA2 and then 48 hours later, cells were pulsed for 30 minutes with IdU and then treated with HU (4 mM, 5 hours). PLA was performed in a native condition as described previously.36 PLA performed with anti-IdU alone served as a negative control. Means ± SEM with representative images from 2 independent experiments are shown. P values were calculated by unpaired, 2-tailed Student t tests. A schematic illustration of the PLA assay is shown.
To examine the recruitment of RAD51 to reversed forks, PLA between an incorporated nucleotide analog and RAD51 has been used in recent studies.35,36 We pulse-labeled the nascent replication tract with IdU, and the potential association between RAD51 and IdU in resected ssDNA was assayed under native conditions using a PLA. After HU treatment, an increased proximity between nascent DNA (IdU) and RAD51 was detected in FANCD2−/−SLFN11−/− cells compared with FANCD2−/− cells (Figure 6C). To precisely measure the impact of SLFN11 on the PLA between IdU and RAD51, we also normalized the length of the IdU-labeled tracts by siDNA2 treatment. We confirmed that the siDNA2 treatment did not drastically alter the results (Figure 6C). Furthermore, signals in the IdU-RAD51 PLA assay were suppressed by wild-type SLFN11 expression compared with the ATPase dead mutant in FANCD2−/−SLFN11−/− cells (supplemental Figure 6C). Taken together, these data suggest that SLFN11 decreases RAD51 recruitment to the reversed fork in a transaction that requires the ATPase domain, which leads to fork degradation by DNA2 or MRE11 nuclease in FANCD2-deficient cells.
Discussion
In this study, we found that SLFN11 deletion rescued the enhanced DNA damage sensitivity associated with the loss of the FA pathway. The affected phenotypes included enhanced cell survival, reduced chromosome breakage, reduced G2 arrest, and reduced CHK1 phosphorylation. Recent studies identified several modulator genes that affect FA phenotypes including CHK1,31 ALDH2,37 BLM,38 USP48,39 PARI,29,40 Lnk,41 and TGF-β.42 Our study identified SLFN11 as a novel, additional type of modulator of FA.
We also found that loss of SLFN11 prevented degradation of stalled replication forks in FANCD2−/− deficient cells during HU or MMC treatment. In keeping with this, Chaudhuri et al32 reported that fork stability confers chemo-resistance in BRCA-deficient cells, and chromosome breakage was also suppressed by fork stabilization. Another possible mechanism for the phenotypic rescue might be HR repair, because SLFN11 inhibits HR repair by regulating RPA loading onto ssDNA. Because inhibition of the NHEJ pathway rescues the FA phenotype,31 there could be a possible relationship of SLFN11 with NHEJ factors. Although we favor the model that the fork stabilization mitigates the FA phenotype, these possibilities need to be tested in future studies.
We propose that the endogenous DNA damage that causes the FA phenotype in patients is exacerbated by the fork degradation via SLFN11 (please see our schematic summary in Figure 7). The fork degradation may aggravate the original impediment, such as aldehyde damage13,37 or R-loops that stall replication forks.43-46 The replication forks arrested at the ICL could be reversed and thus if the degradation is excessive, it may prevent the normal ICL processing such as unhooking. In other words, the inactivation of SLFN11 could make the repair process of the original damage more efficient. We reason that the higher levels of RPA foci may reflect the proposed role of SLFN11 in removing RPA from ssDNA,9 not the increase of ssDNA itself. We note that many studies examined fork degradation in an assay similar to ours using cell lines such as HeLa or U2OS that do not express SLFN11. In other words, the fork degradation can occur in cells without SLFN11 expression. This indicates that cells that lost SLFN11 expression perhaps activate another mechanism that promotes fork degradation. It would be interesting to identify such mechanisms by which SLFN11-negative cell lines promote fork degradation.
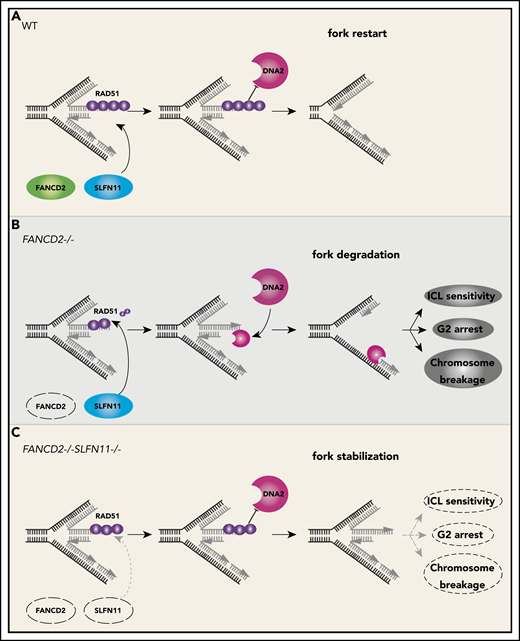
A schematic model of our results. In wild-type (WT) cells (A), the progression of the DNA replication forks is often hindered because of replication stress such as those caused by R-loops or formaldehyde damage. These are proposed endogenous sources of replication stress/DNA damage relevant to FA. The stalled forks undergo remodeling termed “fork reversal,” resulting in the 4-way structure. The reversed fork is digested by nucleases DNA2 or MRE11, leading to genome instability, but the degradation is prevented by FA proteins, including RAD51, FANCD2, and others. We propose SLFN11 is acting against fork protection by RAD51. In FA cells such as FANCD2−/− background (B), SLFN11 expression results in excess fork degradation and inefficient ICL repair, which causes the indicated FA phenotypes. The deletion of SLFN11 (C) places more RAD51 on the nascent DNA, enabling efficient fork protection and reversal of the FA phenotype.
A schematic model of our results. In wild-type (WT) cells (A), the progression of the DNA replication forks is often hindered because of replication stress such as those caused by R-loops or formaldehyde damage. These are proposed endogenous sources of replication stress/DNA damage relevant to FA. The stalled forks undergo remodeling termed “fork reversal,” resulting in the 4-way structure. The reversed fork is digested by nucleases DNA2 or MRE11, leading to genome instability, but the degradation is prevented by FA proteins, including RAD51, FANCD2, and others. We propose SLFN11 is acting against fork protection by RAD51. In FA cells such as FANCD2−/− background (B), SLFN11 expression results in excess fork degradation and inefficient ICL repair, which causes the indicated FA phenotypes. The deletion of SLFN11 (C) places more RAD51 on the nascent DNA, enabling efficient fork protection and reversal of the FA phenotype.
SLFN11 is expressed throughout the human body, and the expression levels appear to be high in hematopoietic stem cells. Thus, SLFN11 should have a profound impact on progression of the FA pathophysiology. If the SLFN11 function can be inhibited, this may provide a means to prevent progression of bone marrow failure in FA. It is notable that a group of FA patients who express CHK1 at relatively low levels show an attenuated MMC-induced G2 arrest and milder bone marrow failure. However, this has also resulted in myelodysplasia/leukemia, perhaps because of surviving cell clones with damaged genomes.31 Because SLFN11 inhibition should attenuate levels of DNA damage itself by suppressing fork degradation, it potentially is a better therapeutic strategy than CHK1 inhibition.
In summary, our results shed new light on the mechanism underlying fork stability and ICL sensitivity in FA cells. SLFN11 may be an integral agent of FA pathophysiology that enhances the DNA damage response and determines cell fate decisions in hematopoietic stem cells in which SLFN11 is highly expressed. It seems worth considering SLFN11 as a therapeutic target for developing novel treatments of FA.
All relevant data are available from the authors upon reasonable request.
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Acknowledgments
The authors thank James Hejna for critical reading and English editing; Junko Murai, Hiroyuki Sasanuma, and Andres Canela for discussions; Masami Tanaka and Kumi Johchi for technical and secretarial help; Toshiyasu Taniguchi, Hitoshi Kurumizaka, Makoto Nakanishi, and Feng Zhang for reagents; and Hiroshi Harada for allowing us to use the FACSCanto.
This work was supported by Japan Society for the Promotion of Science (JSPS) Kagaku Kenkyuhi Hojokin (KAKENHI) Grants, Takeda Science Foundation, Uehara Memorial Foundation, Astellas Foundation for Research on Metabolic Disorders, and Kyoto University Research Fund (Core Stage Back-Up). A.-K.B. was supported by National Institutes of Health grants 5R01 GM074917 and 1R01 GM134681 and C.B.R. is a trainee on Cancer Biology Training grant T32 CA009138 and a Scholar of the Achievement Rewards for College Scientists (ARCS) Foundation. Radiation Biology Center, Graduate School of Biostudies, Kyoto University, is a joint usage research center certified by the Ministry of Education, Culture, Sports, Science and Technology (MEXT), Japan.
Authorship
Contribution: M.T. planned the study; Y.O. carried out most of the experiments and analyzed the data with help from M.A., A.M., Y.T., A.L.M., Y.K., and A.T.-K.; C.B.R., A.S., and A.-K.B. generated the hTERT RPE-1 FANCD2−/− cell line; M.T.K. provided critical plasmids; and Y.O. and M.T. wrote the paper with help from C.B.R., A.S., and A.-K.B.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Minoru Takata, Graduate School of Biostudies, Kyoto University, Yoshidakonoe-cho, Sakyo-ku, Kyoto 606-8501, Japan; e-mail: takata.minoru.8s@kyoto-u.ac.jp.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal