

TO THE EDITOR:
Philadelphia chromosome (Ph)-like/BCR-ABL1–like acute lymphoblastic leukemia (ALL; Ph-like ALL) defines a subgroup of B-cell precursor ALL (B-ALL) lacking the BCR-ABL1 fusion, with a similar gene-expression profile to BCR-ABL1+ ALL and high risk of relapse.1-3 Ph-like ALL represents a genetically heterogeneous group, including a number of fusions, which involve the ABL-class genes: PDGFRB/A, CSF1R, ABL1, and ABL2.4-6 Preclinical studies have shown that leukemic cells from patients with these fusions respond well to tyrosine kinase inhibitors (TKIs),4 which has been confirmed in a number of patients, particularly those with EBF1-PDGFRB fusions.7-10 Here, we focus on the genetic and clinical features of a rare subset of ABL-class patients with the SSBP2-CSF1R fusion. In particular, we reveal the diversity of cytogenetic changes, giving rise to the fusion, highlighting that awareness of these variants is important for its accurate detection in light of TKI treatment options.
Patients in this study originated from 4 international study groups in the United Kingdom, The Netherlands, Germany, and the United States. All participating centers obtained local ethical committee approval and written informed consent in accordance with the Declaration of Helsinki. Demographic and clinical details are summarized in supplemental Table 1 (available on the Blood Web site). Cytogenetic analysis of diagnostic bone marrow was performed in local laboratories. Interphase fluorescence in situ hybridization (FISH) was carried out on the same samples, using commercially available PDGFRB break-apart (BA) probes. Involvement of SSBP2 and CSF1R was confirmed using bespoke probes (supplemental Figure 1). Copy-number abnormalities (CNAs) were determined using single-nucleotide polymorphism (SNP) arrays or array comparative genomic hybridization (aCGH). Expression of the SSBP2-CSF1R fusions was confirmed by reverse transcription–polymerase chain reaction (RT-PCR), whole-transcriptome (RNA sequencing [RNA-Seq]), or targeted RNA-Seq. Patients were assigned to the Ph-like subgroup according to results from low-density gene-expression array card or gene-expression profiling using U133A/U133 Plus 2.0 microarrays (Affymetrix, Santa Clara, CA) and predictive analysis of microarrays, as previously described.1,5,11
Here, we describe the variable genetic presentation among 16 B-ALL patients with fusion of SSBP2 at 5q14 to CSF1R at 5q33. Genetic data are provided in supplemental Table 2. Significantly, the fusion arose from a number of different abnormalities involving chromosome 5.
Seven patients (#1-7) showed balanced translocations: t(5;5)(q14;q33) (Figure 1A). In 4 of these cases (#1, #2, #3, #5), FISH using a PDGFRB BA probe indicated the presence of a rearrangement involving either PDGFRB or CSF1R, as both are located in close proximity on the long arm of chromosome 5 (5q). FISH analysis using in-house BA probes confirmed rearrangements of the CSF1R and/or SSBP2 genes in 4 of these patients tested with the PDGFRB BA probe (#1, #2, #3, #5), as well as 1 among the remaining cases (#7) (Figure 1C-D; supplemental Figure 2). The presence of the fusion transcript, SSBP2-CSF1R, was confirmed in all 6 of these cases tested by RT-PCR or RNA-Seq (supplemental Figure 3).
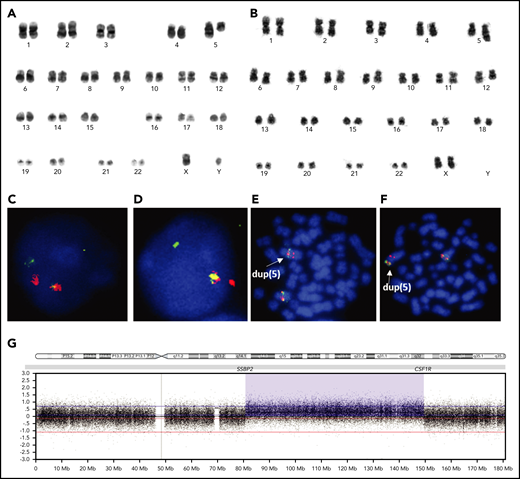
Karyograms, FISH, and SNP 6.0 profile of SSBP2-CSF1R+patients. (A) Karyogram from the diagnostic bone marrow of patient 3 showing that both copies of chromosome 5 are abnormal, consistent with the balanced translocation, t(5;5)(q14;q33). (B) Karyogram from the relapsed bone marrow of patient 9 showing that 1 copy of chromosome 5 is abnormal, consistent with dup(5)(q14q33). (C-D) FISH using SSBP2 and CSF1R BA probes, respectively, confirming the balanced rearrangement in patient 1. (E-F) FISH using SSBP2 and CSF1R BA probes, respectively, showing a partial duplication of both probes on the duplicated 5q in patient 9. (C-F) Original magnification ×100; DAPI, Spectrum Orange and Spectrum Green stain. (G) SNP array profile of chromosome 5 in patient 9, showing duplication of the long arm of chromosome 5, with breakpoints in SSBP2 at 5q14 and CSF1R at 5q33. This duplication was conserved between diagnosis and relapse.
Karyograms, FISH, and SNP 6.0 profile of SSBP2-CSF1R+patients. (A) Karyogram from the diagnostic bone marrow of patient 3 showing that both copies of chromosome 5 are abnormal, consistent with the balanced translocation, t(5;5)(q14;q33). (B) Karyogram from the relapsed bone marrow of patient 9 showing that 1 copy of chromosome 5 is abnormal, consistent with dup(5)(q14q33). (C-D) FISH using SSBP2 and CSF1R BA probes, respectively, confirming the balanced rearrangement in patient 1. (E-F) FISH using SSBP2 and CSF1R BA probes, respectively, showing a partial duplication of both probes on the duplicated 5q in patient 9. (C-F) Original magnification ×100; DAPI, Spectrum Orange and Spectrum Green stain. (G) SNP array profile of chromosome 5 in patient 9, showing duplication of the long arm of chromosome 5, with breakpoints in SSBP2 at 5q14 and CSF1R at 5q33. This duplication was conserved between diagnosis and relapse.
A single patient presented with a balanced paracentric inversion of 5q, inv(5)(q15q33) (#8), who also expressed the SSBP2-CSF1R fusion by RT-PCR.
Four patients (#9-12) showed duplication of 5q, dup(5)(q14q33), either in their karyotype (#9, #10), and/or by aCGH (#11)/SNP array (#9, #12). In 2 cases (#9, #11), FISH using BA probes for PDGFRB/CSF1R and SSBP2 showed signal patterns consistent with duplication (Figure 1E-F).
Arrays identified the breakpoints of the duplications in patients #9 and #11 to be located within SSBP2 and CSF1R (Figure 1B,G); however, in patient #12, SNP array showed the proximal breakpoint to be within SSBP2, whereas the distal breakpoint was telomeric of CSF1R, as confirmed by FISH (supplemental Figure 4B). However, despite both techniques indicating 3 intact copies of CSF1R, expression of the SSBP2-CSF1R fusion (e16-e12) was confirmed by targeted RNA-Seq (supplemental Figure 4C), in line with the 2 other duplications tested by RT-PCR (#9, #11).
As previously shown by Boer et al,6 the SSBP2-CSF1R fusion in patient #13, positive by FISH and RT-PCR, was associated with a series of gains and losses along 5q, indicating that complex rearrangements produced the fusion in this case.
The chromosomal rearrangement underlying the fusion was unknown in 3 cases of SSBP2-CSF1R identified by RT-PCR (#14-16).
As expected, all cases tested (n = 10) showed a Ph-like gene-expression profile. Among the 11 cases with SNP array/aCGH results, deletions of IKZF1 were the most common secondary CNA (n = 7).
Clearly, these 16 cases were highly selected; thus, to establish the true incidence of SSBP2-CSF1R fusions among B-ALL, unbiased FISH screening of the complete childhood ALL trial, UKALL2003, comprising 2791 patients, found only a single case (#3) within the B-other-ALL subgroup.12 Similarly, Boer et al6 reported only 2 patients with SSBP2-CSF1R fusion among a B-ALL cohort of 574 cases, confirming it to be extremely rare at an incidence of 0.1% to 0.5% in childhood B-ALL and 1% to 2% in Ph-like ALL.4,6,13
There was an equal gender distribution within the cohort. Median age was 10.5 years (range, 2-29 years) and median white cell count was 17.9 × 109/L (range, 2.6 × 109/L to 301.8 × 109/L), with 13 of 15 children classified as NCI high risk.
Although treated on a range of protocols, all patients achieved complete remission (CR). Among 14 patients with available minimal residual disease (MRD) data, 7 remained positive at the end of induction (EOI). Among 9 patients classified as high risk by the National Cancer Institute (NCI) and treated on the high-risk arms of their respective protocol, 5 remained MRD+ at EOI.
The outcome of the cohort is shown in Figure 2. Six patients remain alive in long-term first CR, whereas 3 patients relapsed (#3, #4, #9). Relapse was not related to the type of chromosomal abnormality. All 3 relapsed patients had been treated on high-risk protocols at diagnosis, although only 2 (#3, #9) were MRD+ at EOI. Patient #3 relapsed on consolidation therapy, failed to achieve CR2, and subsequently died. Patient #9 suffered an isolated bone marrow relapse 1 month after the end of treatment. She was treated according to the ALLR3 trial high-risk arm, achieved CR2, and became MRD− by day 35. Detection of the SSBP2-CSF1R fusion prompted the addition of imatinib (400 mg per day) to her regimen, with the intention of maintaining remission until unrelated donor stem cell transplant. She died 11 weeks after relapse from infection (Escherichia coli septicemia). The MRD− patient #4 remains alive in CR2. It is of interest to note that MRD values at EOI did not always correlate with outcome, reinforcing the variability in outcome for these patients.
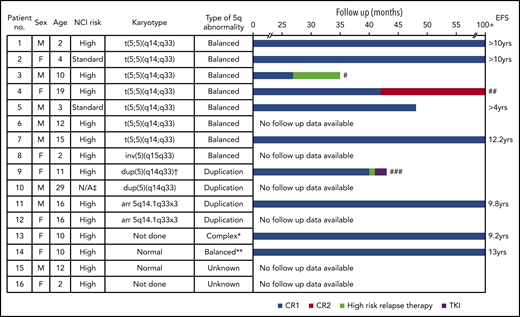
Features and outcome for patients with SSBP2-CSF1R fusions. Bar chart shows follow-up for patients in months to event or censoring. ‡Adult patient. †Karyotype at relapse. *aCGH suggests chromothripsis. **No CNAs of chromosome 5 were observed by SNP array of patient 14, indicating likely balanced rearrangement. #Death without CR2. ##Patient remains in CR2. ###Death from infection in CR2. CR1, complete remission 1; CR2, complete remission 2; EFS, event-free survival; F, female; M, male; N/A, not applicable.
Features and outcome for patients with SSBP2-CSF1R fusions. Bar chart shows follow-up for patients in months to event or censoring. ‡Adult patient. †Karyotype at relapse. *aCGH suggests chromothripsis. **No CNAs of chromosome 5 were observed by SNP array of patient 14, indicating likely balanced rearrangement. #Death without CR2. ##Patient remains in CR2. ###Death from infection in CR2. CR1, complete remission 1; CR2, complete remission 2; EFS, event-free survival; F, female; M, male; N/A, not applicable.
This study has reinforced the rarity of SSBP2-CSF1R fusions in childhood B-ALL and confirmed their restriction to the Ph-like subgroup, and has highlighted the diversity of genetic mechanisms by which the SSBP2-CSF1R fusion may occur: through translocation, inversion, duplication, or other complex rearrangements involving 5q31∼33. Although RNA-based techniques, including RT-PCR and RNA-Seq, have accurately identified the fusion in all cases tested, in view of its rarity, few study groups have the resources to apply these techniques routinely. Thus, for definitive accuracy of detection, an integrated genetic approach, incorporating >1 technique, is preferable.
Unlike other ABL-class fusions, notably EBF1-PDGFRB,9 some SSBP2-CSF1R+ patients responded well to risk-adapted therapy and achieved long-term event-free survival.
Many clinical study groups now advocate the addition of TKI to treatment schedules when a patient is identified with an ABL-class fusion at diagnosis. Understanding the full range of genetic mechanisms generating these fusions to enable effective screening is crucial. Larger collaborative studies are in progress to validate the prognosis and treatment options for the range of ABL-class fusions.
Requests for data may be e-mailed to the corresponding author.
The online version of this article contains a data supplement.
Acknowledgments
The authors thank member laboratories of the UK Cancer Cytogenetic Group for providing cytogenetic data and material. Primary childhood leukemia samples used in this study were provided by the Blood Cancer UK Childhood Leukaemia Cell Bank and the Dutch Childhood Oncology Group Biobank.
This work was supported by Blood Cancer UK (formerly Bloodwise, United Kingdom) and by the Oncode Institute (M.L., J.M.B., M.L.d.B.).
Authorship
Contribution: C.S. and C.J.H. designed the study; C.S., C.J.H., K.R., J.M.B., and A.V.M. analyzed and interpreted data; G.G., D.S., A.V., C.M., R.H., Z.T., R.D., G.E., G.C., B.S., M.L., and M.L.d.B. provided genetic and clinical data; and all authors approved the final manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Christine J. Harrison, Leukaemia Research Cytogenetics Group, Translational and Clinical Research Institute, Newcastle University Centre for Cancer, Faculty of Medical Sciences, Newcastle University, Level 6, Herschel Building, Brewery Lane, Newcastle upon Tyne NE1 7RU, United Kingdom; e-mail: christine.harrison@newcastle.ac.uk.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal