

TO THE EDITOR:
Axicabtagene ciloleucel (axi-cel) is an autologous anti-CD19 chimeric antigen receptor T-cell (CAR-T) therapy approved for the treatment of patients with relapsed or chemo-refractory large B-cell lymphomas (LBCL). Axi-cel induced durable remissions with a ZUMA-1 (Safety and Efficacy of KTE-C19 in Adults With Refractory Aggressive Non-Hodgkin Lymphoma) overall response rate (ORR) and complete response rate (CR) of 83% and 58%, respectively, with 39% progression-free survival (PFS) 2 years postinfusion.1,2 However, the majority of axi-cel–treated patients will ultimately experience disease progression. Only limited data exist on patients with progressive disease (PD) after anti-CD19 CAR-T therapy for LBCL.3,4 Because axi-cel currently accounts for the majority of CAR-T infusions in the United States, data regarding therapeutic options after progression are of great interest.5,6
The US Lymphoma CAR-T Cell Consortium is a group of 17 US centers certified for treatment of patients with axi-cel. We previously reported on 275 axi-cel patients treated with standard-of-care axi-cel, finding a best ORR and CR rate of 82% and 64%, respectively.7 Median PFS was 8.3 months, and nonrelapse mortality was 4.4%. Here we report on outcomes of patients within the cohort who experienced PD. The 17 centers of the consortium each obtained independent institutional review board approval for this retrospective study. The study was conducted in accordance with the guidelines of the International Conference on Harmonisation and the Declaration of Helsinki.
The date of PD was defined as the date of clinical or radiologic relapse. After a median follow-up of 12.9 months, 136 patients (49%) experienced PD after axi-cel treatment. In these patients, best response to axi-cel treatment was 36% CR and 31% partial response (PR); 28% were primary refractory. Patients with PD were more likely to be younger, male, had received more prior lines of therapy, and had a high-risk International Prognostic Index and Eastern Cooperative Oncology Group performance status of 2 to 4 before axi-cel treatment compared with patients who did not have PD (supplemental Table 1, available on the Blood Web site). Patients who would have been ineligible for ZUMA-1 based on comorbidities at leukapheresis were enriched in the PD cohort. Rates of grade 3 or higher cytokine release syndrome and neurotoxicity and pre–axi-cel bridging therapy did not differ between PD and non-PD patients. Median time to progression after axi-cel infusion was 91 days (95% confidence interval [CI], 83-93).
The impact of CD19 loss in patients with LBCL treated with CAR T has not been well described. Demonstration of CD19-positive disease was not required prior to ZUMA-1 enrollment, and pretreatment CD19 status was not associated with response.2 However, recent data reported CD19-negative disease at axi-cel PD in ∼25% of patients.8,9 In our cohort, tissue biopsy to confirm progression was performed in 85 (63%) of 136 patients. Measurement of CD19 expression was assessed by using flow cytometry and/or immunohistochemistry at each center. Cutoff for CD19 positivity was determined locally and varied widely between our centers (supplemental Table 2). Of those biopsied, 61 (72%) had CD19 assessed, and 18 (30%) of 61 were CD19 negative, only 1 of whom was CD19 negative before axi-cel treatment. A multivariable logistic regression model assessing impact of age, sex, and axi-cel response (either CR or PR) on CD19 negativity at PD found patients with CD19-negative PD were more likely to be younger (odds ratio for 1-year decrease of 1.06; 95% CI, 1.01-1.11; P = .02) and female (odds ratio, 6.67; 95% CI, 1.75-33.3). Response to axi-cel was not associated with CD19-negative lymphoma on biopsy (P = .1). CD19-negative status at PD was not associated with inferior overall survival (OS) from the time of progression (139 vs 218 days; P = .4).
To assess the impact of baseline variables on post-PD OS, we fit a multivariable Cox proportional hazards model. Variable clustering was performed via the Hoeffding D statistic, and variable selection was performed via the lasso method (glmnet package in R software, version 3.6.2). Median OS from date of PD was 180 days (95% CI, 105-242) (Figure 1A). Patients with response to axi-cel had increased survival after axi-cel progression (hazard ratio [HR], 0.45; 95% CI, 0.29-0.71; P = .0005); patients with severe cytokine release syndrome (grade 3 or higher) (HR, 5.39; 95% CI, 2.48-11.7; P = 2.2 × 10−5) and bridging therapy between leukapheresis and axi-cel infusion (HR, 2.11; 95% CI, 1.32-3.39; P = .002) had decreased survival (Figure 1B-D). In patients who initially responded to axi-cel (n = 91), a Cox model of time to axi-cel PD as a continuous variable trended toward improved survival post-PD (HR for 1 month increase in time to PD of 0.86; 95% CI, 0.73-1.01; P = .07).
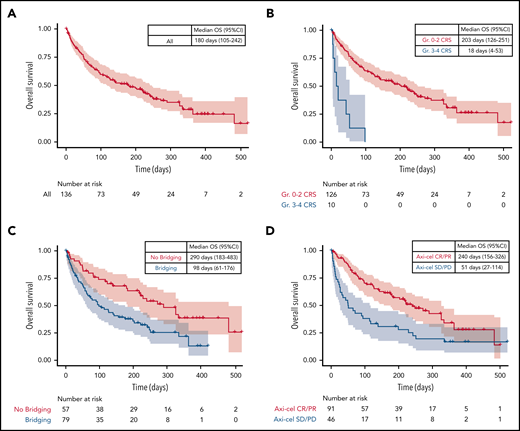
Factors affecting survival after axi-cel progression. (A) Unstratified Kaplan-Meier analysis of OS after axi-cel progression. (B-D) OS stratified according to cytokine release syndrome (CRS), bridging therapy and response to axi-cel. Gr, grade; SD, stable disease.
Factors affecting survival after axi-cel progression. (A) Unstratified Kaplan-Meier analysis of OS after axi-cel progression. (B-D) OS stratified according to cytokine release syndrome (CRS), bridging therapy and response to axi-cel. Gr, grade; SD, stable disease.
Data regarding subsequent therapy after axi-cel treatment were available in 135 (99%) PD patients. Selection of treatment was determined by the treating physician. To allow for analysis of combined modalities, therapies were categorized according to systemic therapy if given in conjunction with localized therapy (eg, concurrent checkpoint inhibitor and radiation); if multiple systemic therapies were combined, primary therapy was determined by the more frequently used therapy within the cohort (supplemental Table 3). One hundred patients (74%) received further therapy, and 35 (26%) received supportive care. The most-used therapies in first salvage post–axi-cel were: checkpoint inhibitor based (n = 30), lenalidomide based (n = 27), chemotherapy (n = 17), and radiation (n = 10). Other therapies included targeted treatments such as venetoclax (n = 1), brentuximab vedotin (n = 2) or ibrutinib (n = 2), novel therapies (n = 8), steroids (n = 1), second CAR-T on clinical trial (n = 1), and allogeneic stem cell transplant (n = 1). In total, 8 patients proceeded to allogeneic stem cell transplant after axi-cel PD, 3 of whom remain in CR.
First salvage therapies post–axi-cel PD received by ≥10 patients were selected for further analysis. Median time to initiating therapy after progression was 21 days and did not differ according to therapy. Overall, best response rates for these patients were 29% ORR, with 17% CR, and median PFS was 55 days (95% CI, 47-86). Response rates according to therapy are presented in Table 1. A univariate log-rank test comparing checkpoint inhibitor–based therapy vs a combination of radiation-, chemotherapy-, and lenalidomide-based therapy reported a trend toward increased PFS in patients treated with checkpoint inhibitor–based therapy (P = .1) (supplemental Figure 1). Response to checkpoint inhibitors did not differ according to aggressive lymphoma histology (supplemental Figure 2), despite pembrolizumab only being approved for the treatment of relapsed/refractory PMBL.
Treatment outcomes for first therapy given after axi-cel progression
| Therapy | CR | ORR | Median PFS (95% CI), d | Median OS (95% CI), d |
|---|---|---|---|---|
| Checkpoint inhibitor based (n = 28*) | 18% | 46% | 88 (35-282) | 331 (168-477) |
| Chemotherapy (n = 17) | 12% | 18% | 51 (21-64) | 104 (51-231) |
| Lenalidomide based (n = 27) | 19% | 19% | 48 (33-84) | 139 (45-NE) |
| Radiation (n = 10) | 20% | 30% | 58 (20-149) | 220 (20-NE) |
| Therapy | CR | ORR | Median PFS (95% CI), d | Median OS (95% CI), d |
|---|---|---|---|---|
| Checkpoint inhibitor based (n = 28*) | 18% | 46% | 88 (35-282) | 331 (168-477) |
| Chemotherapy (n = 17) | 12% | 18% | 51 (21-64) | 104 (51-231) |
| Lenalidomide based (n = 27) | 19% | 19% | 48 (33-84) | 139 (45-NE) |
| Radiation (n = 10) | 20% | 30% | 58 (20-149) | 220 (20-NE) |
NE, not evaluable.
Two patients received checkpoint inhibitors but did not have response data available.
In this subgroup analysis of the US Lymphoma CAR-T Consortium cohort, we report the largest experience of treatment outcomes after axi-cel failure for LBCL. The OS data included here establish a benchmark for future trials in patients with progression after standard-of-care CAR T for LBCL. Potential survival benefit in patients with longer response to axi-cel suggests differences in tumor biology in those with progression at later time points. As with prior studies, in this large cohort, we found CD19 loss in ∼30% of cases at the time of progression. Standardization of CD19 assessment will likely improve our understanding of mechanisms of CAR-T failure in the future. In those who received therapy post–axi-cel, PFS was poor, with a median PFS of first therapy of only 55 days. Although checkpoint inhibitor–based therapy seemed to be the most effective of the therapies used, enthusiasm was tempered due to the lack of OS benefit.
There are several limitations to this study beyond its retrospective nature. Therapy decisions, including whether to offer treatment, were not prospectively suggested. The choice of therapy could be influenced by multiple factors, including physician preference, cost, insurance coverage, and distance from treating center. Patient factors, such as cytopenias, also may have affected treatment choice and duration of therapy after axi-cel progression.10 In the future, monitoring CAR-T cell blood expansion or quantifying tumor burden pre–axi-cel could suggest mechanisms of axi-cel failure and better guide choice of subsequent therapy. The optimal management after CAR-T progression requires prospective studies.
Please contact the corresponding author for original data.
The online version of this article contains a data supplement.
Acknowledgment
This study was supported by a Stanford Blood and Marrow Transplantation support grant from the National Institutes of Health, National Cancer Institute (P01 CA049605).
Authorship
Contribution: J.Y.S., S.D., A.P.R., B.T.H., and D.B.M. contributed to the study concept; J.Y.S., S.D., M.D.J., B.T.H., and D.B.M. were responsible for collection and assembly of data; J.Y.S., S.D., A.P.R., B.T.H., and D.B.M. were responsible for study design; and all authors provided study materials or patients, analyzed and interpreted data, contributed to writing of the manuscript, and gave final approval of the manuscript.
Conflict-of-interest disclosure: M.D.J. reports consultancy for Kite/Gilead, and Novartis. S.D. has served on the advisory board for Kite/Gilead and Atara Biotherapeutics. L.J.N. has received honorarium from Bayer, Celgene, Genentech, Gilead/Kite, Janssen, Juno, Novartis, TG Therapeutics, and Spectrum Therapeutics; and received research support for Celgene, Genentech, Janssen, TG Therapeutics, and Karus Therapeutics. J.M. reports consultancy for Merck, Celgene/Juno, Pharmacyclics/Janssen, Bayer, Genentech, Kite/Gilead, Bristol Myers Squibb, Alexion, Seattle Genetics, Pfizer, Fosunkite, Innovent, BeiGene, and Kyowa; been on the speakers bureau for Pharmacyclics/Janssen, Bayer, Genentech, Kite/Gilead, Kyowa, Seattle Genetics, Acrotech, BeiGene, Verastem, AstraZeneca, and Celgene; and received research support from Kite Pharma, Celgene, Portola, Incyte, Genentech, Pharmacyclics, Seattle Genetics, Janssen, and Millennium. A.G. has received research support from Kite/Gilead and Amgen; and has been an advisory board member/consultant for Kite/Gilead, Amgen, Celgene, EUSA, and Wugen. Y.L. reports consultancy for Kite, Gilead, Novartis, JUNO, Celgene, Janssen, Bluebird Bio, and Legend BioTech; and been a member of the Data and Safety Monitoring Board for Sorrento. O.O. is a scientific advisor for Kite/Gilead and Pfizer. K.A.D. has received research support from Kite/Gilead, Juno Therapeutics, and Genentech. S.S.N. has received research support from Kite/Gilead, Merck, BMS, Cellectis, Poseida, Karus, Acerta, and Unum Therapeutics; has served as an advisory board member/consultant for Kite/Gilead, Merck, Celgene, Novartis, Unum Therapeutics, Pfizer, Precision Biosciences, Cell Medica, Calibr, Allogene, Incyte, and Legend Biotech. F.L.L. serves as a scientific advisor for Kite/Gilead, Novartis, BMS/Celgene, Amgen, Allogene, GammaDelta Therapeutics, Calibr, and Wugen; reports consultancy for Cellular BioMedicine Group Inc.; has received research funding from Kite/Gilead; and has institutional patents, pending and granted, in his name for improvements on CAR-T therapy. B.T.H. reports research funding and consultancy with Kite/Gilead. D.B.M. reports consultancy for Kite/Gilead, Juno/Celgene, and Novartis; and research support from Kite/Gilead. The remaining authors declare no competing financial interests.
Correspondence: David Miklos, Division of Blood and Marrow Transplantation, Stanford University, 300 Pasteur Dr, Stanford, CA 94305; e-mail: dmiklos@stanford.edu.
